

Summary
Gorillas are humans’ closest living relatives after chimpanzees, and are of comparable importance for the study of human origins and evolution. Here we present the assembly and analysis of a genome sequence for the western lowland gorilla, and compare the whole genomes of all extant great ape genera. We propose a synthesis of genetic and fossil evidence consistent with placing the human-chimpanzee and human-chimpanzee-gorilla speciation events at approximately 6 and 10 million years ago (Mya). In 30% of the genome, gorilla is closer to human or chimpanzee than the latter are to each other; this is rarer around coding genes, indicating pervasive selection throughout great ape evolution, and has functional consequences in gene expression. A comparison of protein coding genes reveals approximately 500 genes showing accelerated evolution on each of the gorilla, human and chimpanzee lineages, and evidence for parallel acceleration, particularly of genes involved in hearing. We also compare the western and eastern gorilla species, estimating an average sequence divergence time 1.75 million years ago, but with evidence for more recent genetic exchange and a population bottleneck in the eastern species. The use of the genome sequence in these and future analyses will promote a deeper understanding of great ape biology and evolution.
Humans share many elements of their anatomy and physiology with both gorillas and chimpanzees, and our similarity to these species was emphasised by Darwin and Huxley in the first evolutionary accounts of human origins1. Molecular studies confirmed that we are closer to the African apes than to orangutans, and on average closer to chimpanzees than gorillas2 (Fig. 1a). Subsequent analyses have explored functional differences between the great apes and their relevance to human evolution, assisted recently by reference genome sequences for chimpanzee3 and orangutan4. Here we provide a reference assembly and initial analysis of the gorilla genome sequence, establishing a foundation for the further study of great ape evolution and genetics.
Figure 1. Speciation of the great apes.
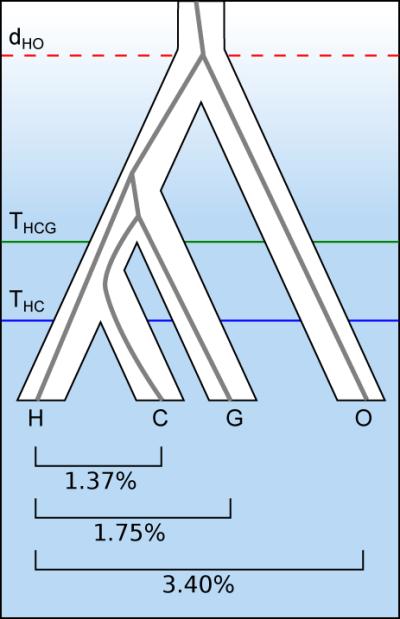
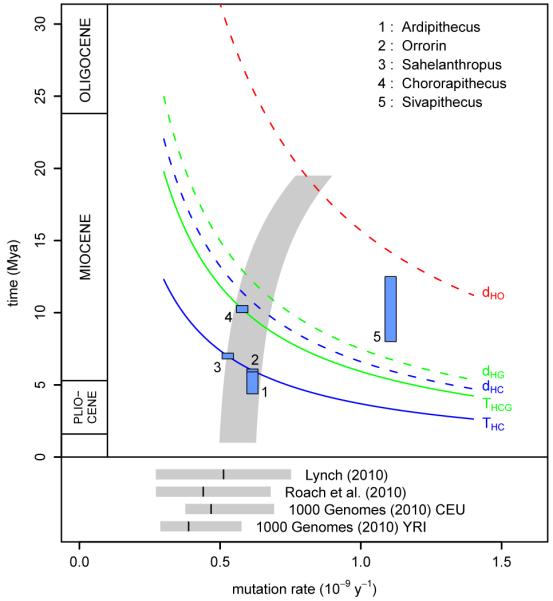
a, Phylogeny of the great ape family, showing the speciation of human (H), chimpanzee (C), gorilla (G) and orangutan (O). Horizontal lines indicate speciation times within the hominine subfamily and the sequence divergence time between human and orangutan. Interior grey lines illustrate an example of incomplete lineage sorting at a particular genetic locus – in this case (((C, G), H), O) rather than (((H, C), G), O). Below are mean nucleotide divergences between human and the other great apes from the EPO alignment. b, Great ape speciation and divergence times. Upper panel: solid lines show how times for the HC and HCG speciation events estimated by CoalHMM vary with average mutation rate; dashed lines show the corresponding average sequence divergence times, as well as the HO sequence divergence. Blue blocks represent hominid fossil species: each has a vertical extent spanning the range of dates estimated for it in the literature13,50, and a horizontal position at the maximum mutation rate consistent both with its proposed phylogenetic position and the CoalHMM estimates (including some allowance for ancestral polymorphism in the case of Sivapithecus). The grey shaded region shows that an increase in mutation rate going back in time can accommodate present-day estimates, fossil hypotheses, and a mid-Miocene speciation for orangutan. Lower panel: estimates of the average mutation rate in present-day humans10-12; grey bars show 95% confidence intervals, with black lines at the means.
Recent technological developments have dramatically reduced the costs of sequencing, but the assembly of a whole vertebrate genome remains a challenging computational problem. We generated a reference assembly from a single female western lowland gorilla (Gorilla gorilla gorilla) named Kamilah, using 5.4 Gbp of capillary sequence combined with 166.8 Gbp of Illumina read pairs (see Methods Summary). Genes, transcripts and predictions of gene orthologues and paralogues were annotated by Ensembl5, and additional analysis found evidence for 498 functional long (> 200 bp) intergenic RNA transcripts. Table 1 summarizes the assembly and annotation properties. An assessment of assembly quality using finished fosmid sequences found that typical (N50) stretches of error-free sequence are 7.2 kbp in length, with errors tending to be clustered in repetitive regions. Outside RepeatMasked regions and away from contig ends, the total rate of single-base and indel errors is 0.13 per kbp. See Supplementary Information for further details.
Table 1.
