

Abstract
β-Site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1) is an aspartyl protease best known for its role in generating the amyloid-β peptides that are present in plaques of Alzheimer's disease. BACE1 has been an attractive target for drug development. In cultured embryonic neurons, BACE1-cleaved N-terminal APP is further processed to generate a fragment that can trigger axonal degeneration, suggesting a vital role for BACE1 in axonal health. In addition, BACE1 cleaves neuregulin 1 type III, a protein critical for myelination of peripheral axons by Schwann cells during development. Here, we asked whether axonal degeneration or axonal regeneration in adult nerves might be affected by inhibition or elimination of BACE1. We report that BACE1 knock-out and wild-type nerves degenerated at a similar rate after axotomy and to a similar extent in the experimental neuropathies produced by administration of paclitaxel and acrylamide. These data indicate N-APP is not the sole culprit in axonal degeneration in adult nerves. Unexpectedly, however, we observed that BACE1 knock-out mice had markedly enhanced clearance of axonal and myelin debris from degenerated fibers, accelerated axonal regeneration, and earlier reinnervation of neuromuscular junctions, compared with littermate controls. These observations were reproduced in part by pharmacological inhibition of BACE1. These data suggest BACE1 inhibition as a therapeutic approach to accelerate regeneration and recovery after peripheral nerve damage.
Introduction
Axonal transection in peripheral nerves is followed by degeneration of the distal axonal stump. The interrupted axons of the proximal stump retain the potential for subsequent regeneration (Ramon y Cajal, 1928). The extent of functional restoration depends on the nature of the injury, the species, and the age of the animals. In the most favorable lesions—nerve crush rather than nerve cut—using young rodents, regenerated peripheral nervous system (PNS) axons reinnervate the target tissues after relatively short periods. However, after comparable nerve injuries in humans, axonal regeneration is slow and often functionally incomplete (for review, see Höke, 2006; Gordon et al., 2009; Griffin et al., 2010). Axonal regeneration is influenced by the intrinsic growth state of neurons (Hammarlund et al., 2009), local axonal protein synthesis (Yoo et al., 2010), cytoskeletal organization (Ertürk et al., 2007), growth factors (Geremia et al., 2010), extracellular matrix, and the clearance of myelin debris from the injured nerve (Schäfer et al., 1996; Brushart et al., 1998; Mears et al., 2003; Vargas et al., 2010). The contribution of myelin debris in inhibiting axonal regeneration is extensively documented in the CNS (Filbin, 2003). In turn, these factors variably influence the latency period before initiation of axonal growth, rate of axonal outgrowth, specificity of target reinnervation, and the speed of recovery (Ramon y Cajal, 1928; McQuarrie, 1978; Brushart, 1993; Jacob and McQuarrie, 1993; Seijffers et al., 2007). In damaged human nerves that require long distance regeneration, shortening the latency period is unlikely to substantially contribute to faster recovery. However, manipulation of molecular pathways that speed the rate of axonal regeneration would be a highly desirable therapeutic approach (Griffin et al., 2010). Various signaling pathways have been suggested to improve axonal regeneration in the PNS (for review, see Chen et al., 2007; Seijffers et al., 2007; Shim and Ming, 2010), but no molecular or pharmacological therapy demonstrating efficacy exists for injured nerves in human.
β-Site amyloid precursor protein (APP) cleaving enzyme 1 (BACE1) is a transmembrane aspartyl protease that cleaves several membrane proteins, including APP implicated in Alzheimer's disease. BACE1 cleaves APP to generate a soluble N-terminal fragment, N-APPβ, and a C-terminal fragment that is further processed by the γ-secretase complex to generate amyloid-β peptides (Wong et al., 1997; Vassar et al., 1999). BACE1 knock-out peripheral nerves are hypomyelinated (Hu et al., 2006; Willem et al., 2006), most likely as a result of reduced cleavage and signaling of neuregulin 1 type III (Michailov et al., 2004; Taveggia et al., 2005).
In cultured neurons, reduction of APP by genetic deletion and by RNA interference has been shown to increase neurite outgrowth (Young-Pearse et al., 2008). Paradoxically, increasing soluble APP also increases neurite outgrowth (Araki et al., 1991; Perez et al., 1997).
BACE1 activity has been reported to affect axonal health. During withdrawal of nerve growth factor from cultured embryonic neurons, BACE1-dependent proteolytic fragment of N-APP has been reported to induce axonal degeneration (Nikolaev et al., 2009). This recent work supports the idea that a reduction in the level of N-APP by inhibition of BACE1 activity could preserve axons.
Here, we investigated nerve fiber degeneration and regeneration in injured sciatic nerve of mice with reduced BACE1 activity.
Materials and Methods
Animals.
BACE1 knock-out (KO) mice and control wild-type (WT) littermates used in this study were on a mixed 129/BL6 line and for paclitaxel treatment on a C57 background, as previously described (Cai et al., 2001; Savonenko et al., 2008). For a subset of experiments, BACE1 KO mice were crossed to mice expressing yellow fluorescent protein (YFP) driven by the thy1.2 neuronal promoter (line YFP-H) (Feng et al., 2000). In these animals, a small proportion (3–10%) of neurons in the ventral horn and dorsal root ganglia express YFP. YFP-positive axons were examined in the sciatic nerves of BACE1 KO-YFP mice and WT-YFP littermates. A total of 220 mice (8–12 weeks of age) were used in different aspects of this study. In each experiment, approximately equal numbers of females and males were used and we saw no bias for female or male. The animal surgeries and experimental protocols were approved by the Animal Care and Use Committee of the Johns Hopkins University School of Medicine.
Sciatic nerve injury.
Mice were deeply anesthetized with isoflurane and left sciatic nerves in the mid thigh or at the sciatic notch level were exposed and crushed by pulling tight a loop of 8-0 nylon black sutures for 30–40 s. The crush sites were marked with a 10-0 nylon black suture going through the epineurium only. For sciatic nerve transection, a small ∼2 mm piece of nerve was dissected out and the two nerve stumps were turned away from each other to prevent axonal regeneration to distal stumps. The distal stumps and contralateral nerves were collected 1–15 d after transection.
Paclitaxel administration.
Paclitaxel (LC Laboratories) was dissolved in 100% ethanol. Equal volumes of paclitaxel solution and cremophor (Sigma-Aldrich) were vigorously vortexed for 10 min. Ice-cold saline (80% of the final volume) was added to freshly made paclitaxel/cremophor solution immediately before injection. The 30 mg/kg body weight of paclitaxel in this solution was injected intravenously via tail vein at three times per week for 2 weeks. Mice (n = 3 per genotype) had no significant weight loss.
Acrylamide administration.
Acrylamide (Sigma-Aldrich) was dissolved in drinking water at 250 ppm. Animal were intoxicated with acrylamide for 4 weeks. Sciatic nerves and hindfeet were harvested from intoxicated WT and BACE1 KO mice (n = 4 per genotype) and processed for plastic sectioning or immunohistochemistry as described below.
Axonal regeneration in vivo assays.
Regeneration into the distal stump was assessed by several complementary techniques. We analyzed the distribution of the axotomy-induced neuronal protein GAP43, a marker that labels the proximal stump and regenerating sprouts of crushed axons and is enriched in the growth cones. In longitudinal sections of the crushed nerves, we also assessed the distribution within the distal stump of two other axonally transported markers—Neurobiotin and the neuronally expressed YFP (Feng et al., 2000). We also examined electron microscopy of transverse sections of the nerve at defined intervals after axotomy. Finally, we measured the time required for axons of different classes to reach and reinnervate their targets. These techniques allowed us to assess separately the latency to the onset of regeneration, the rate of outgrowth, and the time to reinnervation of targets. Technical details of each method are described below.
Neurobiotin labeling.
In a subset of experiments, we used the exogenous tracer Neurobiotin (Vector Laboratories), which is rapidly transported intra-axonally. This method labels axons, including sprouts of regenerating axons entering the distal stump (Lapper and Bolam, 1991; Jacquin et al., 1992). Nerves were first crushed as described above, and then 2.5% Neurobiotin was injected via glass micropipettes (25 μm tip) into nerves proximal to the original crush sites at 5 d later. Three to 4 h later, animals were perfused as detailed below and nerves were collected. Neurobiotin transported along regenerating axons was detected histochemically.
Sciatic nerve transplantations.
We performed reciprocal transplantation of WT nerve segments into BACE1 KO recipients and of BACE1 KO segments into WT recipients. In each transplantation, a 10 mm nerve segment from the donor nerve was removed. The left sciatic nerve of each mouse recipient was transected and the donor nerve segment was promptly transplanted into the host sciatic nerve and sutured in place with 10-0 nylon in proximal-distal orientation. Two weeks later, the nerves were collected and 2 mm segments in the middle of donor nerves and host nerves were processed for examination of axonal regeneration in transverse plastic sections.
