

Abstract
The conserved TREX-2 transcription-export complex integrates transcription and processing of many actively-transcribed nascent mRNAs with the recruitment of export factors at nuclear pores and also contributes to transcriptional memory and genomic stability. We report the crystal structure of the Sac3–Thp1–Sem1 segment of Saccharomyces cerevisiae TREX-2 that interfaces with the gene expression machinery. Sac3–Thp1–Sem1 forms a novel PCI-domain complex characterized by the juxtaposition of Sac3 and Thp1 winged helix domains, forming a platform that mediates nucleic acid binding. Structure-guided mutations underline the essential requirement of the Thp1–Sac3 interaction for mRNA binding and for the coupling of transcription and processing with mRNP assembly and export. These results provide insight into how newly synthesized transcripts are efficiently transferred from TREX-2 to the principal mRNA export factor and, identify how Sem1 stabilizes PCI domain-containing proteins and promotes complex assembly.
Eukaryotic gene expression requires coordination of a series of nuclear events, including transcription and pre-mRNA processing, that culminate in the export of mature mRNA through the nuclear pore complexes (NPCs) after which translation can begin1-3. Although a host of proteins act in concert to co-transcriptionally package the mRNA transcript into messenger ribonucleoprotein particles (mRNP) prior to export 2, the TREX and TREX-2 TRanscription and EXport complexes play a central role by coupling transcription and processing, with mRNA export4. The TREX complex is formed by the association of Yra1 and the DEAD-box helicase Sub2 with the THO complex, and contributes to recruiting and loading of the principal yeast mRNA transport factor (Mex67-Mtr2, metazoan NXF1-NXT1; also known as Tap-p15) onto pre-mRNA1,5-9. The TREX-2 complex facilitates the association of a number of actively-transcribing genes, such as GAL, with NPCs in a process termed “gene gating”10-13 and also contributes to transcriptional memory and genomic stability14-20. Saccharomyces cerevisiae TREX-2 consists of Sac3, Thp1, Sus1 and Cdc31, together with a newly-identified co-factor, Sem1, a small negatively charged protein that may either associate with TREX-2 or influence its stability13,21-24. Although the precise role of Sem1 in TREX-2 is unknown, Sem1 and its human homologue, DSS1, associate with a wide range of conserved complexes including the 19S proteosome lid, and the CSN, eIF3, BRCA2, and Integrator complexes25.
During gene gating, TREX-2 mediates the relocation of active genes to the pores through binding to both the nuclear face of NPCs and the Spt-Ada-Gcn5 acetyltransferase (SAGA) complex12. The resultant generation of transcripts in the immediate vicinity of NPCs facilitates export by increasing the entry efficiency of mRNPs into the NPC transport channel26. TREX-2 also participates in the post-transcriptional NPC-gene tethering that can give rise to the transcriptional memory seen with several yeast genes14-17, although interactions between Mlp1 and Mex67 and/or Nab2, and between histone Htz1 and several nucleoporins, may also contribute to this function18. Importantly, deletion of TREX-2 components results in significant growth impairment and widespread mRNA export defects13,21-24, together with defects in transcriptional elongation and RNA-DNA hybridization, which results in the formation of R-loops that generate transcriptional impairment and genetic instability19,20.
TREX-2 is based on a Sac3 scaffold to which Thp1, Sus1, and Cdc31 are bound13,21,22. Sus1 and Cdc31 bind to the “CID” domain within the C-terminal portion of Sac3 and are required for the association of TREX-2 with NPCs21,22. The Sac3 CID domain forms a long, gently undulating α-helix around which one Cdc31 and two Sus1 chains are wrapped22. In vivo studies using engineered mutations that selectively disrupt the binding of individual chains of Sus1 and Cdc31 to Sac3, indicate that Sus1 and Cdc31 function synergistically to promote the NPC-TREX-2 association22. However, it is unclear how NPC-bound TREX-2 associates with other components of the gene expression machinery to integrate mRNA processing with nuclear export or influence transcriptional memory and genomic stability. The N-terminal region of Sac3 (residues 1-572) is critical for this function and binds both Thp1 and the major mRNA transport factor Mex67–Mtr2. Deletion of either this Sac3 region or Thp1 causes mRNA export and growth defects in yeast21,23. It also remains unclear whether Sem1 binds directly to TREX-2 and, if it does so, to which components it binds and with what stoichiometry23,24. The functions of TREX-2 in mRNA export are conserved from yeast to humans, with the human Sac3 homologue, GANP, linking the nuclear export of specific mRNPs with transcription and processing27.
We report here the 2.9 Å resolution crystal structure of S. cerevisiae Sac3 residues 253-551 complexed with Thp1 and Sem1, together with the 2.1 Å resolution structure of the homologous human PCID2–DSS1 complex, and use structure-guided mutagenesis to define how this region of TREX-2 contributes to mRNA export in vivo. Sac3 and Thp1(PCID2) have PCI folds based on an N-terminal superhelical domain formed by a stack of α-helices, and capped by a C-terminal α/β winged helix domain. Sem1(DSS1) binds primarily to Thp1(PCID2), where it appears to have a stabilizing role. Unexpectedly, the complex is intricately arranged so that the juxtaposition of winged helix domains on Sac3 and Thp1 forms an architectural platform that is critical for nucleic acid binding. A series of engineered Sac3 and Thp1 variants containing structure-guided mutations delineate the essential contributions made by both the Thp1–Sac3 interaction and subsequent nucleic acid binding to the coupling of mRNA export to transcription and pre-mRNA processing. These results provide insight into how newly synthesized transcripts are efficiently transferred from TREX-2 to the principal mRNA export factor, and also indicate how Sem1 can actively stabilize PCI domain-containing proteins and promote complex assembly.
RESULTS
Sem1 stabilizes Thp1 and promotes interaction with Sac3
Pull-down assays were used to monitor assembly of the TREX-2 complex and interactions between bacterially-expressed Sac3, Thp1, and Sem1 (Fig. 1a). When expressed alone, Thp1 remained insoluble irrespective of the use of a range of solubilizing tags (Fig. 1a, lane 1), but its solubility improved dramatically when coexpressed with Sem1 (Fig. 1a, lane 2). The solubility of Sac3 was not influenced by Sem1 coexpression, indicating that Thp1, but not Sac3, required Sem1 association for its solubility (Fig. 1a, lanes 3 and 4). Furthermore, Sem1 was not pulled down by Sac3 unless Thp1 was present (Fig. 1a, lanes 4 and 5), indicating that Sem1 stabilizes Thp1 and in turn facilitates formation of a stoichiometric Sac3–Thp1–Sem1 complex in vitro.
Figure 1.