Assembly and annotation statistics
| Assembly | Annotation | ||
|---|---|---|---|
| Total length | 3,041,976,159 bp | Protein-coding genes | 20,962 |
| Contigs | 465,847 | Pseudogenes | 1,553 |
| Total contig length | 2,829,670,843 bp | RNA genes | 6,701 |
| Placed contig length | 2,712,844,129 bp | Gene exons | 237,216 |
| Unplaced contig length | 116,826,714 bp | Gene transcripts | 35,727 |
| Max contig length | 191,556 bp | lincRNA transcripts | 498 |
| Contig N50 | 11.8 kbp | ||
| Scaffolds | 22,164 | ||
| Max scaffold length | 10,247,101 bp | ||
| Scaffold N50 | 914 kbp |
We also collected less extensive sequence data for three other gorillas, to enable a comparison of species within the Gorilla genus. Gorillas survive today only within several isolated and endangered populations whose evolutionary relationships are uncertain. In addition to Kamilah, our analysis included two western lowland gorillas, Kwanza (male) and EB(JC) (female), and one eastern lowland, Mukisi (male).
Speciation of the great apes
We included the Kamilah assembly with human, chimpanzee, orangutan and macaque in a 5-way whole genome alignment using the Ensembl EPO pipeline6 (Table ST3.2). Filtering out low-quality regions of the chimpanzee assembly and regions with many alignment gaps, we obtained 2.01 Gbp of 1:1:1:1 great ape orthologous alignment blocks, to which we then applied a coalescent inference model, CoalHMM, to estimate the timescales and population sizes involved in the speciation of the hominines (African great apes; see Table ST1.1 for terminology), with orangutan as an outgroup (Supplementary Information).
Two issues need to be addressed in interpreting the results from CoalHMM (Table ST4.2). Firstly, the results themselves are obtained in units of sequence divergence rather than years, and so need to be scaled by an appropriate yearly mutation rate. Secondly, as with any model, CoalHMM makes several simplifying assumptions whose consequences we need to understand in the context of realistic demography. We discuss these issues in turn.
Using a rate of 10−9 mutations per bp per year, derived from fossil calibration of the human-macaque sequence divergence and as used in previous calculations, CoalHMM’s results would correspond to speciation time estimates THC and THCG of 3.7 and 5.95 Mya respectively (Fig. 1b). These dates are consistent with other recent molecular estimates7,8, but are at variance with certain aspects of the fossil record, including several fossils which have been proposed—though not universally accepted13—to be hominins, and therefore to postdate the human-chimpanzee split (Fig. 1b). Indeed the relationship between molecular and fossil evidence has remained difficult to resolve despite the accumulation of genetic data9. Direct estimates of the per-generation mutation rate in modern human populations, based on the incidence of disease-causing mutations10 or sequencing of familial trios11,12, indicate that a lower value of 0.5-0.6 × 10−9 bp−1y−1 is plausible (based on average hominine generation times of 20 to 25 y). This would give substantially older estimates of approximately 6 and 10 Mya for THC and THCG, potentially in better agreement with the fossil record.
However this timetable for hominine speciation must also be reconciled with older events such as the speciation of orangutan, which is thought to have occurred no earlier than the Middle Miocene (12-16 Mya), as fossil apes prior to that differ substantially from what we might expect of an early great ape14. This is possible if we allow for mutation rates changing over time, with a mutation rate of around 1 × 10−9 bp−1y−1 in the common ancestor of great apes, decreasing to lower values in all extant species (Fig. 1b). Comparable changes in mutation rate have been observed previously in primate evolution on larger timescales, including an approximately 30% branch length decrease in humans compared to baboons since their common ancestor15. A decrease within the great apes is also a predicted consequence of the observed increase in body sizes over this time period and the association of small size with shorter generation times in other primates16, and is consistent with deviations from a molecular clock seen in sequence divergences of the great apes and macaque (Table ST3.3). We discuss these and other constraints on estimates of great ape speciation times in the Supplementary Information. However we note that Sahelanthropus and Chororapithecus remain difficult to incorporate in this model, and can be accommodated as hominin and gorillin genera only if most of the decrease occurred early in great ape evolution.
An alternative explanation for the apparent discrepancy in fossil and genetic dates (leaving aside the issue of whether fossil taxa have been correctly placed) is that ancestral demography may have affected the genetic inferences. Certainly CoalHMM’s model does not fit the data in all respects. Perhaps most importantly, it assumes that ancestral population sizes are constant in time and that no gene flow occurred between separated populations, approximations that may not hold in reality. Simulations (details in Supplementary Information) suggest that an ancestral population bottleneck would have had limited impact on the inference of THC, its influence being captured largely by changes in the model’s effective population size. Under conditions of genetic exchange between populations after the main separation of the chimpanzee and human lineages, the speciation time estimated by CoalHMM represents an average weighted by gene flow over the period of separation. This means in some cases it can be substantially older than the date of most recent exchange. However it would only be more recent than the speciation time inferred from fossils if there had been strong gene flow between populations after the development of derived fossil characteristics. To the extent that this is plausible, for example as part of a non-allopatric speciation process, it constitutes an alternative explanation for the dating discrepancy without requiring a change in mutation rate.
In summary, although whole genome comparisons can be strongly conclusive about the ordering of speciation events, the inability to observe past mutation rates means that the timing of events from genetic data remains uncertain. In our view, possible variation in mutation rates allows hominid genomic data to be consistent with values of THC from 5.5 to 7 Mya and THCG from 8.5 to 12 Mya, with ancestral demographic structure potentially adding inherent ambiguity to both events. Better resolution may come from further integrated analysis of fossil and genetic evidence.
Incomplete lineage sorting and selection
The genealogy relating human (H), chimpanzee (C) and gorilla (G) varies between loci across the genome. CoalHMM explicitly models this and infers the genealogy at each position: either the standard ((H,C),G) relationship or the alternatives ((H,G),C) or ((C,G),H), which are the consequences of incomplete lineage sorting (ILS) in the ancestral HC population. We can use the pattern of ILS to explore evolutionary forces during the HCG speciation period. Across the genome we find 30% of bases exhibiting ILS, with no significant difference between the number sorting as ((H,G),C) and ((C,G),H). However, the fraction of ILS varies with respect to genomic position (Fig. 2a) by more than expected under a model of genome-wide neutral evolution (Fig. SF5.1). This variation reflects local differences in the ancestral effective population size Ne during the period between the gorilla and chimpanzee speciation events, most likely due to natural selection reducing Ne and making ILS less likely. Within coding exons mean ILS drops to 22%, and the suppression of ILS extends out to several hundred kbp from coding genes, evident even in raw site patterns before any model inference (Fig. 2b). An analysis of ILS sites in human segmental duplications suggests that assembly errors do not contribute significantly to this signal (Supplementary Information). We therefore attribute it to the effects of linkage around selected mutations, most likely in the form of background selection17, observing that it is greater around genes with lower dN/dS ratios (Fig. SF8.4). Given that more than 90% of the genome lies within 300 kbp of a coding gene, and noting the similar phenomenon reported for recent human evolution11, this supports the suggestion that selection has affected almost all of the genome throughout hominid evolution18.
