


Abstract
We developed a technique called GREM (Genomic Repeat Expression Monitor) that can be applied to genome-wide isolation and quantitative analysis of any kind of transcriptionally active repetitive elements. Briefly, the technique includes three major stages: (i) generation of a transcriptome wide library of cDNA 5′ terminal fragments, (ii) selective amplification of repeat-flanking genomic loci and (iii) hybridization of the cDNA library (i) to the amplicon (ii) with subsequent selective amplification and cloning of the cDNA-genome hybrids. The sequences obtained serve as ‘tags’ for promoter active repetitive elements. The advantage of GREM is an unambiguous mapping of individual promoter active repeats at a genome-wide level. We applied GREM for genome-wide experimental identification of human-specific endogenous retroviruses and their solitary long terminal repeats (LTRs) acting in vivo as promoters. Importantly, GREM tag frequencies linearly correlated with the corresponding LTR-driven transcript levels found using RT–PCR. The GREM technique enabled us to identify 54 new functional human promoters created by retroviral LTRs.
INTRODUCTION
Repetitive elements form a great portion of most eukaryotic genomes and large-scale studies of their transcriptional activity are now attracting increasing interest. Many genomic repeats have originated from insertions of transposable elements. Retroelements (REs), which proliferate via RNA intermediates, are known to be the only transpositionally active group of transposable elements in mammals. In vertebrates, REs occupy up to 30–40% of the genome (1–4). Being mobile carriers of transcriptional regulatory modules, REs can affect regulation of host genes, in particular those involved in embryo development, thus being probable candidates for playing a role in speciation processes (5).
It was recently demonstrated that REs can drive the transcription of unique host non-repetitive sequences (6,7). Many kinds of genomic repeats are known to be transcribed in vivo (8,9). However, a significant portion of such expressed repeats was found within larger transcripts driven from upstream genomic promoters. Conventional and popular methods for transcriptome analysis such as RT–PCR, differential display (10,11), subtractive hybridization (12–14), serial analysis of gene expression (15) and microarray hybridization do not allow to distinguish between read-through transcripts and those due to the intrinsic promoter activity of genomic repeats. Different modifications of the 5′ rapid amplification of cDNA ends (RACE) technique allow one to precisely locate transcription start sites (16), but cannot be used for quantitative and large-scale transcriptome screenings. We aimed to develop a transcriptome-wide strategy that would make it possible to detect intrinsic promoter activity of repetitive elements. To this end, we tried to combine the advantages of 5′-RACE and nucleic acid hybridization techniques.
Here, we describe an approach termed GREM (Genomic Repeat Expression Monitor), which is based on hybridization of total pools of cDNA 5′ terminal parts to genome-wide pools of repetitive elements flanking DNA, followed by selective PCR amplification of the resulting hybrid cDNA–genome duplexes. A library of cDNA/genomic DNA hybrid molecules obtained in such a way can be used as a set of tags for individual transcriptionally active repetitive elements. The method is both quantitative and qualitative, as the number of such tags is proportional to the content of mRNA driven from the corresponding promoter active repetitive element.
We applied GREM for the genome-wide recovery of promoter active human-specific endogenous retroviruses. HERV-K (HML-2) is the only family of endogenous retroviruses known to contain human-specific members (17,18). This group, whose members not only retained their transcriptional activity (19), but also probably still possess some infectious potential (20,21), is thought to be among the most biologically active retroviral families of the human genome (22–24). A major part of endogenous retroviruses have undergone homologous recombination between their LTR sequences, and this family is now represented mostly by solitary LTRs (25,26). Human-specific HERV-K (HML-2) LTRs share a significant sequence identity and form a well-defined cluster (named the HS family) on a phylogenetic tree (17,18). The HS family is characterized by diagnostic nucleotide substitutions within the consensus sequence of HS LTRs (17). The HS family contains 156 mostly (∼86%) human-specific LTR sequences. The HS family members are represented by parts of full-sized HERV-K (HML-2) proviruses (11.5% of individual HS representatives), truncated proviruses (5.2%) or solitary LTRs (83.3%). We describe here the results of the first genome-wide identification of those LTRs serving as in vivo human-specific promoters in germ-line tissue and report the first comprehensive genomic map of transcriptionally active HS LTRs.