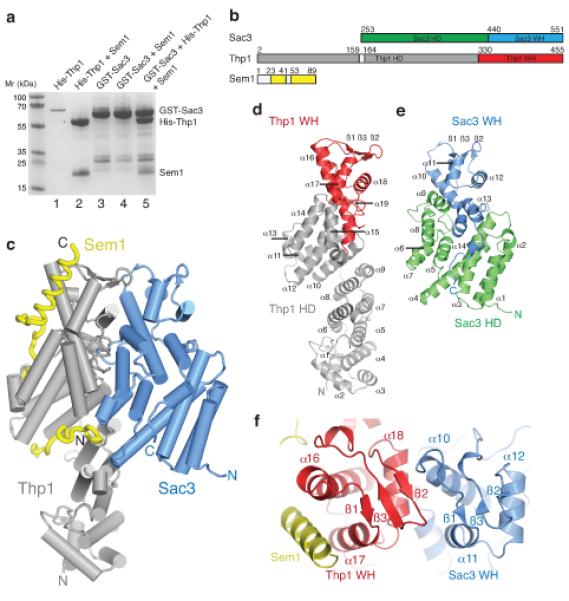
The structure of the Sac3–Thp1–Sem1 complex (a) SDS PAGE of pull-down assays used to monitor the assembly of the Sac3–Thp1–Sem1 complex. His-Thp1 is insoluble (lane 1 – the band at ~70kD is an impurity) unless coexpressed with Sem1 (lane 2), whereas GST-Sac3 is soluble when expressed alone (lane 3) and does not bind Sem1 upon coexpression (lane 4). The Sac3–Thp1–Sem1 complex is formed when GST-Sac3 and Thp1–Sem1 lysates are mixed (lane 5). Lanes 1-2 are His-tag pull-downs and lanes 3-5 are GST-pull-downs. (b) Domain organization in the Sac3–Thp1–Sem1 complex. (c) Overview of the Sac3–Thp1–Sem1 complex with α-helices represented as cylinders and Sem1 in worm format. (d,e) Ribbon views of Thp1 and Sac3, highlighting the secondary structural elements of the superhelical domains (HD, gray, green) and the winged helix domains (WH, red, blue). (f) Sac3–Thp1–Sem1 complex rotated ~90° about the horizontal compared with (c) highlighting the Sac3 and Thp1 winged helix domains.
Crystal structure of the Sac3–Thp1–Sem1 complex
Crystals obtained using vapor diffusion of Sac3 residues 250-563 (Sac3-M) bound to full-length Thp1 and Sem1 diffracted to 2.9 Å resolution using synchrotron radiation (Table 1). The asymmetric unit contained two copies of the complex and had a high solvent content of ~70 %. The final model generated by Se-Met SIR phasing followed by iterative cycles of refinement and rebuilding contained Sac3 residues 253-551, Thp1 residues 2-159 and 164-455, and Sem1 residues 23-41 and 53-89. Density for the remaining residues was absent from the final map (Fig. 1b,c).
Table 1. Crystallographic data.
| Sac3 (250-563)-Thp1-Sem1 | PCID2 (201-399)-DSS1 | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| native | Se peak | native | Se peak | Se inflection | Se remote | |
| Data collection | ||||||
| Space group | P4122 | P3121 | ||||
| Cell dimensions | ||||||
| a, b, c (Å) | 164.77, 164.77, 276.79 | 166.72, 166.72, 284.65 | 70.52, 70.52, 95.42 | 70, 70, 95 | ||
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 120 | 90, 90, 120 | ||
| Wavelength (Å) | 0.9795 | 0.9803 | 1.54179 | 0.9803 | 0.9805 | 0.9689 |
| Resolution (Å) | 20-2.9 (3.06-2.9)* | 30-4.2 (4.43-4.2) | 30-2.12 (2.23-2.12) | 60.98-3.4 (3.58-3.4) | 60.8-3.4 (3.58-3.4) | 60.65-3.4 (3.58-3.4) |
| R pim | 6.7 (42.8) | 4.8 (23.5) | 3.1 (36.4) | 8.6 (18.6) | 8.6 (21.8) | 8.1 (19.2) |
| I / σI | 7.4 (1.9) | 12.5 (3.5) | 14.1 (2.3) | 8.0 (4.2) | 8.3 (3.8) | 8.8 (4.4) |
| Completeness (%) | 99.7 (100) | 99.7 (100) | 91.0 (94.6) | 86.4 (69.8) | 86.5 (70.3) | 87.0 (71.3) |
| Redundancy | 7.0 (7.0) | 14.1 (14.6) | 7.3 (5.6) | 9.8 (10.5) | 10 (10.4) | 9.9 (10.4) |
| No. reflections | 591,489 (85,034) | 422,691 (63,007) | 106,955 (12,010) | 34,358 (4,212) | 34,866 (4,128) | 34,730 (4,284) |
| Refinement | ||||||
| Resolution (Å) | 20-2.9 | 30-2.12 | ||||
| Rwork / Rfree | 20.8 / 23.1 | 20.5 / 23.1 | ||||
| No. atoms | ||||||
| Protein | 26387 | 3753 | ||||
| Water | - | 33 | ||||
| B-factors | ||||||
| Protein | 49.4 | 46.8 | ||||
| Water | - | 45.6 | ||||
| r.m.s deviations | ||||||
| Bond lengths (Å) | 0.011 | 0.003 | ||||
| Bond angles (°) | 0.91 | 0.65 | ||||
Values in parentheses are for highest-resolution shell.
Figure 1c and Supplementary Movie 1 illustrate the structure of the Sac3–Thp1–Sem1 complex. Thp1 and Sac3 have PCI folds25 and are each based on an N-terminal right-handed superhelical domain capped by a C-terminal winged helix domain analogous to that observed in the eIF3k subunit of the eIF3 complex and the Csn7 component of the CSN complex28,29. A single Sem1 chain wraps intimately around Thp1, but makes little contact with Sac3, demonstrating that Sem1 is a stoichiometric component of the TREX-2 complex. The Thp1 superhelical domain forms a right-handed superhelix of 15 α-helices that is capped by a winged helix domain formed by helices α16-18 and β-strands 1-3 (Fig. 1d), Sac3-M has a comparable PCI fold with a right-handed superhelical domain formed by helices α1-9 and a winged helix domain formed by helices α10-12 and β-strands 1-3 (Fig. 1e,f and Supplementary Discussion). A striking feature of the complex is the spatial orientation of the winged helix domains. In both Thp1 and Sac3 these winged helix domains each have an αβααββ motif and associate to form a 6-stranded β-sheet at the surface of the complex (Fig. 1f). This arrangement generates a prominent winged helix dimer that, together with interactions between helices within the superhelical domains of Sac3 and Thp1, constitutes the extensive interaction interface between the two proteins.
Sem1 binds primarily to Thp1
Sem1 makes extensive contacts with Thp1, burying 2741 Å2 of surface area, but makes only minor contact to Sac3, burying 293 Å2 (Supplementary Movie 2). Sem1 residues 53-89 make extensive interactions across the surface of the Thp1 superhelical domain and form a strongly conserved C-terminal helix (Supplementary Fig. 1) that binds in the cleft formed between helices α16 and α17 of the winged helix domain (Fig. 2a,b). The conserved Sem1 Trp60 and Trp64 are buried in surface pockets formed between residues on Thp1 helices α11, α12, and α16, whereas conserved hydrophobic residues (Phe73, Leu77, Leu81) lock the Sem1 helix into the hydrophobic cleft of the winged helix domain (Fig. 2b). The Sem1 helix and Trp60 have excellent density (Supplementary Fig. 2a) and low B-factors, suggesting that they provide the Sem1 binding register, whereas the negative charges of the aspartic and glutamic acids strengthen the interaction by forming ionic interactions and hydrogen bonds with the positively charged surface of Thp1. This intimate burial of the Sem1 C-terminus is consistent with C-terminally tagged Sem1 losing both its ability to bind Thp1 in yeast and its mRNA export function24. Sem1 residues 23-41 bind in a positively charged cleft formed by the TPR-like helices of the Thp1 superhelical domain (Fig. 2c,d). In this cleft, aspartates and glutamates from Sem1 form extensive ionic interactions and hydrogen bonds with several Thp1 helices (Fig 2c). Furthermore, the ring of conserved Sem1 Phe35 is buried. This region of Sem1 also makes limited contact with Sac3 helices α4 and α6, but these Sem1 residues are poorly conserved and probably contribute little to the overall binding. No electron density was observed for the poorly-conserved residues (1-22) at the Sem1 N-terminus, and between the N and C-terminal binding sites (residues 42-52).
Figure 2.
