

Abstract
Fetal hemoglobin (HbF) is a major modifier of disease severity in sickle cell anemia (SCA). Three major HbF quantitative trait loci (QTL) are known: the Xmn I site upstream of Gγ-globin gene (HBG2) on chromosome 11p15, BCL11A on chromosome 2p16, and HBS1L-MYB intergenic polymorphism (HMIP) on chromosome 6q23. However, the roles of these QTLs in SCA patients with uncharacteristically high HbF are not known. We studied 20 African American SCA patients with markedly elevated HbF (mean 17.2%). They had significantly higher minor allele frequencies (MAF) in two HbF QTLs, BCL11A and HMIP, compared with those with low HbF. A 3-bp (TAC) deletion in complete linkage disequilibrium (LD) with the minor allele of rs9399137 in HMIP was also present significantly more often in these patients. To further explore other genetic loci that might be responsible for this high HbF, we sequenced a 14.1 kb DNA fragment between the Aγ(HBG1) and δ-globin genes (HBD). Thirty-eight SNPs were found. Four SNPs had significantly higher major allele frequencies in the unusually high HbF group. In silico analyses of these 4 polymorphisms predicted alteration in transcription factor binding sites in 3.
Keywords: Sickle cell anemia, Fetal hemoglobin, HbF quantitative trait loci
HbF inhibits deoxy-HbS polymerization. Patients with elevated HbF have fewer vasoocclusive complications and prolonged survival [1]. Three major HbF QTL are known. The C>T polymorphism (rs7482144) at nucleotide –158 upstream of HBG2 is associated with increased HbF in some SCA patients [2]. Polymorphisms in intron 2 of BCL11A represented by rs766432 was associated with HbF in healthy Northern Europeans [3], African Americans with SCA [4, 5], Chinese with β-thalassemia trait and Thai’s with HbE-β thalassemia [5]. BCL11A polymorphisms correlate highly with HbF levels in SCA, accounting for 7–12% of the HbF variance [6]. The HMIP polymorphisms are distributed in three LD blocks [7]. HMIP block 2 represented by rs9399137 is most significantly associated with HbF expression and might function as a distal regulatory element [8,9].
We studied a selected group of 20 African American SCA patients with exceptionally high HbF (mean 17.2%) which differed by more than 4 times the standard deviation of 30 other patients with low HbF (mean 5.0%; Table IA). All study subjects’ HBB underwent nucleotide sequencing to ascertain that they were HbS homozygotes. Multiplex ligation-dependent probe amplification (MLPA) was carried out to ensure that they did not harbor hereditary persistence of fetal hemoglobin (HPFH) 1, HPFH 2, Black (δβ)0- and Black (Aγδβ)0-thalassemia deletions [10]. Furthermore, their HBG2 and HBG1 promoters were also sequenced to be certain that they did not have promoter HPFH single nucleotide mutations [11,12]. They were unlikely to be on hydroxyurea based on their MCV being less than 100 fL.
Table IA.
Hematologic results of high and low HbF study groups.
| N | Age | Male/Female | Hb (g/dL) | MCV (fL) | HbF (%) | |
|---|---|---|---|---|---|---|
| High HbF group | 20 | 16.3 ± 8.3 (6 – 30) | 8/12 | 9.0 ± 1.3 (5.7 – 11.7) | 87.9 ± 9.0 (77 – 99) | 17.2 ± 4.8 (11 – 28.9) |
| Low HbF group | 30 | 19.3 ± 9.8 (5 – 49) | 10/20 | 8.6 ± 1.4 (5.2 – 11.4) | 81.4 ± 11.1 (65 – 100) | 5.0 ± 2.5 (0.5 – 8.8) |
Values are shown as mean ± SD; Values shown between parentheses represent range of values.
In addition we conducted a subset analyses in 56 patients with unusually high HbF (mean 20.7%) which differed by more than 11 times the standard deviation of 489 patients with low HbF (mean 3.1%; table IB). These patients were selected from 1,086 subjects from the Cooperative Study of Sickle Cell Disease (CSSCD) who were previously investigated in a genome-wide association study (GWAS) of HbF [13].
Table IB.