Implanting of Alzet pumps for infusion of BACE1 inhibitor.
Alzet pumps were filled, primed, and implanted according to our previously published protocol (Farah, 2004; Farah and Easter, 2005). Briefly, Alzet pumps (model 2001 of Alzet osmotic pumps; Durect Corporation) were filled with 200–220 μl of BACE1 inhibitor IV (Calbiochem) at concentrations of 1–4 mg/ml or with WAY 258131 (Malamas et al., 2010) at doses of 10–30 mg/ml. The latter compound was synthesized by the NeuroTranslational team at the Brain Science Institute at Johns Hopkins University. Both inhibitors were dissolved in DMSO. According to the manufacturer's data, the pump continuously supplied 1–30 μg/h inhibitor for 7 d. To minimize the delay between implantation and the onset of release, the filled pumps were incubated in 1× PBS at 37°C for 4 h immediately before implantation. The mice were anesthetized by inhalation of isoflurane, and an incision, ∼1.5 cm long, was made in the skin on top of the interscapular space. A filled Alzet pump was implanted subcutaneously into each mouse and incisions were closed with 9 mm stainless-steel wound clips.
Tissue processing.
For fluorescent microscopy and immunostaining, mice were deeply anesthetized with 10% chloral hydrate and killed by transcardial perfusion with 1× PBS followed by 2% paraformaldehyde in 0.1 mm phosphate buffer, pH 7.4, for 10–15 min. Tissues were then postfixed in the same fixative for 4–16 h at 4°C. Sciatic nerves and gastrocnemius muscles were cryoprotected in 30% sucrose in 1× PBS for overnight and quickly frozen in cold 2-methylbutane. Tissues were frozen sectioned on a cryostat at thickness of 10 or 20 μm for sciatic nerves and 50 μm for gastrocnemius muscles. In addition, the hindfeet were decalcified, cryoprotected in 30% sucrose in 1× PBS, and sectioned at 50 μm thickness. Sciatic nerves were sectioned longitudinally using a Leica cryostat (model CM3050S; Leica Microsystems). Gastrocnemius muscles and foot were cross-sectioned by a freezing sliding microtome (Microm HM 450; Thermo Fisher Scientific).
To visualize YFP-positive degenerating and regenerating axons, animals were perfused as described above and sciatic nerves were collected. Whole-mounted sciatic nerves were imaged under a confocal microscope and Z-stack images were collected through the depth of the nerves.
For immunocytochemistry, nonspecific antibody binding was blocked by 5% goat serum/0.3% Triton (Sigma-Aldrich) in 1× PBS for 1–2 h at room temperature. Sections were then incubated with primary antibodies (Table 1) overnight at 4°C. After 3× PBS washes, sections were incubated with appropriate secondary antibodies at room temperature for 1 h, washed three times in PBS, mounted (Prolong Antifade kit; Invitrogen), and coverslipped. Appropriate secondary, anti-mouse, -rat, or -rabbit antibodies conjugated with Alexa Fluor 488 or 647 (Invitrogen) were used. For immunoperoxidase labeling, sections were treated with 0.3% H2O2 before staining to quench activity of endogenous peroxidase. Primary antibodies were detected with ABC kit. Images of immunostained tissues were acquired by either standard light microscope, laser-scanning confocal microscope (model LSM 510; Carl Zeiss), or AxioImager with an Apotome (Carl Zeiss).
Table 1.
Primary antibodies used in this study
| Primary antibody | Source | Dilution |
|---|---|---|
| β-Tubulin type III | Covance | 1:1000 |
| GAP43 | Millipore | 1:2000 |
| Iba1 | Wako | 1:1000 |
| CD68 | Serotec | 1:1000 |
| CRGP | Millipore | 1:1000 |
| MBP | Millipore | 1:2000 |
| Neurobiotin | Vector Laboratories | 1:1000 |
| Phosphorylated neurofilament (NF 160) | Abcam | 1:2000 |
| Synaptophysin | Millipore | 1:1000 |
For ultrastructural analyses, nerves were postfixed in OsO4 and embedded in Epon. Cross sections, at 1 μm thickness, were stained for toluidine blue and examined under light microscopy with 100× oil-immersion objective. Seventy-nanometer-thin sections were obtained and stained for citrate/uranyl acetate. Electron micrographs were acquired using a Hitachi 7600 or Zeiss Libra transmission electron microscope.
Quantification.
Under electron microscope, we identified 100 degenerating fibers per genotype (50 per nerve). The degenerating fibers were systematically chosen by scanning the EM section in lines from left to right and imaging fibers at 8000–12,000×. For each degenerating myelinated fiber (defined by having degenerating myelin within a single basal lamina), we counted the number of axonal sprouts growing beneath the basal lamina, and thus these were axonal sprouts per Bungner's band.
At the light-microscopic level, regenerating axons were counted at 10 and 15 d after crush in 1 μm toluidine blue-stained transverse plastic sections taken 6–8 mm distal to the crush site. Axons were counted in each of four fields of view at 100× that were randomly chosen for each nerve (n = 3 for each WT and BACE1 KO nerves). A total number of regenerating axon per nerve was calculated by multiplying the average density of axons in the sampled area of each nerve by the cross-sectional area of that nerve.
Reinnervation of neuromuscular junctions were examined in WT and BACE1 KO gastrocnemius muscles 10 d after sciatic nerve crush at midthigh level. Muscles sections (at 50 μm thickness) were stained for synaptophysin (to mark presynaptic terminals) and α-bungarotoxin (to mark postsynaptic receptors). Two hundred neuromuscular junctions per genotype (n = 3 animal per genotype) were scored as denervated (no overlap of presynaptic and postsynaptic staining), partially reinnervated (some overlap of presynaptic and postsynaptic staining), and fully reinnervated (overlap of presynaptic and postsynaptic staining comparable with unoperated nerves).
Dorsal root ganglia explants.
Postnatal day 4 (P4) to P5 dorsal root ganglia (DRGs) were dissected out of WT and BACE1 KO mice, and were maintained in Neurobasal medium containing 2% B27 supplement and 50 ng/ml NGF (all reagents from Invitrogen). In some of the WT DRG explants, 50 nm to 1 μm BACE1 inhibitor IV was added daily to the media. At least eight explants were used for each condition. Axonal outgrowth of the DRGs was monitored daily under phase-contrast microscopy, and images at 2 and 4 d after culture were acquired with Openlab software. The axon lengths were measured from the borders of the explants borders to the tips of axons using ImageJ (http://rsb.info.nih.gov/ij/). An average of all axonal lengths for each explant was then determined as described previously (Nguyen et al., 2009).
Macrophage culturing and in vitro phagocytosis.
Mice (8–10 weeks of age) were killed by CO2 inhalation. Peritoneal cells were extracted by injection of RPMI 1640 media plus 10% FCS (both from Invitrogen) into the peritoneal cavity, followed immediately by withdrawal. Cells were plated on coverslips in the same media for overnight. Polystyrene beads with 2 μm diameters were coated with mouse IgG (5 mg/ml; Equitech Bio) for 1 h at 37°C. Coated beads were centrifuged onto adherent macrophages at a ratio of 10:1 for 1 min at 50 × g. After 3 or 5 min incubation at 37°C, noningested beads were washed three times with cold 1× PBS and cells were fixed in 3.7% formalin solution. Ten fields of view at 20× were photographed by bright field, and beads ingested by each cell were counted in 200 macrophages per animal (n = 3 per genotype).
Statistical analysis.
All statistical analyses were performed with Student's t test. SigmaPlot software was used to perform the analyses, and any value of p < 0.05 was scored as statistically significant. Graphed data are presented as mean ± SEM.
Results
Lack of axonal protection in Wallerian degeneration and in paclitaxel- and acrylamide-induced neuropathies in BACE1 KO mice
We tested whether genetic deletion of BACE1 could protect axons in adult mice in the fashion similar to that described after growth factor withdrawal during development (Nikolaev et al., 2009). We first asked whether the BACE1 KO mice exhibited delay in Wallerian degeneration after axotomy. As has been previously described (Waller, 1852, Ramon y Cajal, 1928; Stoll et al., 1989; George and Griffin, 1994; Beirowski et al., 2004, 2005), we observed that after transection the axon and the surrounding myelin of the nerve fibers in the distal segment (the nerve segment beyond the injury) underwent Wallerian degeneration (Fig. 1). At 24 h after injury, most axons were intact, but by 48 h after injury, the axonal cytoskeleton and the axoplasm disintegrated into granular and amorphous debris (Fig. 1) (George and Griffin, 1994; Bierowski et al., 2005), and the myelin sheaths in the distal stumps began to segment into the characteristic ovoids. Notably, very few axons of BACE1 KO and WT littermates exhibited an intermediate stage in degradation. Over the next several days, the axonal debris was removed from the distal stump. We found that the initial disintegration of the axoplasm and myelin segmentation followed the same time course in BACE1 KO and WT littermate injured nerves (Fig. 1).