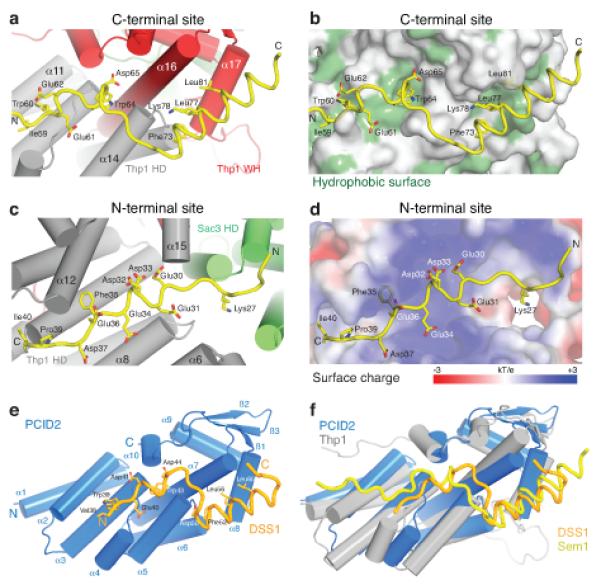
Structural basis for the Thp1–Sem1 interaction. (a,b) Interface between Thp1 and the C-terminal Sem1 binding region with (a) important Sem1 (yellow) sidechains shown as sticks and (b) hydrophobic Thp1 residues highlighted in green. (c,d) Corresponding views of the interface between the Sem1 N-terminal region (yellow) and Thp1 with (c) important sidechains shown as sticks and (d) the electrostatic surface potential of the N-terminal Sem1 binding site on Thp1. (e) PCID2 (residues 205-399, blue) and DSS1 (residues 38-67, orange) complex with the α-helices as cylinders and DSS1 in worm format. DSS1 sidechains that make key interactions within the interface are shown as sticks. (f) Structural alignment of the PCID2–DSS1 complex with the corresponding residues of Thp1 (grey) and Sem1 (yellow) from the Sac3–Thp1–Sem1 complex.
PCID2–DSS1 retains key structural features of Thp1–Sem1
The 2.1 Å resolution crystal structure of the human Thp1 homologue, PCID2 (residues 205-399) complexed with DSS1 (residues 38-67) was also determined (Fig. 2 and Table 1). Although not present in the crystal structure, the region homologous to Sac3 (250-563) in its human homologue GANP (residues 686-981, Supplementary Fig. 1b), retained binding affinity for the PCID2DSS1 complex (Supplementary Fig. 3). As observed for Thp1, coexpression with DSS1 was necessary to obtain soluble PCID2, consistent with a conserved stabilizing function of Sem1(DSS1) in these complexes (Supplementary Fig. 3). PCID2 retained the same pattern of helices followed by a C-terminal winged helix domain observed in Thp1, and the DSS1 C-terminal helix bound to PCID2 mimicking the binding of Sem1 to Thp1 (Fig. 2e,f, Supplementary Fig. 2b and Supplementary Movie 3). The corresponding regions of the two structures are highly homologous and show a strong conservation of the interaction interface between Thp1–Sem1 and PCID2–DSS1 (Supplementary Discussion). Furthermore, this structural homology indicates that the Thp1–Sem1 structure is most likely not altered substantially when it binds to Sac3 (Fig. 2f).
Structural basis for the recruitment of Thp1–Sem1 to Sac3
Our structural data indicate that recruitment of Thp1–Sem1 to Sac3 is mediated primarily by the interface formed between the Sac3 and Thp1 winged helix domains, with Sem1 making a marginal contribution. The Thp1–Sac3 interface is extensive, with a buried surface area of 2318 Å2. Thp1 helices α16, α17, α18 and the α17-α18 loop form the major interaction interface with Sac3 (Fig. 3a). These residues are predominantly within the Thp1 winged helix domain, with helix α18 appearing crucial to the interaction with numerous conserved residues interacting with Sac3 (Fig. 3b). This helix binds to a cleft formed by helix α9 of the Sac3 superhelical domain and α10 of the winged helix domain. Helices α6, α7 and α8 of the Sac3 HD domain also make significant interactions at the base of the interface with Thp1 helices α15 and α16 (Fig. 3c).
Figure 3.
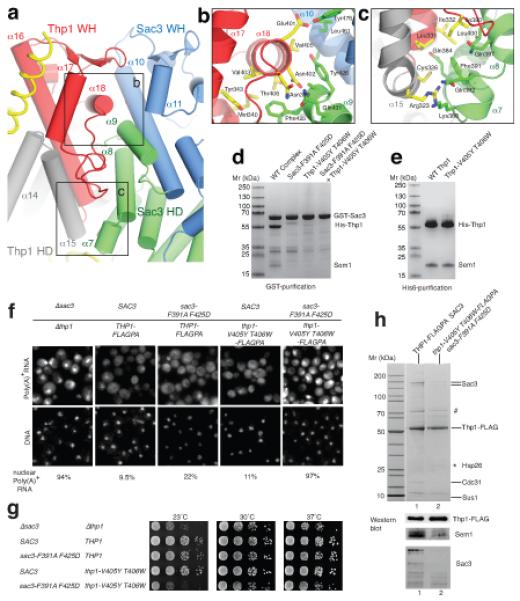
Structure-guided mutations that disrupt the Sac3-Thp1 interaction impair mRNA nuclear export and growth. (a) Arrangement of the winged helix (WH) domains of Thp1 (red) and Sac3 (blue) within the Sac3–Thp1 interaction interface. (b) Interface between helix α18 of the Thp1 winged helix domain and Sac3 helices α9 and α10. Sidechains of key interacting residues shown as sticks. (c) Interactions at the base of the Thp1–Sac3 interface. (d) SDS-PAGE showing impaired complex formation with either Sac3 or Thp1 mutants. (e) His-tag pull-down of coexpressed His-Thp1 and Sem1. (f) In situ hybridization analysis of poly(A)+ RNA export in thp1Δ sac3Δ cells expressing the indicated combinations of wild-type or mutant sac3 and thp1 alleles (see g). The fraction of cells showing nuclear poly(A)+ accumulation is indicated (n=200). (g) Serial dilutions of a yeast sac3Δthp1Δ double disruption strain expressing plasmid-borne SAC3 (TRP1, pRS314-SAC3) and THP1 (LEU2, YCplac111-THP1-FLAG-ProtA) wild-type or mutant alleles grown in on SDC-leu-trp plates at the indicated temperatures. For a negative control, the sac3Δthp1Δ strain was transformed with the empty plasmids pRS314 (TRP1) and pRS315 (LEU2). THP1 wild-type and thp1 mutant alleles were tagged with FLAGPA to facilitate affinity-purification. (h) Tandem affinity-purification of wild-type Thp1-FLAGPA from SAC3 wild-type cells or mutant Thp1-V405Y T406W-FLAGPA co-expressing sac3-F391A F425D. FLAG eluates were analyzed by SDS-PAGE and Coommassie staining, or Western blotting using anti-FLAG, anti-Sem1 and anti-Sac3 antibodies. Indicated bands were identified by mass spectrometry where # indicates Ssa1 plus Sac3, and * indicates bands that could not be identified.
The extensive interface between Thp1 and Sac3 frustrated engineering of single point mutations in either protein that alone could impair their interaction significantly (data not shown). However, a number of double point mutants in Sac3 and Thp1 were constructed that reduced the affinity of Thp1 for Sac3250-563 in vitro. Thus, Sac3250-563-F391A F425D that removed the buried hydrophobic Phe425 within the hydrophobic pocket formed by Thp1 α16, α18 and the α17-α18 loop, together with Phe391 that was buried at the base of the interface (Fig. 3b,c), showed reduced Thp1 binding (Fig. 3d). A second strategy targeted the interactions of Thp1 helix α18, which binds in the cleft formed by helices α9 and α10 of the Sac3 winged helix domain (Fig. 3b). The addition of two bulky side chains (V405Y T406W) to this helix obstructed the intimate association of the binding surfaces and reduced binding to Sac3250-563 (Fig. 3d), while retaining Sem1 binding, indicating that the structure of Thp1 was not altered significantly (Fig. 3e) by the mutations.