Figure 2. Genome-wide ILS and selection.
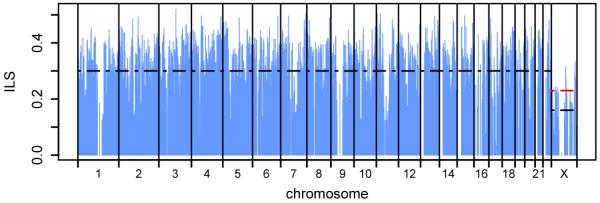
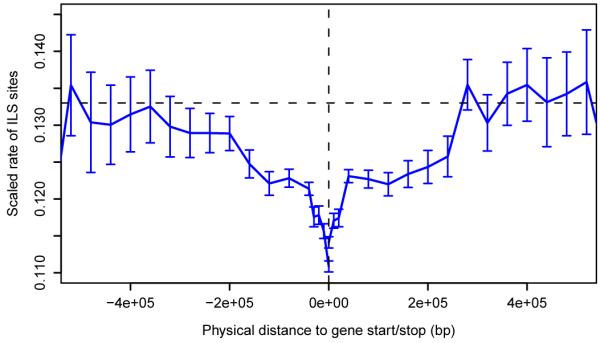
a, Variation in incomplete lineage sorting. Each vertical blue line represents the fraction of ILS between human, chimpanzee and gorilla estimated in a 1 Mbp region. Dashed black lines show the average ILS across the autosomes and on X; the red line shows the expected ILS on X, given the autosomal average and assuming neutral evolution. b, Reduction in ILS around protein coding genes. The blue line shows the mean rate of ILS sites normalised by mutation rate as a function of distance upstream or downstream of the nearest gene (see Supplementary Information). The horizontal dashed line indicates the average value outside 300 kbp from the nearest gene; error bars are s.e.m.
In fitting the transitions between genealogies along the alignment, CoalHMM also estimates a regional recombination rate. This is primarily sensitive to ancestral crossover events prior to HC speciation, yet despite the expectation of rapid turnover in recombination hotspots19, averaged over 1 Mbp windows there is a good correlation with estimates from present-day crossovers in humans (R = 0.49; p < 10−13; Fig. SF5.5), consistent with the conservation of recombination rates between humans and chimpanzees on the 1Mbp scale19.
As expected, we see reduced ILS (Fig. 2a) and HC sequence divergence dHC (Fig. SF6.1) on the X chromosome, corresponding to a difference in Ne between X and the autosomes within the ancestral HC population. Several factors can contribute to this difference20, notably the X chromosome’s haploidy in males, which reduces Ne on X by ¾, enhances purifying selection in males, and reduces the recombination rate, thereby increasing the effect of selection via linkage. However, sequence divergence is additionally affected by the mutation rate, which is higher in males than in females, further reducing the relative divergence observed on X21. Incorporating the ancestral Ne estimates from CoalHMM, we estimate a ratio of 0.87 ± 0.09 between average mutation rates on X and the autosomes on the HC lineage, corresponding to a male/female mutation rate bias α = 2.3 ± 0.4 (details in Supplementary Information). Previous estimates of α in hominids have ranged from 2 to 7 22,23. It is possible that some of the higher values, having been estimated from sequence divergence only and in smaller data sets, were inflated by underestimating the suppression of ancestral Ne on X, in particular due to purifying selection.
Our calculation of α assumes that a single speciation time applies across the genome, attributing differences between the X chromosome and autosomes to the factors mentioned above. Patterson et al.24 proposed an alternative model involving complex speciation, with more recent HC ancestry on X than elsewhere. Given potential confounding factors in demography, selection, mutation rate bias and admixture, our analyses do not discriminate between these models; however if the effective HC separation time on X is indeed reduced in this way it would imply a still lower value of α.
Functional sequence evolution
We looked for loss or gain of unique autosomal sequence within humans, chimpanzees and gorillas by comparing raw sequence data for each in the context of their reference assemblies (Supplementary Information). The total amount is small: 3-7 Mbp per species, distributed genome-wide in fragments no more than a few kbp in length (Table ST7.1). The vast majority (97%) of such material was also found either in orangutan or a more distant primate, indicating loss, and consistent with the expectation that gain is driven primarily by duplication (which our analysis excludes). Some fragments found only in one species overlap coding exons in annotated genes: 6 genes in human, 5 in chimpanzee and 9 in gorilla (Tables ST7.2,3,4), the majority being associated with olfactory receptor proteins or other rapidly-evolving functions such as male fertility and immune response.
We did not assemble a gorilla Y chromosome, but by mapping ~6x reads from the male gorillas Kwanza and Mukisi to the human Y we identified several regions in which human single-copy material is missing in gorilla, comprising almost 10% of the accessible male-specific region. Across the Y chromosome there is considerable variation in the copy number of shared material, and the pattern of coverage is quite different from that of reads from a male bonobo mapped in the same way (Fig. SF7.1). Some missing or depleted material overlaps coding genes (Table ST7.5) including for example VCY, a gene expressed specifically in male germ cells which has two copies in human and chimpanzee but apparently only one in gorilla (Supplementary Information.) The resulting picture is consistent with rapid structural evolution of the Y chromosome in the great apes, as previously seen in the chimpanzee-human comparison25.
Protein evolution
The EPO primate alignment was filtered to produce a high-quality genome-wide set of 11,538 alignments representing orthologous primate coding sequences, which were then scored with codon-based evolutionary models for likelihoods of acceleration or deceleration of the ratio dN/dS of nonsynonymous to synonymous mutation rates in the terminal lineages, ancestral branch, and entire hominine subfamily (Supplementary Information). We find that genes with accelerated rates of evolution across hominines are enriched for functions associated with sensory perception, particularly in relation to hearing and brain development (Table ST8.4G,H). For example, among the most strongly accelerated genes are OTOF (p = 0.0056), LOXHD1 (p < 0.01) and GPR98 (p = 0.0056) which are all associated with diseases causing human deafness (Table ST8.5). GPR98, which also shows significant evidence of positive selection under the branch-site test (p = 0.0081), is highly expressed in the developing central nervous system. The gene with the strongest evidence for acceleration along the branch leading to hominines is RNF213 (branch-site p < 2.9 × 10−9), a gene associated with Moyamoya disease in which blood flow to the brain is restricted due to arterial stenosis26. Given that oxygen and glucose consumption scales with total neuron number27 RNF213 may have played a role in facilitating the evolution of larger brains. Together, these observations are consistent with a major role for adaptive modifications in brain development and sensory perception in hominine evolution.