MATERIALS AND METHODS
DNA sequence analysis
The human-specific HERV-K LTR group (HS) consensus sequence was taken from our previous work (17). LTR flanking regions were investigated with the RepeatMasker program (http://ftp.genome.washington.edu/cgi-bin/RepeatMasker; A. F. A. Smit and P. Green, unpublished data). Homology searches against GenBank were done using the BLAST web server at NCBI (http://www.ncbi.nlm.nih.gov/BLAST) (27). To determine genomic locations of LTR flanking regions, the UCSC genome browser and BLAT searches (http://genome.ucsc.edu/cgi-bin/hgBLAT) were used.
Oligonucleotides
Oligonucleotides were synthesized using an ASM-102U DNA synthesizer (Biosan, Novosibirsk, Russia). Their structures can be found in Table 3 of Supplementary Data.
Tissue sampling
Testicular parenchyma was sampled from a surgical specimen under non-neoplastic conditions. Representative samples were divided into two parts, one of which was immediately frozen in liquid nitrogen and the other was formalin-fixed and paraffin-embedded for histological analysis.
RNA isolation and cDNA synthesis
Total RNA was isolated from frozen samples pulverized in liquid nitrogen using an RNeasy Mini RNA purification kit (Qiagen). All RNA samples were further treated with DNase I to remove residual DNA. Full-length cDNA samples were obtained according to a cap-switch effect-based SMART cDNA synthesis protocol (Clontech, BD Biosciences) using an oligo(dT)-containing primer (CDS), PowerScript reverse transcriptase (Clontech, BD Biosciences) and a riboCS oligonucleotide. When PowerScript reverse transcriptase reaches the 5′ end of the mRNA, the enzyme's terminal transferase activity adds a few additional deoxycytidine nucleotides to the 3′ end of the cDNA. The riboCS oligonucleotide, which contains three guanine ribonucleotide residues at its 3′ end, basepairs with the deoxycytidine stretch, creating an extended template. Reverse transcriptase then switches templates and continues the replication to the end of the oligonucleotide. The resulting full-length single stranded cDNA contains 5′ terminal sequences complementary to the riboCS oligonucleotide. An Advantage 2 Polymerase mix (Clontech), CS and CDS oligonucleotides were used to synthesize the second cDNA strands and to PCR-amplify double-stranded cDNA. Prior to further hybridization in the GREM procedure, 1 µg cDNA was digested with 10 U of AluI restriction endonuclease (Fermentas) for 3 h at 37°C. This enzyme was used because the HS LTR consensus sequence lacks AluI recognition sites.
Selective amplification of genomic regions flanking HS LTRs
Selective amplification of LTR 3′ flanking regions was based on the PCR suppression effect described in detail elsewhere (28–30). Human genomic DNA (1 µg) was digested with 10 U of AluI (Fermentas) restriction endonuclease, ethanol precipitated and dissolved in 20 µl sterile water. Then, 100 pmol of annealed suppression adapters A1A2/A3 were ligated overnight to 300 ng of the digested DNA using 3 U of T4 DNA ligase (Promega) at 16°C. The ligated DNA was purified using Quiaquick purification columns (Quiagen) and eluted with 50 µl water. Of the eluted DNA 1 µl was PCR amplified with the HS LTR-specific primer LTRfor1 and adapter-specific primer A1 using the following cycling program: (i) 72°C, 1′, (ii) 95°C, 1′ and (iii) 95°C, 15″; 65°C, 15″; 72°C, 1′ for 20 cycles. The PCR products were 500-fold diluted and used as templates for nested PCR with the downstream HS LTR-specific primer LTRfor2 and adapter-specific primer A2 under the same cycling conditions, for 22 cycles. The amplified LTR flanking sequences were treated with ExoIII exonuclease (Promega) to generate 5′ protruding termini exactly as described in Refs (30,31).