Figure 1.
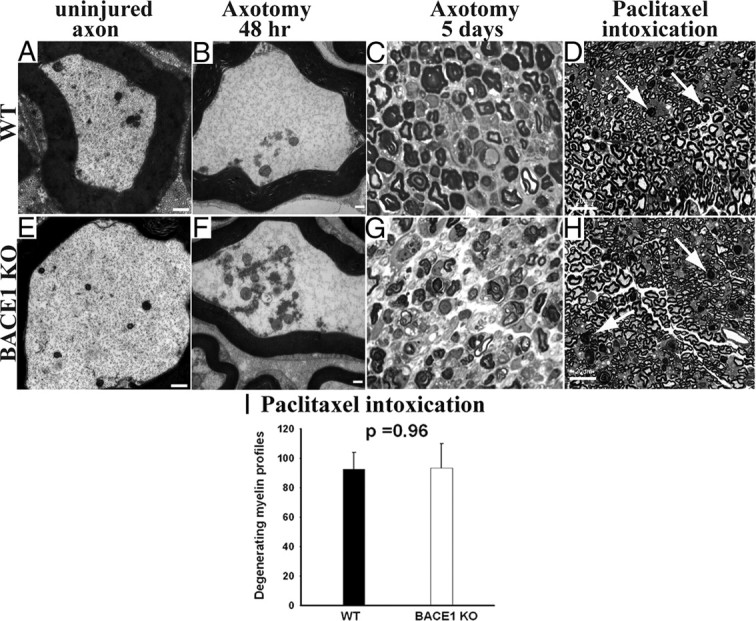
Similar degree of axonal and myelin degeneration in WT and BACE1 KO sciatic nerves after axotomy-induced Wallerian degeneration and intoxication with paclitaxel. A, B, E, and F are electron micrographs, and C, D, G, and H are semithin (1 μm) plastic sections stained with toluidine blue. A, Uninjured WT axon with normal axoplasm. B, WT at 48 h after crush showing conversion of the axonal cytoskeleton to granular debris. C, WT nerve at 5 d after crush showing degeneration of myelin. D, WT nerve intoxicated with paclitaxel. The arrows point to degenerating myelinated fibers. E, Uninjured BACE1 KO axon with normal axoplasm. F, BACE1 KO at 48 h after crush showing conversion of the axonal cytoskeleton to granular debris, similar to WT in B. G, BACE1 KO nerve at 5 d after crush showing degeneration of myelin. H, BACE1 KO nerve intoxicated with paclitaxel. The arrows point to degenerating myelinated fibers. I, Quantification of degenerating myelinated fibers in the whole cross-sectional area of nerves of mice intoxicated with paclitaxel. N = 3 per genotype. Values are mean ± SEM. Scale bars: A, B, D, F, 500 nm; C, F, G, H, 100 μm.
To examine axonal degeneration in neuropathy models, groups of BACE1 KO and WT littermate mice were given paclitaxel, a chemotherapy agent that causes axonal degeneration (Roytta and Raine, 1985, 1986), or the neurotoxin acrylamide (Nguyen et al., 2009). Both qualitatively and quantitatively, we observed a similar degree of myelinated fiber degeneration in WT and BACE1 KO nerves (Fig. 1). Furthermore, unmyelinated axons degenerated comparably in both genotypes. Additionally, absence of BACE1 activity did not protect from the early axoterminal degeneration at neuromuscular junctions in BACE1 KO mice exposed to acrylamide (data not shown).
Accelerated clearance of axonal and myelin debris in BACE1 KO mice
Although the time to initiation of axonal breakdown and to myelin ovoid formation was not different, the subsequent clearance of axonal and myelin debris was markedly faster in the BACE1 KO mice. To evaluate clearance of axonal debris in BACE1 KO nerves, we crossed the BACE1 KO mice to mice expressing YFP in a small subset of the axonal population (Feng et al., 2000). We selected line H from the panel of fluorescent mice lines because it has been previously shown that a small fraction (3%) of the axons are labeled with YFP (Feng et al., 2000; Beirowski et al., 2004, 2005; Vargas et al., 2010). Investigating axonal degeneration in nerves marked with YFP had the advantage of allowing us to analyze whole mounts of nerves, so that the proximal and the whole distal segment of each nerve were visualized throughout the depth of the nerves. At 7 d after cut, little YFP-positive axonal debris remained in the BACE1 KO-YFP nerves. In contrast, much more was present in the littermate WT-YFP nerves (Fig. 2). In addition, at 10 d after crush, axonal debris was more completely cleared in BACE1 KO-YFP nerves after a crush injury than in the WT littermates (Fig. 2).
Figure 2.

Enhanced clearance of axonal debris and enhanced axonal regeneration in YFP-BACE1 KO nerves compared with YFP-WT nerves after axotomy. These images are projections of Z-stacks from whole-mounted nerves to show the YPF-positive axonal debris and YFP-positive regenerated axons through the depth of the nerves. A, YFP-WT nerve at 7 d after transection. Fragmented YFP debris (arrowhead) is present. B, YFP-BACE1 KO nerve at 7 d after transection. YFP debris (arrowhead) is mostly cleared from the nerve. C, YFP-WT nerve at 10 d after crush. Fragmented YFP debris (arrowhead) is present at a segment 10 mm distal to the crush and few regenerated YFP-positive axons (arrow) are growing beneath the debris. D, YFP-BACE1 KO nerve at 10 d after crush. Numerous regenerated YFP-positive axons are present at segment 10 mm distal to crush site and (arrows). There are scanty clumps of YFP-positive axonal debris remaining. Scale bars, 100 μm. Laser-scanning intensity was increased in C and D to reveal small-diameter YFP-positive regenerated axons.
Myelin debris is known to impair axonal regeneration in the CNS, and PNS myelin has a similar effect in vitro and in vivo (Boivin et al., 2007; Barrette et al., 2008; Vargas et al., 2010). We asked whether reduced BACE1 activity affected the clearance of myelin debris from the distal stump. At 5 d after crush, more myelin basic protein (MBP) was degraded in BACE1 KO compared with WT as assessed histologically (Fig. 3) as well as by Western blots (data not shown), and by 15 d after crush clearance of myelin debris was more complete (Fig. 3; compare also Fig. 7A–D). One micrometer plastic sections showed less myelin debris in the distal stump of BACE1 KO nerves, and the remaining myelin ovoids were shorter. These differences correlated with a more rapid transition of macrophages in BACE1 KO nerves to a post-phagocytic foamy character, in which the macrophages contained numerous clear lipid droplets, representing cholesterol ester from digested myelin (Fig. 3D). By 15 d after crush, few macrophages remained in the distal stumps of BACE1 KO mice, and those remaining were predominantly around the endoneurial blood vessels and beneath the perineurium. In contrast, distal stumps of nerves from wild-type mice still had numerous phagocytic macrophages scattered throughout the endoneurial space.
Figure 3.

Enhanced phagocytosis by BACE1 KO macrophages both in vivo and in vitro. A–F are micrographs of distal stumps at 5 d after injury. A and D are stained for Iba1 (ionized calcium-binding adaptor molecule 1), a marker for macrophages. B and E are electron micrographs. C and F are stained for MBP. G and H are bright-field images of cultured peritoneal macrophages. A, The majority of Iba1-positive macrophages in WT nerve are elongated at 5 d after cut. B, Undigested myelin debris and ovoids are present in WT nerve at 5 d after crush. C, WT nerve with abundant elongated MBP-positive myelin debris at 5 d after cut. D, Iba1-positive BACE1 KO macrophages are larger in size and appear activated at 5 d after cut. E, Activated macrophages filled with lipid droplets (arrow) are present in BACE1 KO nerves at 5 d after crush. F, BACE1 KO nerve with broken down MBP-positive myelin debris at 5 d after cut. G, WT macrophages after incubation with IgG-coated beads for 5 min. H, BACE1 KO macrophages after incubation with IgG-coated beads for 5 min. I, Quantification of IgG-coated beads per individual cells after 5 min of phagocytosis. Data are from three independent experiments. Values are mean ± SEM. Scale bars: A, D, 10 μm; B, E, 2 μm; C, F, 20 μm; G, H, 100 μm.
Figure 7.