Ablating Sac3–Thp1 binding inhibits growth and mRNA export
In yeast, single sac3 or thp1 gene disruptions or a double sac3-thp1 knock-out are not lethal, but cause a slower growth phenotype, especially at higher (e.g. 37°C) and lower temperatures (e.g. 23°C), albeit the strength of these growth defects at different temperatures can vary depending on the strain background13,21,22. The in vivo consequences of disrupting the Sac3–Thp1 interaction were tested by expressing the sac3 and thp1 single, and sac3-thp1 double mutant constructs under their own promoter and from single copy plasmids in the corresponding sac3Δ, thp1Δ or sac3Δ-thp1Δ null strains. Epitope-tagged wild-type and mutant thp1 alleles, modified at the 3′-end of the gene with the FLAG-TEV-ProtA construct (THP1-FLAGPA or thp1-FLAGPA) were used, which allowed tandem affinity-purification of TREX-2 from wild-type and mutant cells and analysis of potential defects in TREX-2 assembly and/or stability. Individual sac3 or thp1 mutations that reduced the binding of Thp1–Sem1 to Sac3250-563 in vitro did not impair the interaction with full-length Sac3 in vivo sufficiently to cause a significant growth defect, albeit there was a small increase in the number of cells showing nuclear accumulation of poly(A)+ mRNA (Fig. 3f). However, the combination of these sac3 and thp1 alleles (sac3-F391A F425D; thp1-V405Y T406W) impaired cell growth at 23°C and caused mRNA export defects equivalent to the sac3Δthp1Δ double disruption mutant (Fig. 3f,g). Notably, when Thp1-FLAGPA was affinity-purified from this quadruple mutant, the Thp1-V405Y T406W mutant protein could be obtained in significant amounts, but the other TREX-2 subunits Sac3-F391A F425D, Cdc31, Sus1 and Sem1 did not co-enrich (Fig. 3h). These data indicate that TREX-2 assembly is effectively eliminated in vivo in the quadruple sac3-thp1 mutant and highlight the essential contribution made by the binding of Thp1–Sem1 to Sac3 to the function of TREX-2 in mRNA export.
Sac3–Thp1–Sem1 forms a platform that binds nucleic acids
Winged helix domains are common nucleic acid binding motifs that interact with dsDNA, ssDNA and RNA30 and previous work has indicated that Thp1 may bind nucleic acids in vitro31. The affinity of the Sac3–Thp1–Sem1 complex and its individual components for nucleic acids was assessed by electromobility gel shift assays (EMSA). Whereas the Sac3–Thp1–Sem1 complex bound polyA25 RNA to generate a slowly migrating complex at the top of the gel, its individual components (Thp1–Sem1 and ProteinA-tagged-Sac3250-563) did not associate efficiently with RNA (Fig. 4a,b). Nucleic acid binding could be reconstituted by incubating the individual components prior to the addition of RNA, indicating they were functional and the ProteinA-tag of Sac3250-563 did not interfere with RNA binding (Fig. 4b). Taken together, these data indicate that complex formation between Sac3–Thp1–Sem1 creates an architectural platform mediating nucleic acid association.
Figure 4.
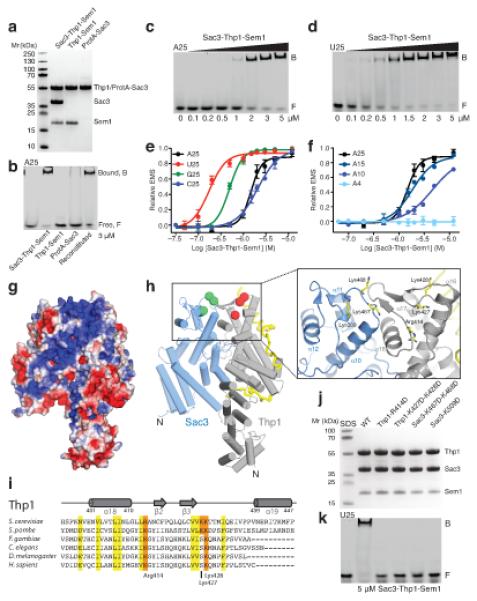
The juxtaposed winged helix domains of the Sac3–Thp1–Sem1 complex bind nucleic acids. (a) SDS-PAGE of purified proteins used in EMSA analysis. (b) EMSA of DY547-labeled polyA25 incubated with the indicated components of the Sac3–Thp1–Sem1 complex. The reconstituted complex lane consists of ProtA-Sac3+Thp1–Sem1 formed by pre-incubating the individual components prior to the assay. (c) EMSA of DY547-labeled polyA25 RNA incubated with increasing concentrations of the Sac3–Thp1–Sem1 complex. (d) EMSA of DY547-labeled polyU25 RNA incubated with increasing concentrations of the Sac3–Thp1–Sem1 complex. (e,f) Quantitation of the EMSA binding of the Sac3–Thp1–Sem1 complex to the indicated nucleic acids with each data point derived from at least two separate assays with error bars representing the standard deviation. (g) Electrostatic surface potential (kT/e between −3 and +3) of the Sac3–Thp1–Sem1 complex highlighting the electropositive stripe (blue) on the Sac3 and Thp1 winged helix domains and the outer surface of Sac3. (h) Thp1–Sac3–Sem1 complex orientated as in (g) with spheres at the Cα position of selected positive residues of Thp1 (red) and Sac3 (green). (i) Sequence alignment of the Thp1 winged helix domain highlighting the conserved positive residues (R414, K427 and K428). (j) SDS-PAGE of the proteins used in (k). (k) EMSA of DY547-labeled polyU25 RNA incubated with wild-type Sac3–Thp1–Sem1 complex or the indicated switched charge mutants.
The affinity of Sac3–Thp1–Sem1 for different types and lengths of nucleic acids was determined by EMSA (Fig. 4c-f, Supplementary Fig. 4, and Supplementary Table 1). The complex associated with an optimal RNA length of 25 bases, with an affinity of 175 nM (polyU25), and a significant decrease in affinity was observed for lengths less than 10 bases. The Sac3–Thp1–Sem1 complex had comparable affinity for both ssDNA and RNA and also associated with dsDNA with a similar affinity to a ssDNA fragment of the same sequence (Supplementary Fig. 4a-c). The Sac3–Thp1–Sem1 complex had a higher affinity for polyU RNA than other bases, suggesting a level of specificity in the binding to RNA.
In the complex, the juxtaposition of the Sac3 and Thp1 winged helix domains generates a positively charged stripe that runs from the junction between the two winged helix domains and down the surface of Sac3 and Thp1 (Fig. 4g). Within this positively charged stripe, Thp1 contains three highly conserved basic residues (Arg414, Lys427, Lys428) that are solvent exposed and do not appear to have a critical structural role (red spheres in Fig. 4h). Arg414 is positioned between helix α18 and the outer β2 strand of the Thp1 winged helix domain and is strongly conserved (Fig. 4h-i). Lysines 427 and 428 are solvent exposed on the second wing of the winged helix domain between the central β3 strand and helix α19, and are also strongly conserved (Fig. 4h-i). Sac3 also contains basic residues within this stripe including Lys467 and Lys468, positioned between the first helix α10 and β1 strand of the Sac3 winged helix domain, and Lys509. Although less obviously conserved, these residues are solvent exposed within the junction between the winged helix domains (green spheres in Fig. 4h). Variants were engineered in which these basic residues were mutated to aspartic acid. Thp1-R414D, and the double mutant K427D K428D, dramatically reduced the affinity of the Sac3–Thp1–Sem1 complex for nucleic acids (Fig. 4j-k). Similar impairment was seen with Sac3-K509D and the K467D K468D double mutant (Fig. 4j-k), indicating that these residues contribute to a novel nucleic acid binding surface generated by the juxtaposition of the Sac3 and Thp1 winged helix domains in the complex.