GREM technique
The technique includes hybridization of PCR amplified genomic sequences flanking repetitive elements (HS LTRs in our case) with cDNA, followed by selective amplification and cloning of hybrid DNA duplexes (see Figure 2). ExoIII-treated LTR flanking sequences (100 ng), obtained as described above, were mixed with 300 ng of cDNA in 4 µl of hybridization buffer (0.5 M NaCl, 50 mM HEPES, pH 8.3, 0.2 mM EDTA), overlaid with mineral oil, denatured at 95°C for 5 min and hybridized at 68°C for 14 h. The final mixture was diluted with 36 µl of dilution buffer (50 mM NaCl, 5 mM HEPES, pH 8.3, 0.2 mM EDTA), and 1 µl of the diluted hybridization mixture was PCR-amplified with 0.2 µM adapter-specific primer A2 and 0.2 µM cDNA 5′end-specific primer CS under the following conditions: (i) 72°C for 5 min to fill in the ends of DNA duplexes, (ii) 95°C for 15″, 65°C for 15″, 72°C for 1′30″, 8 cycles. The PCR products were 500-fold diluted and reamplified by nested PCR for 20 cycles (95°C, 15″, 65°C, 15″, 72°C, 1′30″) with 0.2 µM nested adapter-specific primer A4 and 0.2 µM HS LTR 3′end-specific primer LTRfor3. The final PCR products were cloned in Escherichia coli using a pGEM-T vector system (Promega) and sequenced by the dye termination method using an Applied Biosystems 373 automatic DNA sequencer.
RT–PCR
All RT–PCR experiments described in this section were reproduced at least three times using independent cDNA preparations. For RT–PCR control of LTR transcriptional status, we used pairs of primers, one of which was specific to the 3′ terminal part of a particular HS LTR (for sequences see Table 4 of Supplementary Material), and the other specific to a unique sequence within the corresponding genomic LTR 3′ flanking region. Prior to the RT–PCR analysis, the priming efficiency of the primers was pre-examined by genomic PCRs at temperatures varying depending on the primer combination used. These PCRs were done for 19, 22, 25 and 28 cycles, with 40 ng of the human genomic DNA template isolated from testicular parenchyma. The RT–PCR was done with cDNA samples of the same tissue, an equivalent of 20 ng total RNA being used as template in each PCR reaction performed in a final volume of 40 µl. Aliquots (5 µl) of the reaction mixture after 21, 24, 27, 30, 33, 36 and 39 cycles of the amplification were analyzed by electrophoresis in 1.5% agarose gels. In all cases, the transcriptional status was determined from the number of PCR cycles needed to detect a PCR product of the expected length and the PCR product concentration measured using a Photomat system and the Gel Pro Analyzer software.
RESULTS
GREM approach features
We have developed GREM (Genomic Repeat Expression Monitor), a transcriptome-wide approach that makes it possible to focus on the repetitive elements' own promoter activity and to eliminate the background of read-through sequences. The resulting library of GREM clones can be used as a set of tags for individual transcriptionally active repetitive elements. This approach combines the advantages of both 5′ RACE and nucleic acid hybridization and uses the fact that REs acting as promoters initiate the transcription from within themselves, and the corresponding transcripts contain RE sequences at their 5′ termini. This is true for retroviral LTRs, LINEs and SINEs (32–34). With this in mind, we tried to specifically isolate the transcripts containing RE sequences at their 5′ termini. We showed that the number of individual tags in the library was proportional to the content of mRNA driven by the corresponding promoter active repetitive element. We used GREM to study whole genome patterns of transcripts produced by the HS LTR family members.
Transcription of proviral LTRs may result in two types of products: RNA of viral genes (if driven from the 5′ LTR, see Figure 1), or RNA of unique non-viral sequences that flank the proviral insertions at the 3′ end, provided that the 3′ LTR has a promoter capacity.
Figure 1.