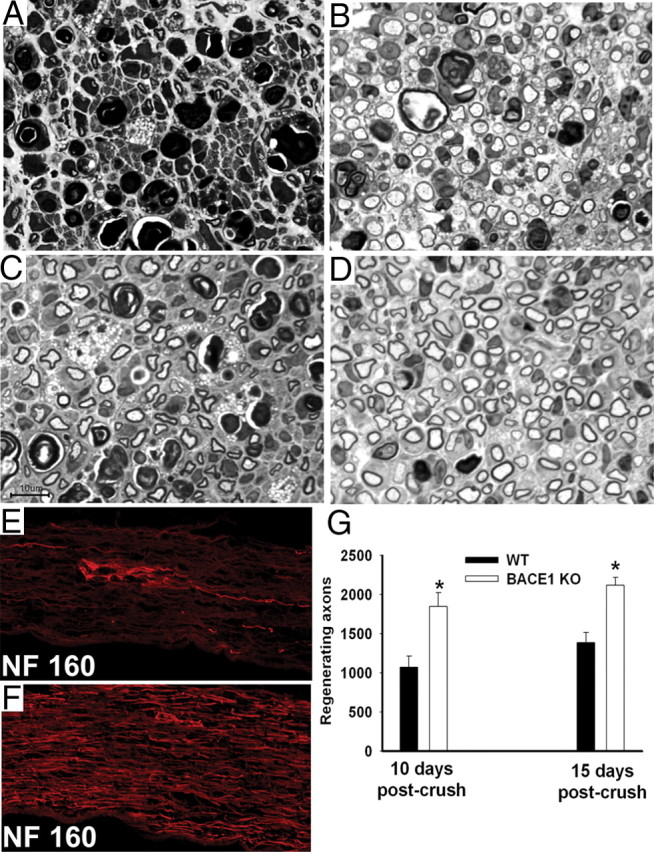
Significantly more regenerated axons at 10 and 15 d after crush in the distal segments of crushed BACE1 KO versus WT sciatic nerve. A–D are semithin (1 μm) plastic sections stained with toluidine blue, and E and F are frozen sections stained for phosphorylated neurofilament (NF 160). A, In WT nerve, a large amount of myelin debris in various stages of degradation is present at 10 d after crush with some regenerating axons. B, In BACE1 KO nerve, large number of regenerating axons, some foamy macrophages, and myelin debris are present at 10 d after crush. C, In WT nerve, regenerating axons are present at 15 d after crush. D, In BACE1 KO nerve, significantly more regenerating axons are present at 15 d after crush (compare C, D). E, WT nerve segment at 10–12 mm distal to the crush site. A few NF 160-positive axons are regenerating. F, BACE1 KO nerve segment at 10–12 mm distal to the crush site. Significant numbers of NF 160-positive axons are regenerating. G, Axon counts at 10 and 15 d after crush. At both time points, BACE1 KO nerves have significantly more regenerated axons than WT nerves. *p < 0.05. n = 3 nerves per genotype at each time point. Values are mean ± SEM. Scale bar: A–D, 10 μm.
The faster clearance of axonal debris and the evolution of macrophage changes suggested a difference in phagocytosis by macrophages in the BACE1 KO animals. Electron-microscopic analysis confirmed the characteristics of faster myelin degeneration in the earlier appearance of foamy post-phagocytic macrophages and the earlier clearance of myelin debris in the BACE1 KO animals (Fig. 3). To further examine macrophage behavior, we removed and cultured noninduced peritoneal macrophages and conducted in vitro phagocytosis assays as described by Link et al. (2010). This assay measures the uptake over time of beads coated with IgG as a phagocytosis index. On average, 65.5% of BACE1 KO macrophages ingested ≥4 beads per cell, and 41.8% of WT macrophages ingested ≥4 beads per cell, a difference that was statistically significant (Fig. 3G–I). These data indicate that BACE1 KO macrophages have enhanced phagocytic activity. This is likely to contribute to the faster in vivo clearance of myelin debris in injured BACE1 KO nerves. We conclude the time to initial axonal degeneration was not affected by BACE1 activity but that clearance of both axonal and myelin debris is accelerated in the distal stumps of BACE1 KO mice.
Accelerated axonal regeneration in BACE1 KO mice
In the first 2 d after injury, little axonal growth occurred in either WT littermate or BACE1 KO nerves, as shown by GAP43 transport, Neurobiotin transport, and plastic-embedded transverse sections (data not shown). This interval represents the latency period (Lanners and Grafstein, 1980; McQuarrie, 1985; Jacob and McQuarrie, 1993; Seijffers et al., 2007); our data indicated that inhibition of BACE1 activity does not alter the latency period. Furthermore, when BACE1 KO nerves were condition lesioned the axons had shortening of the latency period in a fashion comparable with that of WT littermate axons (data not shown). However, by day 3 after a single nerve crush, GAP43-positive sprouts had grown farther down the distal stump in the BACE1 KO nerves than in the WT littermate nerves (Fig. 4A,B). In longitudinal sections of the crushed nerves, we also assessed the distribution of the axonally transported marker Neurobiotin. The advantage of this exogenous labeling technique does not depend on the level of expression of endogenous genes such as GAP43. By day 5, the leading front of regenerating sprouts had grown beyond the length of sciatic nerve available in the BACE1 KO, the distal segment of the BACE1 KO nerves had significantly more regenerated axons, and these axons reached further distally, compared with WT littermate nerves (Fig. 4C,D).
Figure 4.

Accelerated rate of axonal outgrowth in BACE1 KO versus WT after crush injury. A and B are longitudinally sectioned sciatic nerves stained for GAP43 at 3 d after crush. In 2 mm segment distal to the crush, BACE1 KO nerve (B) has more regenerating axons that reach further than those axons of WT littermate nerve (A). C and D are longitudinal sections of nerves filled with Neurobiotin to anterogradely label regenerating axons at 5 d after crush. BACE1 KO nerve (D) has significantly more regenerated axons that reach further distally compared with WT littermate nerve (C). Scale bars: A, B, 100 μm; C, D, 500 μm. The stars in C and D indicate approximate sites of crush.
Analyses of electron micrographs confirmed that BACE KO injured axons regenerated faster than WT. In addition, we found more axonal sprouts in the distal stump. We examined transverse sections of nerves at defined intervals 5 d after crush injury in both WT and BACE1 KO mice (Fig. 5), and observed significantly more axonal sprouts with large (>1 μm) diameter in BACE1 KO nerve at every examined level (Fig. 5B). The sprouts with diameter <1 μm in BACE1 KO nerves were more often clustered in polyaxonal pockets (Fig. 5D). In contrast, distal segments of WT nerve had significantly fewer axonal sprouts and fewer polyaxonal pockets (Fig. 5A,C).
Figure 5.

Extensive axonal regeneration and large polyaxonal pockets in the distal segment of crushed BACE1 KO sciatic nerve at 5 d after crush. A–F are electron micrographs. A, WT nerve. Abundant myelin debris and ovoids and few regenerating sprouts are present (arrow). B, BACE1 KO nerve. Myelin debris is cleared and numerous regenerating sprouts (arrows) are present. C, WT nerve. Few polyaxonal pockets are present. D, BACE1 KO nerve. Numerous polyaxonal pockets (arrows) are present. E, WT nerve. A degenerating myelinated fiber has one (arrow) sprout growing beneath the basal lamina membrane. F, BACE1 KO nerve. Multiple sprouts (arrows) are growing within individual degenerating fibers. G, The BACE1 KO fibers exhibit enhanced number of regenerating sprouts within a single basal lamina membrane compared with WT fibers. For example, 66% of the BACE1 KO fibers have two or more regenerating sprouts compared with 31% of the WT fibers, which have two or more sprouts. Scale bars: A–D, 2 μm.
We next asked whether or not there were more regenerating sprouts associated with the individual Bungner bands. Bungner bands are the denervated Schwann cell bands of the degenerating myelinated fibers of the distal nerve stumps. These Bungner bands could be reliably identified for the first week after nerve crush by the presence of myelin debris within the Schwann cells (Stoll et al., 1989; Schäfer et al., 1996; Griffin et al., 2010). The numbers of axonal sprouts/Bungner band was greater in the BACE1 KO nerves than in WT nerves (Fig. 5) at 5 d after crush. The increase in sprouts/Schwann cell was true both of the degenerating myelinated fibers, identified by their myelin debris, and of small regenerating fibers without myelin debris, many of which represent regenerated Remak bundles (Fig. 5C). We concluded that initial axonal growth is accelerated in BACE1 KO and that the number of sprouts/Bungner band is greater in injured nerves of BACE1 KO mice.
A second ultrastructural feature in some of the BACE1 KO nerves was the prominence of microtubules in the axons. In a minority of premyelinating axons, the change was extreme, with many microtubules collected into a central or paracentral “channel” (Fig. 6). In such fibers, neurofilaments were mostly within a peripheral ring of axoplasm. In most BACE1 KO fibers, the segregation was less dramatic, but there were clusters of microtubules scattered throughout the axoplasm.