Hematologic results of high and low HbF CSCCD groups.
| N | Age | Hb (g/dL) | MCV (fL) | HbF (%) | |
|---|---|---|---|---|---|
| High HbF group | 56 | 15.2 ± 10.9 | 9.6 ± 1.5 | 89.7 ± 8.9 | 20.7 ± 8.2 |
| Low HbF group | 489 | 19.1 ± 10.8 | 8.1 ± 1.1 | 89.2 ± 7.3 | 3.1 ± 1.5 |
Values are shown as mean ± SD; Values shown between parentheses represent range of values.
The MAF of rs7482144, also known as the Xmn I site, on chromosome 11p15 in the unusually high HbF group (10%) is not significantly different from that in the low HbF group (8%) as shown in Table II. The SNP rs5006884, a missense mutation (CTC>TTC or Leu172Phe) in OR51B6 (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?rs=5006884) on chromosome 11p15 was reported to be associated with HbF in SCA in a GWAS [13]. In the present study, the MAF of rs5006884 in the unusually high HbF group (2%) is actually less than that in the low HbF group (10%), even though the difference is not statistically significant (Table II). It should be recognized that the small sample size in the current study does not afford sufficient power to discern possible MAF differences if present at these 2 SNPs between these 2 groups of patients.
Table II.
HbF QTL minor allele frequencies in high and low HbF groups
| Chromosome | SNP | Minor allele | YRI | CEU | Gene | Study groupsn (N=50) | CSCCD groups (N=590) | ||
|---|---|---|---|---|---|---|---|---|---|
| MAF | P | MAF | P | ||||||
| 2 | rs766432 | C | 0.25 | 0.12 | BCL11A | 0.45/0.25 | 0.05 | 0.47/0.2 | 6.399E–10 |
| 6 | rs9399137 | C | 0.042 | 0.25 | HBS1L–MYB | 0.18/0.03 | 0.02 | 0.09/0.03 | 0.006 |
| 6 | rs7775698 | T | 0.32 | 0.22 | HBS1L–MYB | 0.40/0.21 | 0.07 | 0.16/0.19 | 0.52 |
| 11 | rs5006884 | T | 0.25 | 0.22 | OR51B5; OR51B6 | 0.02/0.1 | 0.2 | 0.22/0.1 | 0.00055 |
| 11 | rs7482144 | T | HBG2 | 0.1/0.08 | 1.0 | 0.30/0.1 | 0.002 | ||
YRI, minor allele frequency of subjects from Ibadan, Nigeria based on the HapMap data. CEU, minor allele frequency of northern and Western European subjects, based on the HapMap data. MAF, minor allele frequency. Figure on the left represents MAF of those with high HbF; Figure on the right represents MAF of those with low HbF.
The SNP rs7482144 did not reach genome-wide significance in previous GWAS of HbF in the full CSCCD cohort [13]. In the present study with subsets from the CSCCD cohort, rs7482144 has a significantly higher MAF in the unusually high HbF group (30%) compared to the low HbF group (10%), P = 0.002. The MAF of rs5006884 in OR51B6 in the high HbF group (22%) is significantly higher than that in the low HbF group (10%), P = 0.00055. This SNP reached genome-wide significance in the GWAS of the full CSSCD cohort, however the mean HbF levels in homozygotes for this variant was only 10.6%. The present subset analysis shows that for SCA patients with unusually high HbF the frequency of this mutation is comparable to the general population (Table II).
To examine the BCL11A QTL, three SNPs, rs766432, rs4671393 and rs11886868 were chosen for genotyping [6]. These 3 SNPs are in strong LD. Only data on rs766432 that is most highly correlated with HbF in SCA patients are presented. The MAF in the unusually high HbF group (45%) is significantly higher than that in the low HbF group (25%), P = 0.05 (Table II). The MAF in the subgroups from CSCCD were similar: 47% in the high and 20% in the low HbF groups, P = 6.399×10−10. The allele C of this SNP was associated with increased levels of HbF [13] and the mean HbF levels in subjects homozygous for the C allele was 8.26%. The present subset analysis in the CSSCD subgroups shows that in patients with unusually high HbF the frequency of this mutation is almost twice that of the full cohort.