Schematic representation of solitary (left) and proviral (right) LTRs expression. The transcription driven from 5′ proviral LTRs results in mRNAs of viral genes, whereas the expression of either solitary or 3′ proviral LTRs results in the transcription of host genomic sequences, flanking the 3′ ends of the retroelements.
The GREM technique outlined in Figure 2 consists of three major stages: (i) synthesis of full length cDNA libraries whose clones include specific oligonucleotide adapters exactly tagging the cDNA 5′ ends, (ii) selective PCR amplification of genomic repeat-flanking regions and (iii) hybridization of the genomic repeat-flanking regions to the cDNA with a subsequent PCR amplification of the genome-cDNA heteroduplexes.
Figure 2.
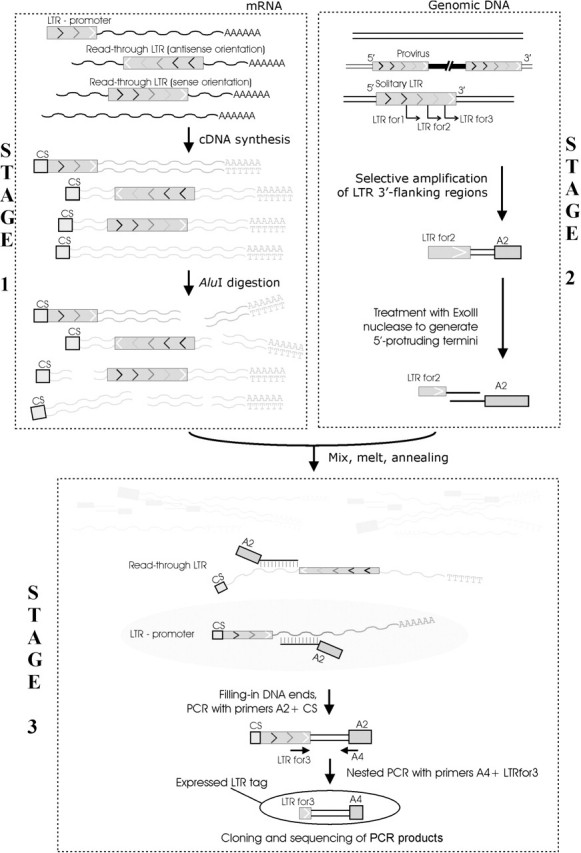
Schematic representation of the GREM technique (for details, see text). The procedure includes three major stages: (Stage 1) genome-wide amplification of the genomic DNA flanking the 3′ ends of target repetitive elements (here, HS LTRs). Treatment of the resulting amplicon with ExoIII generates 5′ protruding ends to be used at the third stage. (Stage 2) A double-stranded oligo d(T)-primed cDNA library is synthesized for tissues where expression of repetitive elements is to be studied. At this stage cDNAs are tagged by a linker oligonucleotide (CS) at the RNA transcription start sites using the ‘cap-switch’ effect. cDNAs are then digested with AluI restriction endonuclease that has no recognition sites within HS LTRs. This step precludes amplification of LTR sequences read-through in the sense orientation. (Stage 3) Finally, the genomic DNA amplicon (Stage 1) is hybridized to the 5′ tagged cDNAs (Stage 2). The protruding DNA ends are filled in with DNA polymerase, and the hybrids obtained (ELTs) are nested PCR amplified with primers specific to the flanking genomic DNA adapter and cDNA 5′ terminal tag sequence, respectively.
The first stage of GREM is aimed at the amplification of full-length cDNAs tagged at the 5′ ends with a specific adapter oligonucleotide (CS in our case). The tagging is achieved owing to the ‘cap-switch’ effect in the process of cDNA synthesis. Having reached the 5′ end of the mRNA template, oligo(dT)-primed reverse transcriptase adds a few additional deoxycytidine nucleotides to the 3′ end of the cDNA. An oligonucleotide with an oligo–ribo(G) sequence at its 3′ end hybridizes to the deoxycytidine stretch to form a primer which allows reverse transcriptase to switch templates and to continue replicating to the end of the oligonucleotide. This technique allows one to precisely tag the cDNA 5′ ends that correspond to transcription start sites (Figure 2). Prior to the hybridization at stage (iii), the cDNA was digested with AluI restriction endonuclease to get shorter fragments and to avoid further background amplification of hybrids with read-through transcripts driven in the sense orientation with respect to the LTR direction (Figure 2, stage 1, step ‘AluI digestion’). AluI was chosen because the HS LTR consensus sequence lacks restriction sites of this frequent-cutter endonuclease. The treatment of cDNA with AluI (Figure 2) suppresses the yield of sense read-through LTR containing products at the following stage (see below).