Figure 6.

BACE1 KO regenerating axons form a central channel of microtubule cluster; WT axons do not. A, Regenerated axon with normally distributed cytoskeleton structures such as microtubules and neurofilament. B, Regenerated BACE1 KO axon with a central channel of microtubules.
Next, we quantitated axonal regeneration after crush injury in measures of both the spatial and temporal patterns of sprout invasion into the distal stump. At defined times, we found more sprouts at each level of the distal stump, and at defined levels we found an earlier and faster increase in sprout numbers with time. As an example of the distribution at a specific time, 10 d after crush there were more axons with diameters >2 μm in transverse sections taken both 4 and 8 mm away from the crush site in BACE1 KO than in WT littermate nerves. As an example of the temporal differences, in the segment 6–8 mm distal to the crush site we quantified regenerating axons with diameters >2 μm in plastic sections of nerves of BACE1 KO and control littermate at 10 and 15 d after crush. The BACE1 KO nerves had on average 42.0% more axons at 10 d and 35.0% more axons at 15 d than WT littermate nerves (Fig. 7). At each time, the axons were larger in the BACE1 KO nerves than in the control nerves, consistent with their earlier arrival. Furthermore, we stained distal nerve segments 10–12 mm away from crush site with antibody against phosphorylated neurofilament (NF160). BACE1 KO nerve had many more neurofilament-positive axons than were present in WT littermate nerves (Fig. 7). Therefore, we concluded that lack of BACE1 activity increases both the numbers of early sprouts/Bungner band and the total number of myelinating axons in the distal stump. These factors appear to act together to produce larger, more mature axons at every given postcrush interval and at each given level of BACE1 KO nerve.
Faster reinnervation of target muscles in BACE1 KO mice
To investigate whether the robust axonal regeneration in BACE1 KO mice leads to accelerated reinnervation of the neuromuscular junctions, we crushed the sciatic nerves of BACE1 KO and WT littermate mice. At 10 d after crush, BACE1 KO muscles showed significantly improved reinnervation over WT muscles (Fig. 8). We also examined muscle reinnervation in the foot and observed mature presynaptic endings as indicated by synaptophysin staining in BACE1 KO compared with WT littermate feet (data not shown), suggesting that reinnervation in the foot occurs faster after crush injury in BACE1 KO mice than WT. These observations demonstrate faster axonal regeneration in BACE1 KO nerves results in faster target reinnervation.
Figure 8.
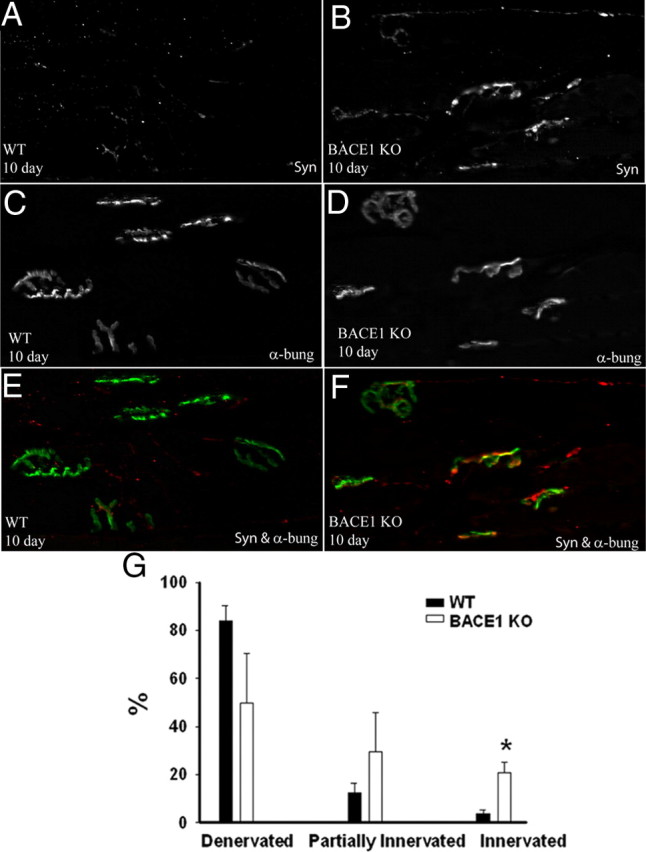
Robust reinnervation of neuromuscular junctions in BACE1 KO versus WT at 10 d after crush. Ten days after nerve crush, the gastrocnemius muscle was harvested from BACE1 KO and WT mice. A and B are stained for synaptophysin; C and D are stained for α-bungarotoxin. E and F are merged images of A and C, and B and D, respectively. Reinnervating presynaptic endings at neuromuscular junctions in BACE1 KO appeared more mature than those in WT. G, Counts of neuromuscular junctions in WT and BACE1 KO mice. BACE1 KO muscles have significantly more reinnervated junctions. N = 3 per genotype. Values are mean ± SEM. *p < 0.05.
Improved debris clearance and increased axonal regeneration using two structurally distinct small molecule BACE1 inhibitors
Next, we investigated whether pharmacological BACE1 inhibitors can reproduce degeneration/regeneration phenotypes observed in injured BACE1 KO nerves. We used two structurally different BACE1 inhibitors: BACE1 inhibitor IV (Stachel et al., 2004) and WAY 258131 (Malamas et al., 2010). The inhibitors were systemically administered via Alzet pumps subcutaneously implanted in the interscapular space for 7 or 15 d. At 7 d after crush, the inhibitor-treated nerves had improved axonal and myelin debris clearance and had significantly more regenerating axons than vehicle-treated mice (Fig. 9). In addition, BACE1 inhibitors increased presynaptic endings in the feet of treated mice as revealed by synaptophysin staining at 2 weeks after crush (data not shown). After inhibitor treatment, however, debris clearance, axonal regeneration, and target reinnervation were not as robust as in BACE1 KO injured animals. We conclude that pharmacological inhibition of BACE1 partly recapitulated degeneration/regeneration phenotypes of BACE1 KO mice.
Figure 9.

Two structurally distinct small molecule BACE1 inhibitors accelerate debris clearance and axonal regeneration after axotomy in WT mice. A, YFP-WT nerve at 7 d after crush. Fragmented YFP debris (arrowheads) is present. Few regenerated YFP-positive axons (arrows) are growing beneath the debris. B, YFP-WT nerve treated with BACE1 inhibitor IV for 7 d after nerve crush. YFP debris (arrowheads) is more fragmented in the nerve. Numerous regenerated YFP-positive axons (arrows) are growing beneath the debris. C, WT nerve at 7 d after crush. A few regenerating axons (arrows) are present. D, WT nerve treated with BACE1 inhibitor IV at 7 d after crush. The nerve has more regenerated axons (arrows) than WT littermate nerve in C. More post-phagocytic macrophages (arrowhead) are present. E, WT foot stained for β-tubulin III to reveal the intraplantar nerves. F, WT foot stained for β-tubulin III 2 weeks after sciatic nerve crush. Regenerated axons have not arrived at the foot yet. G, WT foot stained for β-tubulin III 2 weeks after sciatic nerve crush and treatment with BACE1 inhibitor WAY 258131. β-Tubulin III-positive fibers are regenerated axons in the intraplantar nerves. A and B images are projections of Z-stacks from whole-mounted nerves to show the YFP-positive axonal debris through the depth of the nerve. C and D are electron micrographs. Scale bars: A, B, 100 μm; C, D, 2 μm.
Enhanced regeneration of BACE1 KO axons through WT nerve grafts
We asked whether the accelerated axonal regeneration in injured BACE1 KO nerves was attributable to lack of BACE1 activity in the resident cells of the nerve, such as denervated Schwann cells. Normally in the PNS, BACE1 is predominately a neuronal protein, but BACE1 expression and activity are upregulated in non-neuronal cells under nonphysiological conditions such as injury and stress (Zhang et al., 2007; Tamagno et al., 2008). To examine the cellular basis of accelerated axonal regeneration, we performed reciprocal transplantation of nerve segments between WT and BACE1 KO as diagrammed in Figure 10A. The growth of BACE1 KO axons was significantly accelerated through a wild-type nerve graft, compared with that of wild-type axons growing through BACE1 KO nerve grafts (Fig. 10B,C). These results excluded BACE1 expression in Schwann cells of the distal stump as being responsible for accelerated regeneration. Either BACE1 KO neurons or hematogenous macrophages, or both, could be contributors.
Figure 10.