The QTL in HMIP is best represented by rs9399137 [9]. The MAF of rs9399137 among SCA patients of African descent without European admixture was reported to be 1–2% [14,15]. A GWAS on over 800 African American SCA patients did not detect genome-wide significance of association of this QTL with HbF [13]. The functional motif for this QTL is most likely a 3-bp (TAC) deletion which is in complete LD with the minor allele of rs9399137 [9]. The frequency of this 3-bp deletion is 23% in non-African HapMap populations, but only 5% in Africans [9].
In the current study, the MAF of rs9399137 in the African American SCA patients with unusually high HbF is 18%, significantly higher than that with low HbF (3%), P = 0.02 (Table II). Furthermore, the 3-bp deletion as reported by Farrell et al [9] was found for the first time in these African American SCA patients and it is in complete LD with the minor allele of rs9399137. Among the subset of CSSCD patients, the MAF of rs9399137 in the unusually high HbF group (9%) is also significantly higher than that in the low HbF group (3%), P = 0.006 (Table II). These results raise the possibility that some African American SCA patients with markedly elevated HbF might have inherited the minor allele of chromosome 6q23 QTL due to European genetic admixture.
The minor T allele of rs7775698 tags either an ancestral T nucleotide found mostly in African populations, or a 3-bp deletion often found in European and Chinese populations [9]. In the present study, the MAF of rs7775698 in the high HbF group (40%) is higher than that in the low HbF group (21%). But the difference is not statistically significant (P = 0.07). Among the CSCCD patients, the MAF of rs7775698 in the high HbF group is 16% compared to 19% in the low HbF group (Table II).
We found a 2-bp (CC) deletion plus an (A) insertion 19 bp downstream of rs9399137. This deletion/insertion was present in 33% of chromosome 6 in both high and low HbF groups. It is unlikely that it plays a significant functional role in modulating HbF expression. Its relatively high frequency in both groups makes it a probable haplotype marker in African American SCA patients.
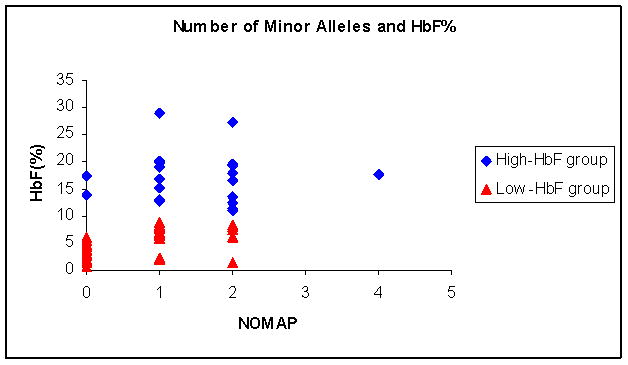
Two of the 3 known HbF major QTLs are present in the unusually high HbF patient study group at a frequency significantly higher than those with low HbF. Their cumulative effect was estimated by assigning to each minor allele at each locus a score of 1, and adding the scores to generate the Number of Minor Allele Present (NOMAP) [16]. There was an average of 2 minor alleles in the unusually high HbF group compared with 1 in the low HbF group (P = 0.001) as shown in Figure 1.
Figure 1.
To explore other possible genetic loci that may modulate HbF expression, we undertook nucleotide sequencing of a 14.1 kb DNA fragment between HBG1 and HBD in 15 high and 15 low HbF patients. This DNA fragment was chosen because BCL11A binds to this intergenic region and it also encompasses the Corfu deletion that in homozygotes is characterized by markedly elevated HbF [17, 18].
Thirty eight SNPs were found in both the high and low HbF groups (Supplementary Table I). SNP rs10128558 as described by Galarneau et al [19] was present in our study groups and found to be in complete LD with Xmn I polymorphism. Its MAF in the high HbF group (10%) is not different from the low HbF group (8%). Based on in silico analysis by TFSEARCH (threshold score 85), 22 of the 38 SNPs are associated with alterations in transcription factor binding including erythroid specific transcription factors. In addition, 4 SNPs have significantly higher major allele frequency in the high compared to the low HbF groups (P < 0.05). Three of these 4 SNPs result in alteration in transcription factor binding sites (Supplementary Table I).