Turning to lineage-specific selection pressures, we find relatively similar numbers of accelerated genes in humans, chimpanzees and gorillas (663, 562 and 535 respectively at nominal p < 0.05, Table ST8.3A) and genome-wide dN/dS ratios (0.256, 0.249, and 0.239 in purifying sites, Table ST8.6) These numbers, which reflect variation in historical effective population sizes as well as environmental pressures, reveal a largely uniform landscape of recent hominine gene evolution - in accordance with previously-published analyses in human and chimpanzee3,28 (Table ST8.7).
Genes with accelerated rates of evolution along the gorilla lineage are most enriched for a number of developmental terms, including ear, hair follicle, gonad, and brain development, and sensory perception of sound. Among the most significantly accelerated genes in gorilla is EVPL (p < 2.2 × 10−5), which encodes a component of the cornified envelope of keratinocytes, and may be related to increased cornification of knuckle pads in gorilla29. Interestingly, gorilla and human both yielded brain-associated terms enriched for accelerated genes, but chimpanzee did not (Table ST8.4A-C). Genes expressed in the brain or involved in its development have not typically been associated with positive selection in primates, but our results show that multiple great ape lineages show elevated dN/dS in brain-related genes when evaluated against a primate background.
We also identified cases of pairwise parallel evolution among hominines. Human and chimpanzee show the largest amount, with significantly more shared accelerations than expected by chance, while gorilla shares more parallel acceleration with human than with chimpanzee across a range of significance thresholds (Figure SF8.3). Genes involving hearing are enriched in parallel accelerations for all three pairs, but most strongly in gorilla-human (Table ST8.4D-F), calling into question a previous link made between accelerated evolution of auditory genes in humans and language evolution28. It is also interesting to note that ear morphology is one of the few external traits in which humans are more similar to gorillas than to chimpanzees30.
Next we considered gene loss and gain. We found 84 cases of gene loss in gorilla due to the acquisition of a premature stop codon, requiring there to be no close paralogue (Table ST8.8); for example, TEX14, an intercellular bridge protein essential for spermatogenesis in mice. Genome-wide analysis of gene gain is confounded by the difficulty in assembly of closely related paralogues. We therefore resequenced, by finishing overlapping fosmids, three gene clusters known to be under rapid adaptive evolution in primates: the growth hormone cluster31, the PRM clusters involved in sperm function and the APOBEC cluster implicated in molecular adaptation to viral defence. In the growth hormone cluster we observed four chorionic somatomammotropin (CSH) genes in gorilla compared to three in humans and chimpanzees, with a novel highly similar pair of CSH-like genes in gorilla that share a 3′ end similar to human growth hormone GH2, suggesting a complex evolutionary history as in other primates31. We saw sequence but not gene copy number changes in the PRM and APOBEC clusters (Supplementary Information).
In several cases, a protein variant thought to cause inherited disease in humans32 is the only version found in all three gorillas for which we have genome-wide sequence data (Table ST8.9). Striking examples are the dementia-associated variant Arg432Cys in the growth factor PGRN and the hypertrophic cardiomyopathy-associated variant Arg153His in the muscle Z disc protein TCAP, both of which were corroborated by additional capillary sequencing (Table ST8.10). Why variants that appear to cause disease in humans might be associated with a normal phenotype in gorillas is unknown; possible explanations are compensatory molecular changes elsewhere, or differing environmental conditions. Such variants have also been found in both the chimpanzee and macaque genomes3,33.
Gene transcription and regulation
We carried out an analysis of hominine transcriptome variation using total RNA extracted and sequenced from lymphoblastoid cell lines (LCLs) of one gorilla, two chimpanzees and two bonobos (Supplementary Information), and published RNA sequence data for eight human individuals34. After quantifying reads mapping to exons and genes in each species, we calculated the degree of species-specific expression and splicing in 9,746 1:1:1 expressed orthologous genes. On average, human and chimpanzee expression were more similar to each other than either was to gorilla (Fig. SF10.2). However this effect is reduced in genes with a higher proportion of ILS sites, which tend to show greater expression distance between humans and chimpanzees (Fig. 3a). More generally, patterns seen in the relative expression distances between the three species showed a significant overlap with those derived from genomic lineage sorting (p = 0.026; Table ST10.4), demonstrating that ILS can be reflected in functional differences between primate species.
Figure 3. Differences in expression and regulation.
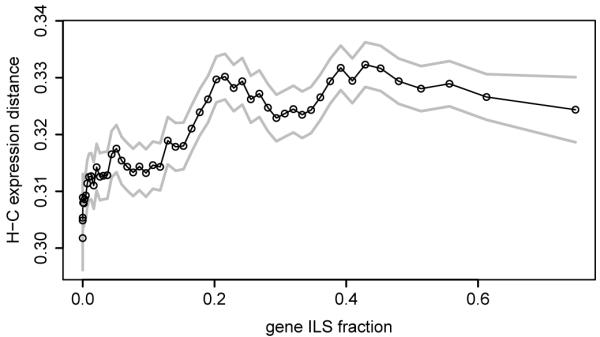
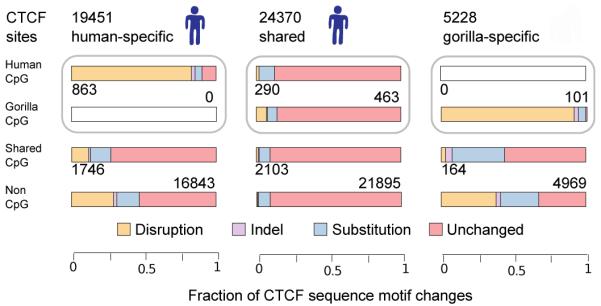
a, Mean gene expression distance between human and chimpanzee as a function of the proportion of ILS sites per gene. Each point represents a sliding window of 900 genes (over genes ordered by ILS fraction); s.d. error limits are shown in grey. b, (top) Classification of CTCF sites in the gorilla (EB(JC)) and human (GM12878) LCLs on the basis of species-uniqueness; numbers of alignable CTCF binding sites are shown for each category; (bottom) sequence changes of CTCF motifs embedded in human-specific, shared and gorilla-specific CTCF binding sites located within shared CpG islands, species-specific CpG islands or outside CpG islands. Numbers of CTCF binding sites are shown for each CpG island category. Gorilla and human motif sequences are compared and represented as indels, disruptions (>4 bp gaps), and substitutions.