Accelerated axonal regeneration of BACE1 KO axons. A, Above is a schematic of a reciprocal sciatic nerve transplantation between BACE1 KO and WT. Below is a representative image of a nerve that was transplanted using this nerve graft technique. The arrows point to where the transplanted segment was sutured in. B, WT nerve segment transplanted into a BACE1 KO host nerve. BACE1 KO axons regenerated through the WT graft well. The arrows point to regenerated BACE1 KO axons. C, BACE1 nerve segment transplanted into a wild-type host. A few WT axons grow through BACE1 graft (arrow). Scale bars: B, C, 10 μm. D, BACE1 KO DRG explants had axons 119.0 and 150.0% longer on average than those of WT DRG explants at 2 and 4 d after culture. BACE1 inhibitor IV (50 nm) increased axonal outgrowth of WT DRG explants by 98.0% at 4 d after culture. Values are mean ± SEM. *p < 0.001.
Accelerated axonal outgrowth in DRGs from BACE KO mice or DRGs treated with BACE1 inhibitors
Here, we asked whether lack of neuronal BACE1 accelerates axonal outgrowth in vitro in explants of DRGs. We established postnatal (P4–P5) primary mouse DRG from WT littermate and BACE1 KO pups. We measured axonal outgrowth at 2 and 4 d after culture. The axons of BACE1 KO DRG explants were on average 119 and 150% longer than those of WT DRG explants at 2 and 4 d after culture, respectively (Fig. 10D). Next, we investigated the effect of BACE1 inhibitor on axonal outgrowth of wild-type DRG explants (Fig. 10D). The addition of 50 nm BACE1 inhibitor IV (IC50 = 29 nm) increased axonal outgrowth of WT DRG explants by 98.0% at 4 d after culture. Together, our in vivo transplantation and in vitro data indicate that reducing, by either genetic deletion or pharmacological compounds, of neuronal BACE1 accelerates axonal outgrowth.
Discussion
We found that reduced BACE1 activity accelerates the rate of axonal regeneration after nerve crush. Furthermore, the data suggest two components of the mechanism: First, the enhancement in regeneration is attributable to speeding of axonal growth rate, rather than to reducing the latency period before growth. Second, in other models both intrinsic neuronal mechanisms and removal of growth-inhibiting external cues such as myelin debris can contribute to enhanced regeneration. BACE1 inhibition appears to work through both of these pathways.
The distinction between axonal growth rate versus time required for neurons to enter an axonal growth state is a fundamental issue. Many previous reports of improved nerve regeneration have depended on shortening of the latency before axonal growth occurs. The classical demonstration of reduced latency is the “conditioning” model: nerves subjected to a previous axotomy undergo neuronal responses that place them into a “growth state,” so that a second crush of that nerve results in prompt initiation of outgrowth, skipping the 2 d delay seen in naive animals. The neuronal growth state depends on the expression of growth-related genes such as ATF3 and can be mimicked by transgenic expression of these genes (Seijffers et al., 2007). In the present studies, we reached the conclusion that the rate of growth rather than the latency to growth was predominantly affected by using multiple complementary techniques that were adopted for studies in the mouse. This strategy of combined approaches may find use in future studies of regeneration in genetically engineered mice, as discussed below.
Neuronal effects
The early stages of Wallerian degeneration were comparable in BACE1 KO and control littermate mice after axotomy, and we found no axonal protection effect from genetic deletion of BACE1 when mice were treated with the axonotoxic compounds paclitaxel or acrylamide. These results contrast with results in embryonic neurons deprived of nerve growth factor (NGF). In NGF withdrawal, axonal degeneration was found to depend on an N-APP fragment. This fragment is generated and acts via a newly identified pathway based on four molecules, BACE1, APP, death receptor 6, and caspase 6 (Nikolaev et al., 2009). These data suggested that the N-APP fragment negatively affects axonal health. Our studies indicate that mechanism of axonal degeneration in Wallerian degeneration and after exogenous toxins is different from the effect of growth factor withdrawal documented by Nikolaev et al. (2009). Consistent with our data, recently Vohra et al. (2010) show that in cultured neurons axonal degeneration caused by mechanical insult or vincristine intoxication occurs independent of APP cleavage and caspase 6 activation.
In BACE1 KO axons, the presumed neuronal contribution is evident in cultured neurons; the BACE1-null neurites grow at faster rates. An attractive possibility is that faster outgrowth could correlate with alterations in the organization of the axonal cytoskeleton. This is suggested by the prominent channels of microtubules seen BACE1 KO sprouts in vivo. Increased microtubule stability produced by low concentrations of Taxol results in faster axonal outgrowth (Ertürk et al., 2007). At any given interval after nerve crush, we also observed more total regenerating axons in crushed BACE1 KO nerves compared with WT littermate nerves, and at early stages BACE1 KO nerve had more sprouts/Schwann cell band. That this difference correlates with better regeneration is perhaps surprising, but it suggests that increased sprout numbers may increase the capacity to explore for optimal growth environments in the distal stump—in effect, to allow more “shots on goal” for regenerating sprouts.
Effect on debris clearance
Genetic deletion of BACE1 resulted in faster clearance of debris from degenerated fibers in the distal stump. BACE1 KO PNS nerves are known to be hypomyelinated (Hu et al., 2006; Willem et al., 2006), a difference that is attributable to reduced neuregulin 1 signaling. The faster clearance of myelin debris from the BACE1 KO mice is undoubtedly in part attributable to less myelin at baseline. However, we also observed faster clearance of axonal debris in BACE1 KO nerves, indicating an enhanced general debris clearance mechanism. Consistent with this observation, short-term pharmacological inhibition of BACE1 resulted in faster debris clearance from WT nerves undergoing Wallerian degeneration. The numbers of macrophages in WT and BACE1 KO nerves were similar at early time points after injury, but the BACE1 KO macrophages appear more effective in phagocytosis. Future studies will address whether after a bone marrow transplantation BACE1 KO macrophages exhibit enhanced phagocytosis in injured WT recipient nerves.
Because elimination of BACE1 contributes to more efficient phagocytosis both in vivo and in vitro, our data suggest that there is likely to be a substrate for BACE1 in macrophages. BACE1 cleaves galactoside 2,6-sialyltransferase I (ST6Gal-I), a transmembrane protein expressed by macrophages (Kitazume et al., 2001; Woodard-Grice et al., 2008). ST6Gal-I could be a substrate of BACE1 that leads to more rapid phagocytosis when it is not cleaved by BACE1. Another attractive known contributor to phagocytosis is the triggering receptor expressed on macrophage 2 (TREM2). Increasing TREM2 expression on macrophages has been shown to speed phagocytosis of myelin in experimental allergic encephalomyelitis (Takahashi et al., 2007). TREM2 on the membrane of macrophages is believed to be the active form and cell surface TREM2 is associated with phagocytosis (Turnbull et al., 2006; Piccio et al., 2007; Hsieh et al., 2009). An attractive possibility is that more full-length TREM2 on the surface of macrophages drives more phagocytosis and BACE1 inhibition leads directly or indirectly to more TREM2 on cell membrane. Whether or not TREM2 is a BACE1 substrate remains to be determined. A third but less attractive candidate is the known BACE1 substrate, P-selectin glycoprotein ligand-1 (PSGL-1) (Lichtenthaler et al., 2003). The BACE1-cleaved fragment of PSGL-1 is believed to play a role in recruitment of leukocytes from the blood to the tissue (McEver and Cummings, 1997; Lichtenthaler et al., 2003) rather than to promote phagocytosis per se. For this reason PSGL-1 is unlikely candidate to play a role in phagocytosis by macrophages.
Here, we showed that cultured BACE1 KO macrophages are more efficient in in vitro phagocytosis activity of IgG-coated beads, a capacity dependent on macrophage Fc receptor. Recent data implicated natural IgG and IgM antibodies to peripheral nerve components in accelerating clearance of myelin debris during Wallerian degeneration (Vargas et al., 2010). Thus, it is possible that BACE1 inhibition in some fashion alters Fc-mediated phagocytosis. Future studies will determine whether BACE1 affects Fc receptor levels on macrophages.
BACE1 in Schwann cells
Denervated Schwann cells have BACE1 activity (data not shown). However, two lines of data argue against Schwann cells being responsible for the observed faster axonal growth. First, 15 d after transplantation of WT nerve grafts into BACE1 KO recipient sciatic nerves, there was more extensive regeneration than when WT or BACE1 KO grafts were transplanted into WT recipient nerves. This indicates that the major component of the effect on regeneration is contributed by recipient cells (neuronal or macrophage) rather than by donor Schwann cells. The second line of evidence is the greater rate of outgrowth that was seen in cultured DRG neurons, especially between 2 and 4 d after explantation. These cultures were from P4–P5 mice, to make them as consonant as possible with the adults used in the in vivo studies. This evidence suggests that at least part of the “recipient” effect is neuronal, because there are few macrophages in these cultures.