Impaired RNA binding generates growth and export defects
sac3 and thp1 mutants that impair DNA or RNA binding in vitro caused significant growth and mRNA export defects in vivo, similar to those observed with the combination of sac3 and thp1 alleles that interfered with the Sac3–Thp1 interaction in vivo (Fig. 5). Importantly, TREX-2 assembly was not disrupted in the thp1 mutants with impaired nucleic acid binding (Fig. 5c), showing that these structure-guided mutations uncoupled TREX-2 nucleic acid binding from complex assembly. This result highlights the contribution made by the TREX-2-nucleic acid association to the yeast mRNA export pathway.
Figure 5.
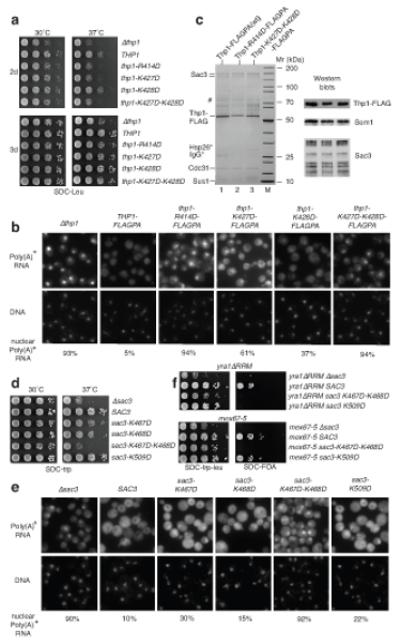
Structure-guided Thp1 and Sac3 mutants with impaired in vitro nucleic acid binding show cell growth and mRNA export defects, but do not inhibit TREX-2 assembly. (a) 10X serial dilutions of yeast thp1Δ expressing plasmid-borne THP1 (LEU2, YCplac111-THP1-FLAG-ProtA) wild-type or mutant alleles grown on SDC-leu plates. The thp1Δ cells were transformed with empty plasmid pRS315 (LEU2). (b) Poly(A)+ RNA export in thp1Δ cells expressing wild-type THP1 or mutant thp1 alleles, using the strains described in (a). The fraction of cells showing nuclear poly(A)+ accumulation is indicated (n=200). DNA stained with DAPI. (c) Tandem affinity-purification of plasmid-derived wild-type Thp1-FLAGPA and mutant Thp1-K414D-FLAGPA or Thp1-K427D K428D-FLAGPA from thp1Δ cells. Final eluates were analyzed by SDS-PAGE or Western blotting using anti-FLAG, anti-Sem1 and anti-Sac3 antibodies. Indicated bands were identified by mass spectrometry. # indicates Ssa1 plus Sac3; * bands not identified. (d) 10X serial dilutions of yeast sac3Δ expressing plasmid-borne SAC3(TRP1, pRS314-SAC3) wild-type or mutant alleles grown on SDC-trp1 plates. The sac3Δ cells were transformed with empty plasmid pRS314 (TRP1). (e) Poly(A)+ RNA export in sac3Δ cells expressing wild-type SAC3 or mutant sac3 alleles. The fraction of cells showing nuclear poly(A)+ accumulation is indicated (n=200). (f) Synthetically lethal interactions between sac3 mutant alleles that had impaired nucleic acid binding in vitro, and yra1-ΔRRM and mex67-5 alleles. pRS314-Sac3 wild-type and mutants were co-expressed with pRS315-yra1ΔRRM or pRS315-mex67-5 in sac3Δ yra1Δ cells (upper panel) or sac3Δ mex67Δ (lower panel) shuffle strains. Cells were grown in 10X serial dilutions on SDC-leu-trp or SDC+FOA plates.
sac3 mutations that showed impaired nucleic acid binding also exhibited genetic interactions with other mRNA export factors in vivo. Both the mex67-5 ts allele or the yra1-ΔRRM allele produce viable cells at 30°C. However, synthetic lethality was observed when either allele was combined with sac3 mutant alleles impaired in nucleic acid binding (Fig. 5f). Although Sac3250-563-K509D impaired RNA binding in vitro (Fig. 4j,k), this was not sufficiently severe to generate a growth defect in vivo and showed only a small export defect (Fig. 5d,e). However, this allele was synthetically lethal with yra1-ΔRRM, but not with mex67-5 (Fig. 5f). These data suggests that in vivo the milder RNA binding defect exerted by the sac3-K509D allele is linked to the function of Yra1 in the export pathway. Overall the in vivo data with the mutants that impaired RNA binding indicate a synergy between the TREX-2 mediated nucleic acid binding function and the Mex67-Yra1 dependent mRNA export pathway.
DISCUSSION
Within TREX-2, Sac3, Thp1 and Sem1 are critical for coupling transcription and pre-mRNA processing with nuclear export. The crystal structure of Thp1–Sem1–Sac3250-563 shows that both Thp1 and Sac3250-563 have analogous PCI folds in which N-terminal superhelical domains are capped by C-terminal winged helix domains. These data, together with the in vivo behavior of structure-based engineered mutations, provide insight into how TREX-2 functions at the interface between transcription and mRNA export.
Implications for integration of the gene expression pathway
Our in vitro and in vivo data indicate RNA binding by the juxtaposed Sac3 and Thp1 winged helix domains in TREX-2 is important for efficient mRNA nuclear export. Mutants in which nucleic acid binding was impaired showed both growth and mRNA export defects. The synthetic growth defects observed between these mutants and mex67-5 or yra1-ΔRRM indicate that TREX-2 binding to mRNA is important in generating export-competent mRNPs.
Our data support a model whereby NPC-bound TREX-2 acts as a scaffold to spatially integrate transcription and mRNA export and provides a structural context for how this could be achieved (Fig. 6). TREX-2 comprises two subregions that make different contributions to its function. Whereas the localization of TREX-2 to NPCs requires its proximal CID domain complexed with Cdc31 and two Sus1 chains13,22, its interaction with the transcription and mRNP assembly machinery is mediated by its distal N- and M-regions21 that bind Thp1–Sem1 and Mex67–Mtr2. Deletion of thp1 or sem1 results in mRNA export defects and generation of mRNPs with elevated Yra1 and Sub2 levels23. These observations, together with the synthetic growth defects observed between TREX-2 mutants with impaired nucleic acid binding and the mex67-5 or yra1-ΔRRM alleles (Fig. 5f), are consistent with the distal region of TREX-2 functioning to load Mex67–Mtr2 and generate export-competent mRNPs23. Mex67–Mtr2 by itself has low affinity for mRNA and its recruitment to mRNPs and generation of export-competent complexes requires Yra1 (refs 1,3). Mex67–Mtr2 binds to the Sac3 N-terminus adjacent to Thp1–Sem1 probably through its NTF2-like and UBA domains binding to Sac3 FG-nup-like repeats21. As illustrated in Figure 6, tethering an mRNA transcript already loaded with Yra1 and Sub2 to the winged helix domains of Sac3 and Thp1 would bring it in close proximity to the Mex67–Mtr2 heterodimer, promoting the generation of an export-competent mRNP in the immediate vicinity of the pore, thereby increasing the rate of mRNA export.