At the second stage, we selectively PCR amplified genomic regions flanking the 3′ termini of HS LTRs. The cDNA hybridization with the amplicon obtained was used to select the cDNA molecules that contain HS LTRs at their 5′ termini. The amplification of genomic flanking regions is a critical step ensuring the specificity of the whole procedure. Nested PCRs result in selective amplification of all target RE-flanking sequences, whereas cDNA amplification would not provide similar selectivity, as the exact locations of transcription start sites within RE sequences may vary for different individual REs (7,35,36) and, therefore, the design of suitable primers for PCR would be problematic.
To amplify genomic LTR flanking regions, we digested human genomic DNA with AluI restriction endonuclease, ligated the fragments obtained to a 45 nt long GC-rich synthetic linker oligonucleotide (A1A2) and performed a series of nested PCR amplifications using HS LTR specific and adapter-specific primers. As mentioned before, the HS LTR consensus sequence lacks AluI restriction sites, whereas this endonuclease normally produces DNA fragments too short to be subject to PCR fragment size selection (37). As shown previously (28–30), the use of GC-rich linkers minimizes background PCR amplification and results in almost 100% selective amplification of the expected fraction of the genome. The amplified LTR flanking sequences were treated with ExoIII exonuclease to generate 5′ protruding termini required at stage (iii) of GREM and to avoid any background cross-hybridization between LTR-containing sequences. We have recently demonstrated (30,31) that ExoIII may be used to remove adapter sequences from hybridizing mixtures. Under the conditions used, ExoIII removes nucleotides slowly enough (∼5 nt/min) to more or less precisely excise ∼30 HS LTR 3′ terminal nucleotides from the amplicons. At the last step, the digested cDNA was hybridized to the LTR 3′ flanking genomic fragments. To selectively amplify the heteroduplexes containing genomic LTR flanking regions and cDNA 5′ terminal fragments generated due to LTR promoter activity, we used PCR with the CS primer against 5′ cDNA tags and A2 primer specific to the adapters ligated to the genomic DNA. This PCR step was followed by an additional nested PCR with primers A4 and LTRfor3 to increase the specificity of amplification (Figure 2).
As a result, only heteroduplexes, but not duplexes of cDNA not relevant to LTR expression or containing read-through LTRs, were amplified. As mentioned above, a potential background of transcripts containing LTRs read-through in the sense direction was supposed to be negligible. A careful inspection of human transcribed sequence databases revealed in total 38 transcripts containing read-through HS LTRs, among them only 4 LTRs in the sense orientation. An ‘in silico’ simulation of AluI digestion suggested a complete removal of all such transcripts from GREM libraries.
The finally obtained amplified heteroduplexes, referred to as Expressed LTR Tags (ELTs), were further cloned and sequenced. Every particular ELT contained a 3′ HS LTR terminal portion, a fragment of the 3′ flanking genomic DNA and an adapter sequence (A4).
Detection of HS LTR promoter activity by the GREM technique
We used GREM to study the HERV-K (HML-2) LTRs promoter activity in normal testicular parenchyma. Of 500 sequenced ELT clones, 395 ELTs were selected after removal of rearranged plasmid and low-quality sequences. An ELT analysis allowed us to unambiguously map corresponding expressed solitary and 3′ proviral LTRs. A total of 54 elements were found to be promoter active in testis. However, unambiguous mapping was impossible in the case of 5′ proviral LTRs because the adjoining proviral sequences were repetitive and very similar (Figure 1). The results of the ELT analysis, presented as the first genome-wide map of promoter active HS LTRs, are shown in Table 1. For five randomly chosen individual solitary LTRs found to be promoter active according to GREM data, we precisely mapped transcription initiation sites using the 5′ RACE approach (7). In all cases, the transcription was driven from the same non-canonical promoter located on the border of the R and U5 regions within the HS LTR consensus sequence.