Effects on axonal outgrowth
In vivo, we used several complementary assays to assess axonal regeneration in crushed nerves, including the distribution of GAP-43 protein and Neurobiotin labeling. These two measures helped separate an effect on latency from that of enhanced outgrowth of regenerating sprouts. With these techniques, we found that, 2 d after nerve crush, most axons had not started growing in either the WT littermates or in the BACE1 KO animals. Rather, they had retracted or degenerated back from the site of the crush slightly, with the largest fibers more affected than the smaller fibers, as previously described in wild-type animals (Ramon y Cajal, 1928). However, by day 3, greater penetration into the distal stump was present in the BACE1 KO animals than in wild-type animals, and this difference increased over the next 4 d (6–7 d after crush is the longest interval after axotomy that it is possible to “catch” the fastest growing fibers within the sciatic nerve of the mouse). After this time, a high proportion of the sprouts have grown beyond the available length of nerve in both the BACE1 KO and littermate animals.
We observed no to little axonal regeneration in nerves of BACE1 KO and WT littermate mice 2 d after crush, indicating before the crush BACE1 KO animals are not in a neuronal “growth state” of the type produced by earlier axotomy in the “conditioning lesion” (Lanners and Grafstein, 1980; McQuarrie, 1985; Jacob and McQuarrie, 1993; Seijffers et al., 2007). In peripheral nerves, a persistent axotomy-like growth state has been achieved by the genetically engineered persistent expression of some axotomy-related “genes” such as the transcription factors ATF3 (Seijffers et al., 2007). Rather, the growth state needs to be induced by axotomy in the BACE1 KO as it does in wild-type animals. The BACE1 KO axons grew faster than WT once the 2 d latency period had passed. In short nerves such as those of the rodents, reduction in the latency period can substantially reduce the time required for growing fibers to reach their targets. However, in long nerves such as those in humans, the time to reinnervation of a target that is several centimeters away from the site of axotomy would be little affected by elimination of the latency period. In contrast, a faster rate of outgrowth would have a substantial effect.
Our work implies that faster rate of outgrowth associated with BACE1 inhibition could be useful in speeding nerve regeneration in human conditions. In humans, axonal regeneration is sufficiently slow that denervated Schwann cells, which provide a permissive microenvironment for regeneration, and target tissues are both at risk for undergoing atrophy and death, precluding functional recovery (Höke, 2006; Gordon et al., 2009; Griffin et al., 2010). This situation underscores the critical need for agents, such as BACE1 inhibitors, that potentially can speed up axonal regeneration. BACE1 has been an attractive drug target for Alzheimer's disease. Over the past 10 years, numerous BACE1 inhibitors have been developed (for review, see Citron, 2004; Silvestri, 2009), but most, if not all, of these inhibitors do not cross the blood–brain barrier well. However, existing BACE1 inhibitors may access the PNS as the barriers are more promiscuous than brain. In addition, the blood–nerve barrier is broken down in the distal stump of injured nerves (George and Griffin, 1994) (for review, see Vargas and Barres, 2007) making drug more assessable. In conclusion, our data support additional evaluation of BACE1 inhibitors as a novel therapeutic approach for nerve injury.
Footnotes
This work was supported by The Robert Packard Center for ALS Research, Muscular Dystrophy Association, Accelerated Translational Incubator Pilot Grant UL1 RR 025005 (The Johns Hopkins University), National Institute of Neurological Disorders and Stroke–National Institutes of Health Grants P01 NS047308 and R01 NS041269, and Adelson Program in Neural Repair and Rehabilitation. We thank Michelle Polley, Osefame Ewaleifoh, Shilpa Sakharkar, Ryan Taub, Zoe Rammelkamp, Karen Kumar, and Ying Wu for technical assistance. We are grateful to Dr. Michael Caterina and Elizabeth Hwang for helping with macrophage in vitro assay. We acknowledge the help of Carol Rubright and Carol Cooke of the Electron Microscope Unit of the Department of Neurology at The Johns Hopkins University School of Medicine.
The authors declare no competing financial interests.
References
- Araki W, Kitaguchi N, Tokushima Y, Ishii K, Aratake H, Shimohama S, Nakamura S, Kimura J. Trophic effect of β-amyloid precursor protein on cerebral cortical neurons in culture. Biochem Biophys Res Commun. 1991;181:265–271. doi: 10.1016/s0006-291x(05)81412-3. [DOI] [PubMed] [Google Scholar]
- Barrette B, Hébert MA, Filali M, Lafortune K, Vallières N, Gowing G, Julien JP, Lacroix S. Requirement of myeloid cells for axon regeneration. J Neurosci. 2008;28:9363–9376. doi: 10.1523/JNEUROSCI.1447-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Berek L, Adalbert R, Wagner D, Grumme DS, Addicks K, Ribchester RR, Coleman MP. Quantitative and qualitative analysis of Wallerian degeneration using restricted axonal labelling in YFP-H mice. J Neurosci Methods. 2004;134:23–35. doi: 10.1016/j.jneumeth.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Beirowski B, Adalbert R, Wagner D, Grumme DS, Addicks K, Ribchester RR, Coleman MP. The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci. 2005;6:6. doi: 10.1186/1471-2202-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin A, Pineau I, Barrette B, Filali M, Vallières N, Rivest S, Lacroix S. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J Neurosci. 2007;27:12565–12576. doi: 10.1523/JNEUROSCI.3027-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brushart TM. Motor axons preferentially reinnervate motor pathways. J Neurosci. 1993;13:2730–2738. doi: 10.1523/JNEUROSCI.13-06-02730.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brushart TM, Gerber J, Kessens P, Chen YG, Royall RM. Contributions of pathway and neuron to preferential motor reinnervation. J Neurosci. 1998;18:8674–8681. doi: 10.1523/JNEUROSCI.18-21-08674.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Chen ZL, Yu WM, Strickland S. Peripheral regeneration. Annu Rev Neurosci. 2007;30:209–233. doi: 10.1146/annurev.neuro.30.051606.094337. [DOI] [PubMed] [Google Scholar]
- Citron M. Beta-secretase inhibition for the treatment of Alzheimer's disease—promise and challenge. Trends Pharmacol Sci. 2004;25:92–97. doi: 10.1016/j.tips.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Ertürk A, Hellal F, Enes J, Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. 2007;27:9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah MH. Cumulative labeling of embryonic mouse neural retina with bromodeoxyuridine supplied by an osmotic minipump. J Neurosci Methods. 2004;134:169–178. doi: 10.1016/j.jneumeth.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Farah MH, Easter SS., Jr Cell birth and death in the mouse retinal ganglion cell layer. J Comp Neurol. 2005;489:120–134. doi: 10.1002/cne.20615. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- George R, Griffin JW. Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model. Exp Neurol. 1994;129:225–236. doi: 10.1006/exnr.1994.1164. [DOI] [PubMed] [Google Scholar]
- Geremia NM, Pettersson LM, Hasmatali JC, Hryciw T, Danielsen N, Schreyer DJ, Verge VM. Endogenous BDNF regulates induction of intrinsic neuronal growth programs in injured sensory neurons. Exp Neurol. 2010;223:128–142. doi: 10.1016/j.expneurol.2009.07.022. [DOI] [PubMed] [Google Scholar]
- Gordon T, Chan KM, Sulaiman OA, Udina E, Amirjani N, Brushart TM. Accelerating axon growth to overcome limitations in functional recovery after peripheral nerve injury. Neurosurgery. 2009;5(4 Suppl):A132–A144. doi: 10.1227/01.NEU.0000335650.09473.D3. [DOI] [PubMed] [Google Scholar]
- Griffin JW, Pan B, Polley MA, Hoffman PN, Farah MH. Measuring nerve regeneration in the mouse. Exp Neurol. 2010;223:60–71. doi: 10.1016/j.expneurol.2009.12.033. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höke A. Mechanisms of disease: what factors limit the success of peripheral nerve regeneration in humans? Nat Clin Pract Neurol. 2006;2:448–454. doi: 10.1038/ncpneuro0262. [DOI] [PubMed] [Google Scholar]
- Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109:1144–1156. doi: 10.1111/j.1471-4159.2009.06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Jacob JM, McQuarrie IG. Acceleration of axonal outgrowth in rat sciatic nerve at one week after axotomy. J Neurobiol. 1993;24:356–367. doi: 10.1002/neu.480240308. [DOI] [PubMed] [Google Scholar]