Figure 6.
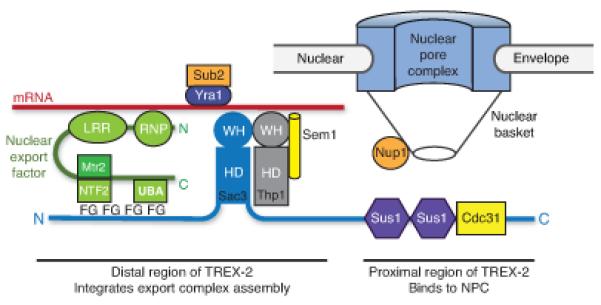
Schematic illustration of how the TREX-2 complex integrates the formation of an export competent mRNP adjacent to NPCs. The proximal region of Sac3 to which Sus1 and Cdc31 are attached binds to components of the nuclear basket such as Nup1 whereas the distal region that contains the winged helix domains of Sac3 and Thp1 binds to mRNA, to which Yra1–Sub2 is also attached. The Mex67–Mtr2 nuclear export factor binds to the FG repeats in the distal region of Sac3, probably via its NTF2 and UBA domains that bind to FG-nucleoporins. This interaction brings the Mex67 LRR and RNP domains close to the mRNA, facilitating the Sub2-mediated remodeling that generates an export-competent mRNP close to the NPC.
TREX-2 plays a critical role in the redistribution of actively transcribed genes in gene gating, through its interactions with both SAGA and NPCs12. Tethering of the transcription machinery to NPCs is proposed to increase the rate of mRNA export26. The role of TREX-2 in generating export competent mRNPs, as proposed in Figure 6, would ensure mRNP packaging is not rate-limiting, allowing cells to take advantage of this redistribution of the transcriptional machinery to NPCs and increase the rate of mRNA export. Although we have not explored this possibility in vivo, because TREX-2 also associates with dsDNA in vitro, the complex could also associate with DNA exposed in actively-transcribing genes, thereby facilitating their association with NPCs.
Sem1 function and implications for other PCI-complexes
Although Sem1 has been proposed to function as the “molecular glue” that holds Sac3 and Thp1 together32, in TREX-2 Sem1 makes few contacts with Sac3 and instead appears to facilitate complex formation by stabilizing Thp1. A stabilizing role for Sem1 would be consistent with studies in sem1Δ strains that showed reduced TREX-2 Thp1 levels24. Sem1 may also stabilize other complexes. Within the 19S proteosome lid33, Sem1 associates with the PCI-proteins Rpn7 and Rpn3 to form a subcomplex with Rpn12. Significantly, Sem1 deletion results in destabilization of the 19S proteosome34,35. Sem1 may stabilize Rpn7 or Rpn3 in a manner analogous to the Sac3–Thp1–Sem1 complex, facilitating binding through winged helix juxtaposition. Indeed, deletion of the central acidic motif of Sem1 that associates with the Thp1 TPR-like domain, precludes association of Rpn3 with the proteosome36. Sem1 binds to Thp1 by a mechanism similar to the binding of Hsp70 and Hsp90 to Hop, with the repeating negatively-charged and hydrophobic residues binding in an extended conformation to the positively charged concave binding surface formed by the Thp1 TPR-like repeats37. This binding mechanism is likely to be conserved in the other PCI-domain complexes that interact with Sem1, which could help identify putative binding sites in the PCI domains of the 19S proteosome lid and the eIF3 and CSN complexes.
The interaction between the Thp1 and Sac3 winged helix domains dominates the interface between these two chains. This structure, combined with the higher level of conservation between winged helix domains relative to the superhelical domain in other PCI-fold proteins38, suggests that interactions between winged helix domains could be the major interaction interface between chains within PCI complexes. In addition to its architectural role in complex formation, this winged helix alignment may create a functional interaction platform analogous to that seen in the Sac3–Thp1–Sem1 complex. Thus, Csn12 and Thp3 also form a stable complex with Sem1 that functions in RNA splicing and transcription elongation through recruitment to transcribed chromatin23,39. Although further work is required to verify the extent to which functional similarity is shared, the homology between Sac3–Thp1–Sem1 and Csn12–Thp3–Sem1 indicates that these complexes may share a nucleic acid binding function mediated by close apposition of their winged helix domains.
In summary, the structure of Sac3–Thp1–Sem1 provides a structural basis for understanding how the TREX-2 complex facilitates the formation of export-competent mRNPs at the nuclear face of NPCs and thereby provides a “fast track” for the nuclear export of a subset of transcripts such as those from the GAL system. The close juxtaposition of the winged helix domains of Sac3 and Thp1 is critical for binding mRNA and thereby bringing it in close apposition to the Mex67–Mtr2 export factor. This, in turn, facilitates a remodeling of the RNP that displaces Yra1 while attaching Mex67–Mtr2 more strongly. The small Sem1 protein functions to stabilize Thp1 and probably functions in an analogous way in other complexes that contain PCI folds.
Supplementary Material
ACKNOWLEDGEMENTS
We are most grateful to our colleagues in Cambridge and Heidelberg, especially Divyang Jani, Sonja Kuhlmann, Neil Marshall, Eugene Valkov and Michelle Halls for their helpful comments and assistance. We also thank Thomas Sorensen and Mark Williams at the Diamond Light Source. A.M.E was supported by a Marie Curie Fellowship and an EMBO Long-term Fellowship. Supported in part by grants from the Medical Research Council (U105178939) and the Wellcome Trust (MS) and the Deutsche Forschungsgemeinschaft (SFB 638/B2) (EH).
APPENDIX
METHODS
Cloning and protein purification
PCR-amplified Thp1 and Sem1 cDNAs were cloned into the first and second MCS of RSFDuet-1 (Novagen) respectively, and Sac3 (250-563) cDNA was cloned into a modified TEV protease-cleavable version of pGEX-4T-1 (GE Healthcare)40. PCR-amplified PCID2 (201-399) and DSS1 cDNAs were cloned into the first and second MCS of RSFDuet-1 (Novagen) respectively. The recombinant proteins were expressed in BL21-CodonPlus(DE3)-RIL cells (Stratagene) by IPTG induction at 20°C. Mutations were introduced using QuikChange site-directed mutagenesis (Stratagene). The Sac3–Thp1–Sem1 complex was purified from the soluble fraction by sequential chromatography on Glutathione Sepharose 4B and Superdex 200 columns (GE Healthcare). The PCID2–DSS1 complex was purified from the soluble fraction by sequential chromatography on HisTrap HP, HiPrep 26/10, and Superdex 200 columns (GE Healthcare) (Discussed in detail in Supplementary Methods).
Crystallization and structure determination
Sac3–Thp1–Sem1 crystals were grown at 19°C by hanging drop vapor diffusion in 2.2 M NaCl, 50 mM citric acid (pH 5.2). Se-Met crystals were grown in 1.95 M NaCl, 50 mM citric acid (pH 5.3). Sac3–Thp1–Sem1 crystals were flash cooled in liquid nitrogen in 2.8 M sodium malonate (pH 5.2). PCID2–DSS1 crystals were grown at 19°C by hanging drop vapor diffusion in 25.5% (w/v) PEG 2K MME, 0.95 M sodium formate, 0.1 M MES (pH 6.5). Se-Met PCID2–DSS1 crystals were grown in 23 % (w/v) PEG 2K MME, 0.8 M sodium formate, 0.1 M MES (pH 6.5). PCID2–DSS1 crystals were flash cooled in liquid nitrogen in mother liquor containing 10% ethylene glycol. Crystallographic data were collected in-house using a Rigaku FR-E+ SuperBright generator equipped with Osmic mirrors and a MarDTB camera, or at the Diamond Light Source, UK (Beamlines I02 and I03).