We also explore species specific variation in splicing35, by calculating the variance in differential expression of orthologous exons within each gene. In total we found 7% of genes whose between-species variance is significant at the 1% level (based on the distribution of within-human variances, Fig. SF10.5). For example, Fig SF10.6 illustrates gorilla-specific splicing in the SQLE gene, involved in steroid metabolism.
We further investigated great ape regulatory evolution by comparing the binding in human and gorilla of CTCF, a protein essential to vertebrate development involved in transcriptional regulation, chromatin loop formation, and protein scaffolding36. We performed ChIP-seq of CTCF in a gorilla LCL (from EB(JC)), and compared this with matched human experiments37, using the EPO alignments to identify species-specific and shared binding regions (Fig. 3b and Supplementary Information). Consistent with previous results reporting strong CTCF binding conservation38, and in contrast to the rapid turnover of some other transcription factor binding sites39, we found that approximately 70% of gorilla CTCF binding regions are shared with human. This compares with around 80% pairwise overlaps between three human LCLs (Fig. SF11.1A). Binding regions that are shared among all three human individuals are three times more likely to be shared with gorilla than individual-specific regions (Fig. SF11.1B).
The genomic changes leading to loss of CTCF binding differ between regions within CpG islands and those in the rest of the genome. Losses of CTCF binding outside CpG islands and within species-specific CpG regions co-occur with sequence changes in the binding motif, but for shared CpG islands most binding losses have no corresponding motif sequence change (Fig 3b). It is possible that DNA methylation differences are driving this effect, as CTCF binding can be abolished by methylation of specific target regions36. Alternatively, CTCF binding within CpG islands may also depend more on other regulators’ binding and less on the CTCF motif itself.
Genetic diversity within Gorilla
Recent studies of molecular and morphological diversity within the Gorilla genus have supported a classification into two species, eastern (G. beringei) and western (G. gorilla)40, with both species further divided into subspecies (Fig. 4a). Although separated today by over 1000 km, it has been suggested that gene flow has occurred between the eastern and western species since divergence41. To investigate this, we collected reduced representation sequence data (Supplementary Information) for another female western lowland gorilla, EB(JC), and a male eastern lowland gorilla, Mukisi.
Figure 4. Gorilla species distribution and divergence.
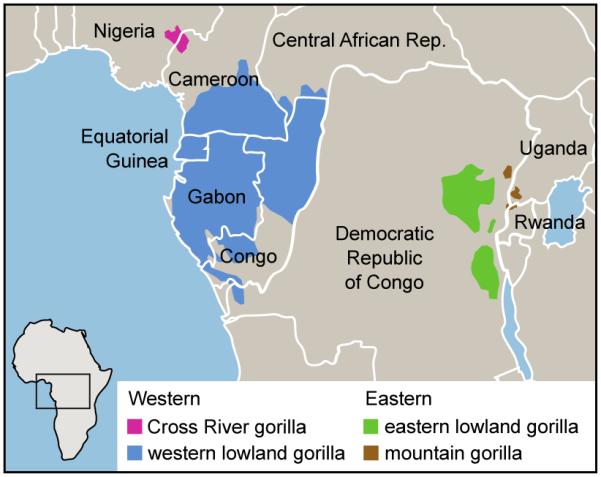


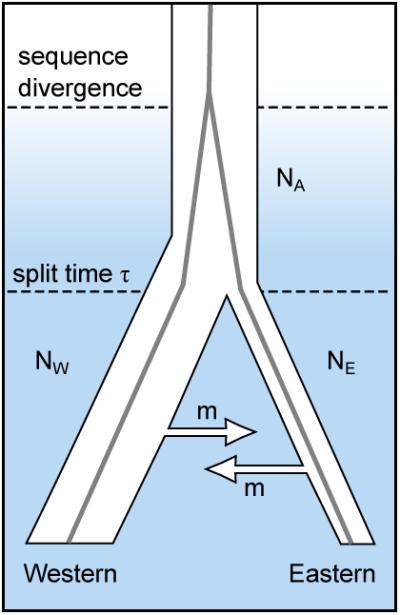
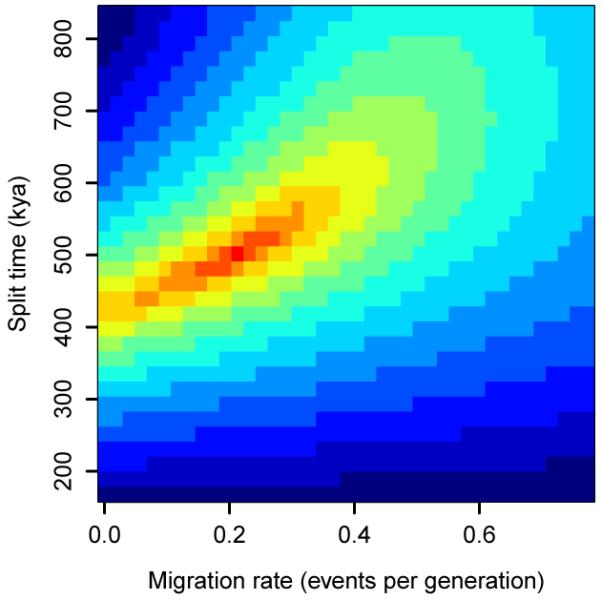
a, Distribution of gorilla species in Africa. The western species (Gorilla gorilla) comprises two subspecies: western lowland gorillas (G. gorilla gorilla) and Cross River gorillas (G. gorilla diehli). Similarly, the eastern species (Gorilla beringei) is subclassified into eastern lowland gorillas (G. beringei graueri) and mountain gorillas (G. beringei beringei). (Based on data in IUCN 2010.) b, Western lowland gorilla Kamilah, source of the reference assembly (photo JR). c, Eastern lowland gorilla Mukisi (photo M. Seres). d, Isolation-migration model of the western and eastern species. NA, NW and NE are ancestral, western and eastern effective populations sizes; m is the migration rate. e, Likelihood surface for migration and split time parameters in the isolation-migration model.