- Jacquin MF, Hu JW, Sessle BJ, Renehan WE, Waite PM. Intra-axonal Neurobiotin injection rapidly stains the long-range projections of identified trigeminal primary afferents in vivo: comparisons with HRP and PHA-L. J Neurosci Methods. 1992;45:71–86. doi: 10.1016/0165-0270(92)90045-f. [DOI] [PubMed] [Google Scholar]
- Kitazume S, Tachida Y, Oka R, Shirotani K, Saido TC, Hashimoto Y. Alzheimer's β-secretase, β-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci U S A. 2001;98:13554–13559. doi: 10.1073/pnas.241509198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanners HN, Grafstein B. Effect of a conditioning lesion on regeneration of goldfish optic axons: ultrastructural evidence of enhanced outgrowth and pinocytosis. Brain Res. 1980;196:547–553. doi: 10.1016/0006-8993(80)90423-0. [DOI] [PubMed] [Google Scholar]
- Lapper SR, Bolam JP. The anterograde and retrograde transport of Neurobiotin in the central nervous system of the rat: comparison with biocytin. J Neurosci Methods. 1991;39:163–174. doi: 10.1016/0165-0270(91)90082-b. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler SF, Dominguez DI, Westmeyer GG, Reiss K, Haass C, Saftig P, De Strooper B, Seed B. The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J Biol Chem. 2003;278:48713–48719. doi: 10.1074/jbc.M303861200. [DOI] [PubMed] [Google Scholar]
- Link TM, Park U, Vonakis BM, Raben DM, Soloski MJ, Caterina MJ. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat Immunol. 2010;11:232–239. doi: 10.1038/ni.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malamas MS, Erdei J, Gunawan I, Turner J, Hu Y, Wagner E, Fan K, Chopra R, Olland A, Bard J, Jacobsen S, Magolda RL, Pangalos M, Robichaud AJ. Design and synthesis of 5,5′-disubstituted aminohydantoins as potent and selective human beta-secretase (BACE1) inhibitors. J Med Chem. 2010;53:1146–1158. doi: 10.1021/jm901414e. [DOI] [PubMed] [Google Scholar]
- McEver RP, Cummings RD. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100(11 Suppl):S97–S103. [PubMed] [Google Scholar]
- McQuarrie IG. The effect of a conditioning lesion on the regeneration of motor axons. Brain Res. 1978;152:597–602. doi: 10.1016/0006-8993(78)91116-2. [DOI] [PubMed] [Google Scholar]
- McQuarrie IG. Effect of conditioning lesion on axonal sprout formation at nodes of Ranvier. J Comp Neurol. 1985;231:239–249. doi: 10.1002/cne.902310211. [DOI] [PubMed] [Google Scholar]
- Mears S, Schachner M, Brushart TM. Antibodies to myelin-associated glycoprotein accelerate preferential motor reinnervation. J Peripher Nerv Syst. 2003;8:91–99. doi: 10.1046/j.1529-8027.2003.03012.x. [DOI] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave KA. Axonal neuregulin-1 regulates myelin sheath thickness. Science. 2004;304:700–703. doi: 10.1126/science.1095862. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Mehta NR, Conant K, Kim KJ, Jones M, Calabresi PA, Melli G, Hoke A, Schnaar RL, Ming GL, Song H, Keswani SC, Griffin JW. Axonal protective effects of the myelin-associated glycoprotein. J Neurosci. 2009;29:630–637. doi: 10.1523/JNEUROSCI.5204-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Perez RG, Zheng H, Van der Ploeg LH, Koo EH. The β-amyloid precursor protein of Alzheimer's disease enhances neuron viability and modulates neuronal polarity. J Neurosci. 1997;17:9407–9414. doi: 10.1523/JNEUROSCI.17-24-09407.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, Colonna M, Panina-Bordignon P. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. 2007;37:1290–1301. doi: 10.1002/eji.200636837. [DOI] [PubMed] [Google Scholar]
- Ramon y Cajal S. Degeneration and regeneration of the nervous system. In: Mays R, editor. London: Oxford UP; 1928. [Google Scholar]
- Röytta M, Raine CS. Taxol-induced neuropathy: further ultrastructural studies of nerve fibre changes in situ. J Neurocytol. 1985;14:157–175. doi: 10.1007/BF01150269. [DOI] [PubMed] [Google Scholar]
- Röyttä M, Raine CS. Tool-induced neuropathy: chronic effects of local injection. J Neurocytol. 1986;15:483–496. doi: 10.1007/BF01611731. [DOI] [PubMed] [Google Scholar]
- Savonenko AV, Melnikova T, Laird FM, Stewart KA, Price DL, Wong PC. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc Natl Acad Sci U S A. 2008;105:5585–5590. doi: 10.1073/pnas.0710373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer M, Fruttiger M, Montag D, Schachner M, Martini R. Disruption of the gene for the myelin-associated glycoprotein improves axonal regrowth along myelin in C57BL/Wlds mice. Neuron. 1996;16:1107–1113. doi: 10.1016/s0896-6273(00)80137-3. [DOI] [PubMed] [Google Scholar]
- Seijffers R, Mills CD, Woolf CJ. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci. 2007;27:7911–7920. doi: 10.1523/JNEUROSCI.5313-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S, Ming GL. Roles of channels and receptors in the growth cone during PNS axonal regeneration. Exp Neurol. 2010;223:38–44. doi: 10.1016/j.expneurol.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestri R. Boom in the development of non-peptidic beta-secretase (BACE1) inhibitors for the treatment of Alzheimer's disease. Med Res Rev. 2009;29:295–338. doi: 10.1002/med.20132. [DOI] [PubMed] [Google Scholar]
- Stachel SJ, Coburn CA, Steele TG, Jones KG, Loutzenhiser EF, Gregro AR, Rajapakse HA, Lai MT, Crouthamel MC, Xu M, Tugusheva K, Lineberger JE, Pietrak BL, Espeseth AS, Shi XP, Chen-Dodson E, Holloway MK, Munshi S, Simon AJ, Kuo L, et al. Structure-based design of potent and selective cell permeable inhibitors of human beta-secretase (BACE-1) J Med Chem. 2004;47:6447–6450. doi: 10.1021/jm049379g. [DOI] [PubMed] [Google Scholar]
- Stoll G, Griffin JW, Li CY, Trapp BD. Wallerian degeneration in the peripheral nervous system: participation of both Schwann cells and macrophages in myelin degradation. J Neurocytol. 1989;18:671–683. doi: 10.1007/BF01187086. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 2007;4:e124. doi: 10.1371/journal.pmed.0040124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo DG, Mattson MP, Tabaton M. Oxidative stress activates a positive feedback between the γ- and β-secretase cleavages of the β-amyloid precursor protein. J Neurochem. 2008;104:683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, Chao MV, Falls DL, Role L, Salzer JL. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 2005;47:681–694. doi: 10.1016/j.neuron.2005.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006;177:3520–3524. doi: 10.4049/jimmunol.177.6.3520. [DOI] [PubMed] [Google Scholar]
- Vargas ME, Barres BA. Why is Wallerian degeneration in the CNS so slow? Annu Rev Neurosci. 2007;30:153–179. doi: 10.1146/annurev.neuro.30.051606.094354. [DOI] [PubMed] [Google Scholar]
- Vargas ME, Watanabe J, Singh SJ, Robinson WH, Barres BA. Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc Natl Acad Sci U S A. 2010;107:11993–11998. doi: 10.1073/pnas.1001948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Vohra BP, Sasaki Y, Miller BR, Chang J, DiAntonio A, Milbrandt J. Amyloid precursor protein cleavage-dependent and -independent axonal degeneration programs share a common nicotinamide mononucleotide adenylyltransferase 1-sensitive pathway. J Neurosci. 2010;30:13729–13738. doi: 10.1523/JNEUROSCI.2939-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller AV. Nouvelles recherches sur la regeneration des fibres nerveuses. CR Acad Sci (Paris) 1852;34:675–679. [Google Scholar]
- Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C. Control of peripheral nerve myelination by the beta-secretase BACE1. Science. 2006;314:664–666. doi: 10.1126/science.1132341. [DOI] [PubMed] [Google Scholar]
- Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- Woodard-Grice AV, McBrayer AC, Wakefield JK, Zhuo Y, Bellis SL. Proteolytic shedding of ST6Gal-I by BACE1 regulates the glycosylation and function of alpha4beta1 integrins. J Biol Chem. 2008;283:26364–26373. doi: 10.1074/jbc.M800836200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S, van Niekerk EA, Merianda TT, Twiss JL. Dynamics of axonal mRNA transport and implications for peripheral nerve regeneration. Exp Neurol. 2010;223:19–27. doi: 10.1016/j.expneurol.2009.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Pearse TL, Chen AC, Chang R, Marquez C, Selkoe DJ. Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev. 2008;3:15. doi: 10.1186/1749-8104-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhou K, Wang R, Cui J, Lipton SA, Liao FF, Xu H, Zhang YW. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem. 2007;282:10873–10880. doi: 10.1074/jbc.M608856200. [DOI] [PubMed] [Google Scholar]