Table 1.
Genome-wide map of promoter active HS LTRs with relative contents of expressed LTR tags (ELTs) for human testicular parenchyma
| LTR ID | GenBanka | Genomic location | ELT content (%)b | Status of LTR | Human specificity (+/−) |
|---|---|---|---|---|---|
| 1 | AL359965 | 1p32.3 | 0.25 | Solitary | + |
| 2 | AL356379 | 1p34.2 | 0.50 | Solitary | − |
| 3 | AL355480 | 1p34.1 | 0.50 | Solitary | + |
| 4 | AL139421 | 1p22.1 | 0.50 | Solitary | + |
| 5 | AL135927 | 1q22 | 5.57 | Solitary | + |
| 6 | AL353807 | 1q23.1 | 0.25 | 3′ proviral | + |
| 7 | AC011811 | 2q37.1 | 0.50 | Solitary | + |
| 8 | AC074019 | 2q36.3 | 0.50 | Solitary | + |
| 9 | AC069420 | 3q27.2 | 32.66 | 3′ proviral | + |
| 10 | AC025548 | 3p21.31 | 0.25 | Solitary | + |
| 11 | AC024626 | 4 | 0.25 | Solitary | + |
| 12 | AC118278 | 4p16.3 | 1.77 | Solitary | + |
| 13 | AC110373 | 4q26 | 1.27 | Solitary | − |
| 14 | AC010267 | 5q23.1 | 0.25 | Solitary | + |
| 15 | AC116309 | 5p13.3 | 2.53 | 3′ proviral | + |
| 16 | AC026424 | 5q13.3 | 0.25 | Solitary | + |
| 17 | AC008648 | 5q35.1 | 0.25 | Solitary | + |
| 18 | AC016577 | 5q33.3 | 1.52 | 3′ proviral | + |
| 19 | AL139090 | 6q15 | 1.01 | Solitary | + |
| 20 | AL009179 | 6p22.1 | 0.50 | Solitary | − |
| 21 | AC026010 | 6q23.2 | 0.76 | Solitary | + |
| 22 | AL451165 | 6p21.31 | 15.19 | Solitary | + |
| 23 | AL138889 | 6p21.31 | 0.25 | Solitary | + |
| 24 | AL590543 | 6q25.1 | 0.25 | Solitary | − |
| 25 | AL589643 | 6q21 | 0.25 | Solitary | + |
| 26 | AC023201 | 6q25.1 | 0.25 | Solitary | + |
| 27 | AL353588 | 6p21.1 | 0.25 | Solitary | − |
| 28 | AC069335 | 7q34 | 1.01 | Solitary | − |
| 29 | AC021973 | 8q24.3 | 0.25 | 3′ proviral | + |
| 30 | AC120036 | 8q11.21 | 0.50 | Solitary | + |
| 31 | AF235103 | 8q24.3 | 0.50 | Solitary | − |
| 32 | AC015640 | 9p22.2 | 0.50 | Solitary | + |
| 33 | AL162412 | 9q21.12 | 0.50 | Solitary | + |
| 34 | AL353766 | 9q31.2 | 0.50 | Solitary | + |
| 35 | AC068707 | 10q11.21 | 1.27 | Solitary | − |
| 36 | AL392107 | 10q24.2 | 0.25 | 3′ proviral | + |
| 37 | BC001407 | 10q21.3 | 2.79 | Solitary | + |
| 38 | AP002754 | 11q12.2 | 0.25 | Solitary | − |
| 39 | AP002513 | 11q13.4 | 1.01 | Solitary | + |
| 40 | AP002793 | 11q12.13 | 0.50 | Solitary | − |
| 41 | AP003385 | 11q13.2 | 0.25 | Solitary | + |
| 42 | AC002350 | 12q24.11 | 0.25 | 3′ provirus | + |
| 43 | U47924 | 12p13.31 | 0.76 | Solitary | + |
| 44 | AL135901 | 13q14.2 | 0.25 | Solitary | − |
| 45 | AC055861 | 15q26.3 | 0.50 | Solitary | + |
| 46 | AC026817 | 15q22.2 | 0.50 | Solitary | + |
| 47 | AC068213 | 15q22.31 | 0.76 | Solitary | + |