Table 2 summarizes the sequence diversity in these individuals and in Kamilah, based on alignment of sequence data to the gorilla assembly. The ratio of homozygous to heterozygous variant rates for EB(JC) (close to 0.5) is consistent with her coming from the same population as Kamilah (Supplementary Information), and her rate of heterozygosity matches Kamilah’s. Mukisi, on the other hand, has twice the rate of homozygous differences from the assembly, consistent with his coming from a separate population. Furthermore, heterozygosity in Mukisi is much lower, suggesting a reduced population size in the eastern species. This agrees with previous studies based on fewer loci41, and also with estimates of present-day numbers in the wild, which indicate that whereas the western lowland subspecies may number up to 200,000 individuals, the eastern population as a whole is around ten times smaller42,43. Because it manifests in genetic diversity, this disparity must have existed for many millennia, and cannot have resulted solely from the current pressure of human activity in central Africa or recent outbreaks of the Ebola virus.
Table 2.
Nucleotide polymorphism in western and eastern gorillas
| Species | heterozygous site rate (%) |
homozygous site rate (%) |
hom:het ratio | |
|---|---|---|---|---|
| Kamilah | western lowland | 0.189 | 0.0015 | - |
| EB(JC) | western lowland | 0.178 | 0.10 | 0.56 |
| Mukisi | eastern lowland | 0.076 | 0.19 | 2.5 |
Rates are based on variants detected by mapping sequence data to the gorilla reference and filtering sites by depth and mapping quality (Supplementary Information). The homozygosity rate for Kamilah is low (and is effectively an error rate) because her sequence was used for assembly. Reduced heterozygosity in Mukisi is not due to familial inbreeding, since there are no long homozygous stretches.
Based on an alignment of the EB(JC) and Mukisi data to the human reference sequence and comparing high confidence genotype calls for the two individuals, we estimate a mean sequence divergence time between them of 1.75 Mya. However the pattern of shared heterozygosity is not consistent with a clean split between western and eastern gorillas (Supplementary Information). Under a model which allows symmetric genetic exchange between the populations after an initial split (Fig. 4d; Supplementary Information), the maximum likelihood species split time is ~0.5 Mya with moderate subsequent exchange of ~0.2 individuals per generation each way between breeding pools, totalling ~5,000 in each direction over 0.5 My (Fig. 4e). Different model assumptions and parameterisations would lead to different values. More extensive sampling and sequencing of both gorilla populations will afford better resolution of this issue.
We also collected whole-genome sequence data from an additional male western lowland gorilla ‘Kwanza’ at 12x, and further whole genome sequence data for (eastern) Mukisi at 7x (Supplementary Information). Differences between the western gorillas and Mukisi represent a combination of inter-individual and inter-species variants. These include 1,615 non-synonymous SNPs in 1,326 genes, seven of which have more than four amino acid differences each (Table ST12.2), among which are two olfactory receptor genes and EMR3, implicated in immune and inflammatory responses44. Nineteen of the genes annotated in Kamilah carry an apparently homozygous premature stop codon in Mukisi. These include the gene encoding the seminal fluid protein SEMG2, implicated in sperm competition and known to be inactivated in some gorillas, where sperm competition is rare45. Both EMR3 and SEMG2 were corroborated by additional sequencing (Tables ST12.3, ST12.4).
Finally, we investigated genomic duplication in gorilla using a whole genome shotgun sequence detection method applied to data from the western gorillas Kamilah and Kwanza (Supplementary Information). This revealed a level of private segmental duplication (0.9 Mbp and 1.5 Mbp in the two gorillas) well outside the range found in pairwise comparisons of humans (Fig. SF13.1), where a value of ~100 kbp is typical between any two individuals46. These results suggest greater copy number diversity in gorillas than in humans, consistent with previous observations in the great apes 47.
Conclusion
Since the Middle Miocene - an epoch of abundance and diversity for apes throughout Eurasia and Africa, the prevailing pattern of ape evolution has been one of fragmentation and extinction48. The present-day distribution of non-human great apes, existing only as endangered and subdivided populations in equatorial forest refugia43, is a legacy of that process. Even humans, now spread around the world and occupying habitats previously inaccessible to any primate, bear the genetic legacy of past population crises. All other branches of the genus Homo have passed into extinction. It may be that in the condition of Gorilla, Pan and Pongo we see some echo of our own ancestors prior to the last 100,000 years, and perhaps a condition experienced many times over several million years of evolution. It is notable that species within at least three of these genera continued to exchange genetic material long after separation4,49, a disposition that may have aided their survival in the face of diminishing numbers. As well as teaching us about human evolution, the study of the great apes connects us to a time when our existence was more tenuous, and in doing so, highlights the importance of protecting and conserving these remarkable species.
Methods summary
Assembly
We constructed a hybrid de novo assembly combining 5.4 Gbp of capillary read pairs with the contigs from an initial short read assembly of 166.8 Gbp of Illumina paired reads. Improvements in long-range structure were then guided by human homology, placing contigs into scaffolds wherever read pairs confirmed collinearity between gorilla and human. Base-pair contiguity was improved by local reassembly within each scaffold, merging or extending contigs using Illumina read pairs. Finally we used additional Kamilah BAC and fosmid end pair capillary sequences to provide longer range scaffolding. Base errors were corrected by mapping all Illumina reads back to the assembly and rectifying apparent homozygous variants, while recording the location of heterozygous sites.
Further details and other methods are described in Supplementary Information.