This study based on a small cohort of carefully selected African American SCA patients with unusually high HbF revealed that the MAF for rs766432 (BCL11A), rs9399137 and 3-bp deletion both within HMIP are much higher than that found in patients with low HbF. These findings also raise the possibility that some African American patients with markedly elevated HbF might have inherited the minor allele of chromosome 6q23 QTL due to European genetic admixture. Validation of these findings in a larger patient cohort and further functional investigations into these and other polymorphisms are warranted.
Materials and Methods
Study Groups
Blood samples referred to the Hemoglobin Diagnostic Reference Laboratory for DNA-based diagnostics at the Boston Medical Center were selected for this study. The samples were collected between 2003 and 2008. Patients younger than 5-year old, the time at which HbF levels stabilized [13] and patients with MCV greater than 100 fL were excluded. Nucleotide sequencing of the HBB and promoters of HBG2 and HBG1 was done after PCR amplification. The presence of HBB deletions was excluded by multiplex ligation dependent probe amplification [20]. This study was approved by the Boston University School of Medicine Institutional Review Board.
CSCCD subsets
We used data from the Cooperative Study of Sickle Cell Disease (CSSCD). From 1,086 cases who underwent GWAS [13] we selected 56 patients with unusually high HbF and 489 patients with low HbF for subset analyses.
QTLs
The Xmn I polymorphism (rs7482144) was genotyped by polymerase chain reaction (PCR) of the HBG2 promoter, followed by restriction enzyme digestion analysis.
Genotyping of SNPs in BCL11A was done by a TaqMan SNP genotyping assay (Applied BioSystems, Foster City, CA) according to the manufacturer’s instruction. Pre-designed probes were ordered for genotyping analyses: rs766432 (C__1025980_10), rs11886868 (C__11363852_10), rs4671393 (C__25926414_10). Amplification was done with 5μl of 2X TaqMan Universal PCR master mix, 0.5μl of 40X primer and TaqMan probe dye mix, and between 10–50 ng of DNA. Cycling conditions consisted of 10 min at 95°C, followed by 40 cycles 15 sec at 92°C, 1 min at 60°C. Allelic discrimination is performed on Applied BioSystem RT-PCR system.
Genotyping of SNPs in HMIP, rs9399137, rs7775698, and 3-bp (TAC) deletion, and SNP rs5006884 in OR51B6 was done by PCR, followed by nucleotide sequencing using the BigDye terminator cycle sequencing kit from Applied BioSystems.
HBG1-HBD nucleotide sequencing
Short (500–600 bp) and overlapping fragments of DNA covering the 14.1 kb region were amplified by PCR. All PCR reactions were performed in a total volume of 20μl. Master mix concentrations and cycling conditions were optimized based on the region being amplified. Usually, each reaction contained 100–250 ng of DNA, 1X PCR buffer (Applied BioSystems), 2 mM MgCl2, 200 μM dNTP, 1 ng of each primer, and 0.5U AmpliTaq polymerase. Cycling conditions consisted of 5 min at 94°C, followed by 30 cycles of 40 sec at 94°C, 40 sec at 55°C and 3 min at 72°C and a 7 min elongation step at 72°C.
Statistical Analysis
Association between HbF and the minor alleles of each QTL was statistically analyzed by the Fisher exact test performed in R (www.r-project.org). Comparison of hematological parameters and correlation between number of minor allele present and HbF in both groups was examined using paired T-test. An overall significance level of 0.05 was set for all statistical analyses.
Supplementary Material
Acknowledgments
We sincerely thank Drs. Hong-yuan Luo, Richard M. Sherva, and John J. Farrell for advice throughout the course of this study. The support and encouragement of Dr. Katya Ravid was very much appreciated.
This work was supported in part by NIH/NIDDK RO1 DK069646, and Training Grant in cardiovascular biology, NIH/NHLBI HL007969.