| 48 | AC018768 | 16p13.2 | 0.25 | Solitary | + |
| 49 | AC012175 | 16p13.3 | 0.25 | Solitary | + |
| 50 | AC012146 | 17p13.2 | 1.52 | Solitary | + |
| 51 | AC008996 | 19q12 | 0.25 | 3′ proviral | + |
| 52 | AL109748 | 21q11.2 | 0.50 | Solitary | + |
| 53 | AC007326 | 22q11.21 | 0.25 | 3′ proviral | + |
| 54 | AL109653 | Xq27.3 | 0.25 | Solitary | + |
| 5′ proviral LTRs | 14.94 | 5′ proviral |
aGenBank accession number corresponding to the LTR; not applicable to 5′ proviral LTRs line.
bThe relative ELT content calculated as a ratio of the number of tags for each individual HS LTR to the total number (395) of all ELTs in the sequenced library.
Linear correlation of LTR transcription levels with the corresponding ELT proportions in the GREM libraries
We further addressed the question of whether there is a correlation between an LTR directed transcript level, measured by RT–PCR, and the frequency of the corresponding ELT occurrence in the GREM libraries. The RT–PCR amplification was done with a primer specific to an LTR 3′ terminal region and directed towards the LTR 3′ end used in pair with one of the unique primers designed against genomic loci located at a distance of 70–300 bp from the LTR 3′ end. First strand cDNAs obtained for testicular parenchyma were used as templates. The transcript levels were measured relative to the housekeeping beta-actin gene transcript level. For a sampling of 20 HS LTRs, the frequencies of ELT occurrence linearly correlated with measured by RT–PCR levels of transcripts directed by the corresponding individual LTRs (Table 2) with a correlation coefficient value of 0.91. Such a correlation suggests that the GREM approach is adequate for both qualitative identification and quantitative characterization of LTRs displaying promoter activity.
Table 2.
Relative LTR transcript levels and frequency of occurrence of the corresponding ELTs for testicular parenchyma
| LTR ID | Transcript levela (percentage of the beta-actin gene transcript level) | ELT frequency (%) |
|---|---|---|
| 4 | 0.26 ± 0.09 | 0.50 |
| 9 | 2.9 ± 0.2 | 32.66 |
| 12 | 0.16 ± 0.05 | 1.77 |
| 16 | 0.17 ± 0.03 | 0.25 |
| 17 | 0.02 ± 0.005 | 0.25 |
| 18 | 0.24 ± 0.03 | 1.52 |
| 22 | 1.4 ± 0.4 | 15.19 |
| 24 | 0.032 ± 0.013 | 0.25 |
| 27 | 0.13 ± 0.04 | 0.25 |
| 37 | 0.35 ± 0.06 | 2.79 |
| 38 | 0.16 ± 0.04 | 0.25 |
| 43 | 0.059 ± 0.014 | 0.76 |
| 47 | 0.12 ± 0.02 | 0.76 |
| 55 | 0.004 ± 0.001 | 0 |
| 56 | 0 | 0 |
| 57 | 0 | 0 |
| 58 | 0.24 ± 0.03 | 3.61 |
| 59 | 0.013 ± 0.004 | 0 |
| 60 | 0.01 ± 0.003 | 0 |
| 5′ proviral LTRs | 1.08 ± 0.09 | 14.94 |
aRelative transcript levels measured by RT–PCR.