Supplementary Material
Acknowledgements
We thank H. Li and E. Birney for discussions, D. Zerbino, J. Stalker, L. Wilming, D. Rajan and H. Clawson for technical assistance, J. Ahringer for comments on the manuscript, K. Leus of the Center for Research and Conservation of the Royal Zoological Society of Antwerp for sample material from Mukisi, and the Marmoset Genome Analysis Consortium for permission to use the unpublished assembly of the marmoset genome. This research was supported in part by Wellcome Trust grants WT062023 (to J.H., K.B., S.F., A.J.V., P.F.), WT089066 (to R.D.), WT077192 (to R.D., S.M., A.K.-K., J.T.S., W.W.), WT077009 (to Y.X., B.Y., Q.A., Y.C., C.T.-S.), WT077198 (to G.K.L.) and 075491/Z/04 (to G.L.); EMBL grants (to P.C.S., P.F.); scholarships from the Gates Cambridge Trust (to G.E.J. and T.D.O’C.); an MRC Special Fellowship in Biomedical Informatics (to A.S.); funding from the Lundbeck Foundation (to A.H.); the Academy of Finland and the Emil Aaltonen Foundation (to T.L.); a Marie Curie fellowship (to T.M.-B.); the European Community's Seventh Framework Programme (FP7/2007-2013)/ ERC Starting Grant (StG_20091118) (to T.M.-B.); an FPI grant from the Spanish Ministry of Education (BES-2010-032251) (to J.P.-M.); a BBSRC Doctoral Training Grant (to S.H.M.); grants from the UK Medical Research Council (to A.H., S.M., C.P.P.); the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health (to J.C.M.); the Danish Council for Independent Research, Natural Sciences, grant no. 09-062535 (to K.M., M.H.S.); a Commonwealth Scholarship (to M.C.W.); the Swiss National Science Foundation, Louis Jeantet Foundation (to E.T.D.); an ERC Starting Grant and an EMBO Young Investigator Award, Hutchinson Whampoa (to D.T.O.); NHGRI support (to W.C.W.); support from BIOBASE GmbH (to E.V.B., P.D.S., M.M., A.D.P., K.S., D.N.C.); US National Science Foundation grant DGE-0739133 (to W.W.); NHGRI U54 HG003079 (to R.K.W.); NIH grant HG002385 (to E.E.E). E.E.E. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Author contributions Manuscript main text: A.S., R.D., C.T-S., N.I.M., G.E.J., P.C.S., A.K-K. Project coordination: A.S., A.S.R., A.K-K., R.D. Project initiation: J.R., R.D., R.K.W. Library preparation and sequencing: I.G., D.J.T., M.A.Q., C.M.C., B.Z., P.dJ., O.A.R., Q.A., B.Y., Y.X., T.A.G., W.C.W. Assembly: A.S., L.W.H., Y.G., J.T.S., J.M., W.W., Z.N. Fosmid finishing: P.H. Assembly quality: A.S., S.Mead., G.L., C.P.P. Annotation: Y.A.T., G.J.L., A.J.V., A.Heg., S.M.S. Primate multiple alignments: J.H., K.B., S.F. Great ape speciation and ILS: J.Y.D., A.S., T.M., M.H.S., K.M., G.E.J. Sequence loss and gain: A.S., S.M., C.T-S., A.T., A.J.V. Protein evolution: G.E.J., S.H.M., N.I.M., B.J.B., T.D.O’C., Y.X., Y.C., N.G. Human disease allele analysis: Y.X., Y.C., C.T-S., P.D.S., E.V.B., A.D.P., M.M., K.S., D.N.C. Transcriptome analysis: T.L., E.T.D. ChIP-seq experiment and analysis: P.C.S., M.C.W., D.S., P.F., D.T.O. Additional gorilla samples: B.Y., Y.X., L.V., C.T-S. Gorilla species diversity and divergence: A.S., A.H., T.M., L.N.A., B.Y., L.V. Gorilla species functional differences: Y.X., Y.C., C.T-S. Segmental duplication analysis: T.M-B., C.A., S.S., E.K., J.P-M., E.E.E.
Author information Accession numbers for all primary sequencing data are given in Supplementary Information. The assembly has been submitted to EMBL with accession numbers FR853080 to FR853106, and annotation is available at Ensembl (http://www.ensembl.org/Gorilla_gorilla/Info/Index).
The authors declare no competing interests.
References
- 1.Huxley TH. Evidence as to Man’s Place in Nature. Williams & Norgate; 1863. [Google Scholar]
- 2.King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 3.ChimpanzeeSequencingandAnalysisConsortium Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. doi:10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 4.Locke DP, et al. Comparative and demographic analysis of orang-utan genomes. Nature. 2011;469:529–533. doi: 10.1038/nature09687. doi:10.1038/nature09687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubbard TJ, et al. Ensembl 2009. Nucleic Acids Res. 2009;37:D690–697. doi: 10.1093/nar/gkn828. doi:10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paten B, Herrero J, Beal K, Fitzgerald S, Birney E. Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome research. 2008;18:1814–1828. doi: 10.1101/gr.076554.108. doi:10.1101/gr.076554.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradley BJ. Reconstructing phylogenies and phenotypes: a molecular view of human evolution. J Anat. 2008;212:337–353. doi: 10.1111/j.1469-7580.2007.00840.x. doi:10.1111/j.1469-7580.2007.00840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgess R, Yang Z. Estimation of hominoid ancestral population sizes under bayesian coalescent models incorporating mutation rate variation and sequencing errors. Molecular biology and evolution. 2008;25:1979–1994. doi: 10.1093/molbev/msn148. doi:10.1093/molbev/msn148. [DOI] [PubMed] [Google Scholar]
- 9.Steiper ME, Young NM. Timing primate evolution: Lessons from the discordance between molecular and paleontological estimates. Evolutionary Anthropology: Issues, News, and Reviews. 2008;17:179–188. doi:10.1002/evan.20177. [Google Scholar]
- 10.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A. 2010;107:961–968. doi: 10.1073/pnas.0912629107. doi:10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.1000GenomesProjectConsortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. doi:10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roach JC, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. doi:10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood B, Harrison T. The evolutionary context of the first hominins. Nature. 2011;470:347–352. doi: 10.1038/nature09709. doi:10.1038/nature09709. [DOI] [PubMed] [Google Scholar]
- 14.Hartwig WC, et al. The Primate Fossil Record. Cambridge University Press; 2002. [Google Scholar]
- 15.Kim SH, Elango N, Warden C, Vigoda E, Yi SV. Heterogeneous genomic molecular clocks in primates. PLoS Genet. 2006;2:e163. doi: 10.1371/journal.pgen.0020163. doi:10.1371/journal.pgen.0020163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleagle JG. Primate Adaptation and Evolution. Second Edition Academic Press; 1998. [Google Scholar]
- 17.Charlesworth D, Morgan MT, Charlesworth B. Mutation Accumulation in Finite Populations. Journal of Heredity. 1993;84:321–325. [Google Scholar]