The Sac3–Thp1–Sem1 structure was determined by SIR phasing of native and Se-Met datasets (Table 1) in AutoSharp41,42 followed by solvent flipping43. The resultant phases were applied to the high-resolution native dataset and the model built manually into the resultant map with Coot44. The high solvent content (~70%) of the crystals greatly enhanced the clarity of the maps after solvent flipping. Iterative cycles of refinement were carried out using PHENIX45 and REFMAC546, with local rebuilding in Coot, gave a model with an R-factor of 20.8% (Rfree 23.1%) and excellent geometry (Table 1). The structure had a final MolProbity47 score of 2.23 (98th percentile). The PCID2–DSS1 structure was determined using the native and Se-Met (Table 1) datasets in PHENIX Autosol incorporating the AutoBuild program45. Iterative cycles of refinement in PHENIX Refine and local rebuilding in Coot gave a model with an R-factor of 20.5% (Rfree 23.1%) and excellent geometry (Table 1). The structure had a final MolProbity47 score of 1.02 (100th percentile).
Protein binding assays
Sac3–Thp1–Sem1 complex assembly was monitored using by pull-downs using Ni-NTA resin (Qiagen) or Glutathione Sepharose 4B (GE Healthcare). Thp1 was cloned into pET28a (Novagen) and for coexpression His-Thp1 and Sem1 were cloned into RSFDuet-1 (Novagen). GST-Sac3 was cloned into pET24d (Novagen) and for coexpression GST-Sac3 and Sem1 were cloned into pETDuet-1 (Novagen). Western blots confirmed both proteins were expressed. The in vitro assembly of GANP–PCID2–DSS1 was monitored using the same vector strategy described above. To monitor Sac3 and Thp1 mutants, cell lysates expressing GST-Sac3 or coexpressing Thp1–Sem1 were mixed and incubated with Glutathione Sepharose 4B resin at 4°C for 1 hr. To monitor the association of Thp1-V405Y T406W with Sem1, cell lysate coexpressing His-Thp1 and Sem1 was incubated with Ni-NTA resin at 4°C for 1 hr. In each case, resin was washed with 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM DTT, 40 mM imidazole. Samples were analyzed by SDS-PAGE and Coomassie stained.
EMSA
EMSA assays were performed by adding 100 nM of 5′-DY547-labeled RNA (Dharmacon) or 100 nM 5′-Cy3-labeled DNA (Sigma) to the indicated protein in binding buffer containing 20 mM Tris, 10 mM NaCl, 2 mM MgCl2, 0.1% (w/v) bovine serum albumin, 2% (v/v) glycerol and 0.1% (w/v) Orange G. Samples were incubated on ice for 30 min before separation on a 6 % DNA retardation gel (Invitrogen) at 4°C. The DY547-RNA or Cy3-DNA was detected using a Typhoon Trio Variable Mode Imager (GE Healthcare) and band-density analyzed with ImageJ and Graphpad Prism.
Strains and plasmids
Yeast strains and plasmids are listed in Supplementary Tables 2 and 3. Gene disruption strains were obtained by homologous integration of PCR-derived DNA fragments fitted with the appropriate selectable marker gene48. Plasmid yCpLac111-THP1-FLAGPA was generated by cloning THP1 including its endogenous promoter into the BamHI/EcoRI sites of plasmid yCLac111. The FLAGPA (FLAG-TEV-ProtA) tag was inserted between the EcoRI and NotI sites. PCR-based site-directed mutagenesis within SAC3 and THP1 was performed using the Finnzymes Phusion polymerase49, with pRS314-SAC321 and yCPLac111-THP1-FLAGPA as templates.
Analysis of nuclear mRNA export by in situ hybridization
Analysis of poly(A)+ RNA export by in situ hybridization was performed using Cy3-labelled oligo-d(T) probes50. Fluorescence microscopy used an Imager Z1 (Carl Zeiss) with a 63× NA 1.4 Plan-Apo-Chromat oil immersion lens (Carl Zeiss) and DICIII, DAPI or HECy3 filter sets. Images were acquired with an AxioCamMRm camera (Carl Zeiss) and AxioVision 4.3 software (Carl Zeiss) at resolution 1388 × 1040 (Binning 1×1, gain factor 1).
Tandem affinity-purification of Thp1 and Sac3
For tandem affinity-purification using the FLAG-TEV-ProtA tandem affinity-tag (FLAGPA), cells expressing FLAGPA constructs were grown in synthetic medium to OD~1 before transfer into YPD medium for two doubling times. Cells were harvested and lysed in 100 mM NaCl, 50 mM Tris-Cl pH 7.5, 2 mM CaCl2, 1.5 mM MgCl2, 1 mM DTT, 0.5 mM DTT, FY complete protease inhibitor, by grinding in liquid nitrogen (Retsch, MM400). Affinity-purification was performed as described51, except for the addition of a second purification step using beads coupled to anti-FLAG antibodies. Eluates were analyzed on 4-12% SDS-PAGE gradient gels (Invitrogen) and either Coomassie stained or analyzed by Western blotting. Anti-Sem1 and anti-Sac3 antibodies were kindly provided by Dr. Jussi Jäntti (Helsinki, Finland) and Dr. Ralf Kölling (Stuttgart, Germany).
Footnotes
AUTHOR CONTRIBUTIONS
A.M.E. and M.S. determined structures and performed in vitro experiments whereas L.D. and E.H. performed in vivo studies. All authors contributed to interpretation of results and writing the manuscript.
Accession codes
The coordinates and structure factors for the 2.9 Å resolution Sac3–Thp1–Sem1 and the 2.1 Å PCID2–DSS1 complexes have been deposited in the Protein Data Bank with accession codes 3T5V and 3T5X, respectively.