References
- 1.Steinberg MH. Management of sickle cell disease. New England Journal of Medicine. 1999;340:1021–1030. doi: 10.1056/NEJM199904013401307. [DOI] [PubMed] [Google Scholar]
- 2.Gilman JG, Huisman TH. Two independent genetic factors in the β-globin gene cluster are associated with high Gγ-levels in the HbF of SS patients. Blood. 1984;64:452–457. [PubMed] [Google Scholar]
- 3.Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nature Genetics. 2007;39:1197–1199. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 4.Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1l-MYB and β-globin loci associate with fetal hemoglobin levles and pain crises in sickle cell disease. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11869–11874. doi: 10.1073/pnas.0804799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sedgewick AE, Timofeev N, Sebastiani P, et al. BCL11A is a major HbF quantitative trait locus in three different populations with β-hemoglobinopathies. Blood Cells, Molecules, and Diseases. 2008;41:255–258. doi: 10.1016/j.bcmd.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thein SL, Menzel S, Lathrop M, et al. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Human Molecular Genetics. 2009;18:R216–223. doi: 10.1093/hmg/ddp401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thein SL, Menzel S, Peng X, et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11346–11351. doi: 10.1073/pnas.0611393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wahlberg K, Jiang J, Rooks H, et al. The HBS1L-MYB intergenic interval associated with elevated HbF levels shows characteristics of a distal regulatory region in erythroid cells. Blood. 2009;114:1254–1262. doi: 10.1182/blood-2009-03-210146. [DOI] [PubMed] [Google Scholar]
- 9.Farrell JJ, Sherva RM, Chen Z-y, et al. A 3-bp deletion in the HBS1L-MYB intergenic region on chromosome 6q23 is associated with HbF expression. Blood. 2011;117:4935–4945. doi: 10.1182/blood-2010-11-317081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thein SL, Wood WG. The molecular basis of β thalassemia, δβ thalassemia, and hereditary persistence of fetal hemoglobin. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin. 2. Cambridge: Cambridge University Press; 2009. pp. 338–356. [Google Scholar]
- 11.Collins FS, Stoeckert CJ, Serjeant GR, et al. Gγβ+ hereditary persistence of fetal hemoglobin: cosmid cloning and identification of a specific mutation 5′ to the Gγ gene. Proceedings of the National Academy of Sciences of the United States of America. 1984;81:4894–4898. doi: 10.1073/pnas.81.15.4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins FS, Metherall JE, Yamakawa M, et al. A point mutation in the Aγ-globin gene promoter in Greek hereditary persistence of fetal haemoglobin. Nature. 1985;313:325–326. doi: 10.1038/313325a0. [DOI] [PubMed] [Google Scholar]
- 13.Solovieff N, Milton JN, Hartley SW, et al. Fetal hemoglobin in sickle cell anemia: genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood. 2010;115:1815–1822. doi: 10.1182/blood-2009-08-239517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Creary LE, Ulug P, Menzel S, et al. Genetic variation on chromsome 6 influences F cell levels in healthy individuals of African descent and HbF level in sickle cell patients. PLos One. 2009;1:e4218. doi: 10.1371/journal.pone.0004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makani J, Menzel S, Nkya S, et al. Genetics of fetal hemokglobin in Tanzanian and British patients with sickle cell anemia. Blood. 2011;117:1390–1392. doi: 10.1182/blood-2010-08-302703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galanello R, Sanna S, Perseu L, et al. Amelioration of Sardinian β0 thalassemia by genetic modifiers. Blood. 2009;114:3935–3937. doi: 10.1182/blood-2009-04-217901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jawaid K, Wahlberg K, Thein SL, et al. Binding patterns of BCL11A in the globin and GATA1 loci and characterization of the BCL11A fetal hemoglobin locus. Blood Cells, Molecules and Diseases. 2010;45:140–146. doi: 10.1016/j.bcmd.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 18.Chakalova L, Osborne CS, Dai Y-F, et al. The Corfu δβ thalassemia deletion disrupts γ-globin gene silencing and reveals post-transcriptional regulation of HbF expression. Blood. 2005;105:2154–2160. doi: 10.1182/blood-2003-11-4069. [DOI] [PubMed] [Google Scholar]
- 19.Galarneau G, Palmer CD, Sankaran VG, et al. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nature Genetics. 2010;42:1049–1051. doi: 10.1038/ng.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redeker EJ, de Visser AS, Bergen AA, et al. Multiplex ligation-dependent probe amplification (MLPA) enhances the molecular diagnosis of aniridia and related disorders. Molecular Vision. 2008;14:836–840. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.