- 18.McVicker G, Gordon D, Davis C, Green P. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet. 2009;5:e1000471. doi: 10.1371/journal.pgen.1000471. doi:10.1371/journal.pgen.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–324. doi: 10.1126/science.1117196. doi:10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 20.Vicoso B, Charlesworth B. Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet. 2006;7:645–653. doi: 10.1038/nrg1914. doi:10.1038/nrg1914. [DOI] [PubMed] [Google Scholar]
- 21.Ellegren H. Characteristics, causes and evolutionary consequences of male-biased mutation. Proc Biol Sci. 2007;274:1–10. doi: 10.1098/rspb.2006.3720. doi:10.1098/rspb.2006.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goetting-Minesky MP, Makova KD. Mammalian male mutation bias: impacts of generation time and regional variation in substitution rates. J Mol Evol. 2006;63:537–544. doi: 10.1007/s00239-005-0308-8. doi:10.1007/s00239-005-0308-8. [DOI] [PubMed] [Google Scholar]
- 23.Presgraves DC, Yi SV. Doubts about complex speciation between humans and chimpanzees. Trends Ecol Evol. 2009;24:533–540. doi: 10.1016/j.tree.2009.04.007. doi:10.1016/j.tree.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patterson N, Richter DJ, Gnerre S, Lander ES, Reich D. Genetic evidence for complex speciation of humans and chimpanzees. Nature. 2006;441:1103–1108. doi: 10.1038/nature04789. doi:10.1038/nature04789. [DOI] [PubMed] [Google Scholar]
- 25.Hughes JF, et al. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature. 2010;463:536–539. doi: 10.1038/nature08700. doi:10.1038/nature08700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamada F, et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet. 2011;56:34–40. doi: 10.1038/jhg.2010.132. doi:10.1038/jhg.2010.132. [DOI] [PubMed] [Google Scholar]
- 27.Herculano-Houzel S. Scaling of brain metabolism with a fixed energy budget per neuron: implications for neuronal activity, plasticity and evolution. PLoS One. 2011;6:e17514. doi: 10.1371/journal.pone.0017514. doi:10.1371/journal.pone.0017514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark AG, et al. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science. 2003;302:1960–1963. doi: 10.1126/science.1088821. doi:10.1126/science.1088821. [DOI] [PubMed] [Google Scholar]
- 29.Ellis RA, Montagna W. The skin of primates. VI. The skin of the gorilla (Gorilla gorilla) Am J Phys Anthropol. 1962;20:79–93. doi: 10.1002/ajpa.1330200210. [DOI] [PubMed] [Google Scholar]
- 30.Streeter GL. Some uniform characteristics of the primate auricle. Anat Rec A Discov Mol Cell Evol Biol. 1922;23:335–341. doi:10.1002/ar.1090230604. [Google Scholar]
- 31.Wallis OC, Zhang YP, Wallis M. Molecular evolution of GH in primates: characterisation of the GH genes from slow loris and marmoset defines an episode of rapid evolutionary change. J Mol Endocrinol. 2001;26:249–258. doi: 10.1677/jme.0.0260249. [DOI] [PubMed] [Google Scholar]
- 32.Stenson PD, et al. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. doi:10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibbs RA, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. doi:316/5822/222 [pii] 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- 34.Montgomery SB, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. doi:10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blekhman R, Marioni JC, Zumbo P, Stephens M, Gilad Y. Sex-specific and lineage-specific alternative splicing in primates. Genome Research. 2010;20:180–189. doi: 10.1101/gr.099226.109. doi:10.1101/gr.099226.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. doi:10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDaniell R, et al. Heritable individual-specific and allele-specific chromatin signatures in humans. Science. 2010;328:235–239. doi: 10.1126/science.1184655. doi:10.1126/science.1184655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunarso G, et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nature Genetics. 2010;42:631–634. doi: 10.1038/ng.600. doi:10.1038/ng.600. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt D, et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328:1036–1040. doi: 10.1126/science.1186176. doi:10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Groves C. Primate Taxonomy. Vol. 350. Smithsonian Institution Press; 2001. [Google Scholar]
- 41.Thalmann O, Fischer A, Lankester F, Paabo S, Vigilant L. The complex evolutionary history of gorillas: insights from genomic data. Mol Biol Evol. 2007;24:146–158. doi: 10.1093/molbev/msl160. doi:10.1093/molbev/msl160. [DOI] [PubMed] [Google Scholar]
- 42.Stokes E, Malonga R, Rainey H, Strindberg S. Western Lowland Gorilla surveys in Northern Republic of Congo 2006-2007. Summary Scientific Report. WCS Global Conservation; 2008. [Google Scholar]
- 43.IUCN IUCN Red List of Threatened Species. (Version 2010.1) 2010 < http://www.iucnredlist.org >.
- 44.Stacey M, Lin HH, Hilyard KL, Gordon S, McKnight AJ. Human epidermal growth factor (EGF) module-containing mucin-like hormone receptor 3 is a new member of the EGF-TM7 family that recognizes a ligand on human macrophages and activated neutrophils. J Biol Chem. 2001;276:18863–18870. doi: 10.1074/jbc.M101147200. doi:10.1074/jbc.M101147200. [DOI] [PubMed] [Google Scholar]
- 45.Jensen-Seaman MI, Li WH. Evolution of the hominoid semenogelin genes, the major proteins of ejaculated semen. Journal of molecular evolution. 2003;57:261–270. doi: 10.1007/s00239-003-2474-x. doi:10.1007/s00239-003-2474-x. [DOI] [PubMed] [Google Scholar]
- 46.Alkan C, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nature Genetics. 2009;41:1061–1067. doi: 10.1038/ng.437. doi:10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gazave E, et al. Copy number variation analysis in the great apes reveals species-specific patterns of structural variation. Genome Research. 2011 doi: 10.1101/gr.117242.110. doi:10.1101/gr.117242.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Begun DR. In: Handbook of Palaeoanthropology Vol. 2: Primate evolution and Human Origins. Henke W, Tattersall I, editors. Springer; 2007. pp. 921–977. [Google Scholar]
- 49.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. doi:10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lebatard AE, et al. Cosmogenic nuclide dating of Sahelanthropus tchadensis and Australopithecus bahrelghazali: Mio-Pliocene hominids from Chad. Proc Natl Acad Sci U S A. 2008;105:3226–3231. doi: 10.1073/pnas.0708015105. doi:10.1073/pnas.0708015105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.