REFERENCES
- 1.Köhler A, Hurt E. Exporting RNA from the nucleus to the cytoplasm. Nat Rev Mol Cell Biol. 2007;8:761–773. doi: 10.1038/nrm2255. [DOI] [PubMed] [Google Scholar]
- 2.Kelly SM, Corbett AH. Messenger RNA export from the nucleus: a series of molecular wardrobe changes. Traffic. 2009;10:1199–1208. doi: 10.1111/j.1600-0854.2009.00944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stewart M. Nuclear export of mRNA. Trends Biochem Sci. 2010;35:609–617. doi: 10.1016/j.tibs.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez-Navarro S, Hurt E. Linking gene regulation to mRNA production and export. Curr Opin Cell Biol. 2011;23:302–309. doi: 10.1016/j.ceb.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Chávez S, et al. A protein complex containing Tho2, Hpr1, Mft1 and a novel protein, Thp2, connects transcription elongation with mitotic recombination in Saccharomyces cerevisiae. EMBO J. 2000;19:5824–5834. doi: 10.1093/emboj/19.21.5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strässer K, et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature. 2002;417:304–308. doi: 10.1038/nature746. [DOI] [PubMed] [Google Scholar]
- 7.Hurt E, Luo M-J, Röther S, Reed R, Strässer K. Cotranscriptional recruitment of the serine-arginine-rich (SR)-like proteins Gbp2 and Hrb1 to nascent mRNA via the TREX complex. Proc Natl Acad Sci USA. 2004;101:1858–1862. doi: 10.1073/pnas.0308663100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reed R, Hurt E. A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell. 2002;108:523–531. doi: 10.1016/s0092-8674(02)00627-x. [DOI] [PubMed] [Google Scholar]
- 9.Stutz F, Izaurralde E. The interplay of nuclear mRNP assembly, mRNA surveillance and export. Trends Cell Biol. 2003;13:319–327. doi: 10.1016/s0962-8924(03)00106-5. [DOI] [PubMed] [Google Scholar]
- 10.Blobel G. Gene gating: a hypothesis. Proc Natl Acad Sci USA. 1985;82:8527–8529. doi: 10.1073/pnas.82.24.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez-Navarro S, et al. Sus1, a functional component of the SAGA histone acetylase complex and the nuclear pore-associated mRNA export machinery. Cell. 2004;116:75–86. doi: 10.1016/s0092-8674(03)01025-0. [DOI] [PubMed] [Google Scholar]
- 12.Cabal GG, et al. SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature. 2006;441:770–773. doi: 10.1038/nature04752. [DOI] [PubMed] [Google Scholar]
- 13.Fischer T, et al. Yeast centrin Cdc31 is linked to the nuclear mRNA export machinery. Nat Cell Biol. 2004;6:840–848. doi: 10.1038/ncb1163. [DOI] [PubMed] [Google Scholar]
- 14.Light WH, Brickner DG, Brand VR, Brickner JH. Interaction of a DNA zip code with the nuclear pore complex promotes H2A.Z incorporation and INO1 transcriptional memory. Mol Cell. 2010;40:112–125. doi: 10.1016/j.molcel.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed S, et al. DNA zip codes control an ancient mechanism for gene targeting to the nuclear periphery. Nat Cell Biol. 2010;12:111–118. doi: 10.1038/ncb2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abruzzi KC, Belostotsky DA, Chekanova JA, Dower K, Rosbash M. 3′-end formation signals modulate the association of genes with the nuclear periphery as well as mRNP dot formation. EMBO J. 2006;25:4253–4262. doi: 10.1038/sj.emboj.7601305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chekanova JA, Abruzzi KC, Rosbash M, Belostotsky DA. Sus1, Sac3, and Thp1 mediate post-transcriptional tethering of active genes to the nuclear rim as well as to non-nascent mRNP. RNA. 2008;14:66–77. doi: 10.1261/rna.764108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egecioglu D, Brickner JH. Gene positioning and expression. Curr Opin Cell Biol. 2011 doi: 10.1016/j.ceb.2011.01.001. doi:10.1016/j.ceb.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.González-Aguilera C, et al. The THP1-SAC3-SUS1-CDC31 complex works in transcription elongation-mRNA export preventing RNA-mediated genome instability. Mol Biol Cell. 2008;19:4310–4318. doi: 10.1091/mbc.E08-04-0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tous C, et al. A novel assay identifies transcript elongation roles for the Nup84 complex and RNA processing factors. EMBO J. 2011;30:1953–1964. doi: 10.1038/emboj.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer T, et al. The mRNA export machinery requires the novel Sac3p-Thp1p complex to dock at the nucleoplasmic entrance of the nuclear pores. EMBO J. 2002;21:5843–5852. doi: 10.1093/emboj/cdf590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jani D, et al. Sus1, Cdc31, and the Sac3 CID region form a conserved interaction platform that promotes nuclear pore association and mRNA export. Mol Cell. 2009;33:727–737. doi: 10.1016/j.molcel.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilmes GM, et al. A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing. Mol Cell. 2008;32:735–746. doi: 10.1016/j.molcel.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faza MB, et al. Sem1 is a functional component of the nuclear pore complex-associated messenger RNA export machinery. J Cell Biol. 2009;184:833–846. doi: 10.1083/jcb.200810059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pick E, Hofmann K, Glickman MH. PCI complexes: Beyond the proteasome, CSN, and eIF3 Troika. Mol Cell. 2009;35:260–264. doi: 10.1016/j.molcel.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 26.Brickner DG, et al. H2A.Z-mediated localization of genes at the nuclear periphery confers epigenetic memory of previous transcriptional state. PLoS Biol. 2007;5:e81. doi: 10.1371/journal.pbio.0050081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wickramasinghe VO, et al. mRNA export from mammalian cell nuclei is dependent on GANP. Curr Biol. 2010;20:25–31. doi: 10.1016/j.cub.2009.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dessau M, et al. The Arabidopsis COP9 signalosome subunit 7 is a model PCI domain protein with subdomains involved in COP9 signalosome assembly. Plant Cell. 2008;20:2815–2834. doi: 10.1105/tpc.107.053801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei Z, et al. Crystal structure of human eIF3k, the first structure of eIF3 subunits. J Biol Chem. 2004;279:34983–34990. doi: 10.1074/jbc.M405158200. [DOI] [PubMed] [Google Scholar]
- 30.Gajiwala KS, Burley SK. Winged helix proteins. Curr. Opin. Struct. Biol. 2000;10:110–116. doi: 10.1016/s0959-440x(99)00057-3. [DOI] [PubMed] [Google Scholar]
- 31.Gallardo M, Luna R, Erdjument-Bromage H, Tempst P, Aguilera A. Nab2p and the Thp1p-Sac3p complex functionally interact at the interface between transcription and mRNA metabolism. J Biol Chem. 2003;278:24225–24232. doi: 10.1074/jbc.M302900200. [DOI] [PubMed] [Google Scholar]
- 32.Faza MB, Kemmler S, Panse VG. Sem1: A versatile “molecular glue”? Nucleus. 2010;1:12–17. doi: 10.4161/nucl.1.1.10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharon M, Taverner T, Ambroggio XI, Deshaies RJ, Robinson CV. Structural organization of the 19S proteasome lid: insights from MS of intact complexes. PLoS Biol. 2006;4:e267. doi: 10.1371/journal.pbio.0040267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isono E, Saeki Y, Yokosawa H, Toh-e A. Rpn7 Is required for the structural integrity of the 26 S proteasome of Saccharomyces cerevisiae. J Biol Chem. 2004;279:27168–27176. doi: 10.1074/jbc.M314231200. [DOI] [PubMed] [Google Scholar]
- 35.Funakoshi M, Li X, Velichutina I, Hochstrasser M, Kobayashi H. Sem1, the yeast ortholog of a human BRCA2-binding protein, is a component of the proteasome regulatory particle that enhances proteasome stability. J Cell Sci. 2004;117:6447–6454. doi: 10.1242/jcs.01575. [DOI] [PubMed] [Google Scholar]
- 36.Wei S-J, et al. Identification of a specific motif of the DSS1 protein required for proteasome interaction and p53 protein degradation. J Mol Biol. 2008;383:693–712. doi: 10.1016/j.jmb.2008.08.044. [DOI] [PubMed] [Google Scholar]
- 37.Scheufler C, et al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 38.Scheel H, Hofmann K. Prediction of a common structural scaffold for proteasome lid, COP9-signalosome and eIF3 complexes. BMC Bioinformatics. 2005;6:71. doi: 10.1186/1471-2105-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jimeno S, et al. New suppressors of THO mutations identify Thp3 (Ypr045c)-Csn12 as a protein complex involved in transcription elongation. Mol Cell Biol. 2011;31:674–685. doi: 10.1128/MCB.01188-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuura Y, Stewart M. Structural basis for the assembly of a nuclear export complex. Nature. 2004;432:872–877. doi: 10.1038/nature03144. [DOI] [PubMed] [Google Scholar]
- 41.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol. Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 42.Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Meth. Enzymol. 1997 doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 43.Abrahams JP, Leslie AGW. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D Biol. Crystallogr. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collaborative Computational Project Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 47.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Longtine MS, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 49.Edelheit O, Hanukoglu A, Hanukoglu I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 2009;9:61. doi: 10.1186/1472-6750-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amberg DC, Goldstein AL, Cole CN. Isolation and characterization of RAT1: an essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev. 1992;6:1173–1189. doi: 10.1101/gad.6.7.1173. [DOI] [PubMed] [Google Scholar]
- 51.Rigaut G, et al. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.