DISCUSSION
Now it is clear that not only protein coding transcripts are essential for normal functioning of eukaryotic cell (38,39). Apart from structural and catalytic RNAs that take part in splicing, translation, X chromosome inactivation and protein sorting, a huge number of evolutionary conserved non-coding RNAs are thought to be involved in gene expression regulation in a wide variety of species (40). REs, which were constantly being ‘domesticated’ by host genomes in evolution, might provide regulatory modules for the expression of such RNAs. They could also cooperate with pre-existing gene structures to form new splice sites or regulatory RNAs (4). A comprehensive analysis of such an RE-controlled diversity of RNAs will be undoubtedly required for further functional characterization of the human genome. Focusing on human-specific REs would allow to identify candidate regulators that emerged in human genome evolution and contributed to the human–chimpanzee divergence (41).
HS LTR expression
A detailed functional analysis of individual promoter active LTRs revealed in this study is under way in our laboratory. Here we only mention that not only 5′ proviral LTRs, whose transcriptional activity is absolutely required for viral gene expression, but also 3′ proviral and solitary LTRs could serve as active promoters in human testicular parenchyma in vivo. As seen from Tables 1 and 2, some of the latter elements were transcribed at strikingly high levels, as for example solitary LTRs 5, 22 and 37, and 3′ proviral LTR 9. Interestingly, the transcriptional activity of even almost sequence identical promoter competent HS elements greatly differed ranging from ∼0.004 to ∼3% of the beta-actin transcript level (almost a 1000-fold range according to RT–PCR and in good agreement with the GREM data). Therefore, the LTR status (solitary, 3′ or 5′ proviral) per se cannot explain why the transcript levels are so different for different individual LTRs, and other hypotheses, probably based on chromatin structure-dependent transcriptional regulation, should be considered to clarify the situation.
GREM technique, rationale and potentials
In this article we describe the first application of a new technique aimed at genome- and transcriptome-wide detection of promoter active repetitive elements. As demonstrated here by the example of HS LTRs, GREM allows one to correctly identify RE-driven transcripts and, therefore, promoter active REs. Moreover, the technique can be also used to quantitatively estimate the contribution of individual repetitive elements to the transcriptome. The GREM protocol contains a stage of DNA hybridization and several PCR amplification steps, and therefore we tried to minimize possible bias effects. In particular, the well-known PCR fragment size selection effect was practically excluded by shortening DNA fragments to 100–300 bp with frequent-cutter AluI enzyme. Another possible problem of PCR selection in favor of GC-rich sequences was solved using highly processive DNA polymerases (Clontech Advantage Polymerase Mix). Finally, the time–temperature conditions of hybridization in this study were chosen to provide reassociation of ∼99% hybridizing molecules [for reassociation kinetics formulas, see (42)]. The theoretical considerations above were supported by a linear correlation of GREM tag frequencies with RT–PCR-measured contents of corresponding transcripts. Thus, being an adequate technique for large-scale transcriptome analyses, GREM provides a unique advantage of the selection of RE-promoted transcripts free of sense and antisense read-through background. This was in addition confirmed by an ‘in silico’ GREM library construction, where the final pool of GREM tags lacked all 38 known HS LTR read-through cDNAs (see above). Theoretically, for genomic repeats other than HS LTRs, a small number of read-through transcripts in the sense orientation, initiated at 100–300 bp upstream of REs (located closer than the closest frequent-cutter endonuclease restriction site), may appear as false-positive clones. However, a simple RT–PCR test with a primer specific to a genomic sequence located immediately upstream of the repetitive element would definitely answer the question whether the transcription is initiated from within the RE (Table 3).
Table 3.
Genomic primer sets used for PCR amplification
| Name | Sequence (5′–3′) | Accessiona |
|---|---|---|
| Oligonucleotides used for GREM procedure | ||
| (1) LTR specific primers | ||
| LTRfor1 | gtcttgtgaccctgacacatcc | — |
| LTRfor2 | cctccatatgctgaacgctg | — |
| LTRfor3 | ggggcaacccacccctac | — |
| (2) Suppression adapter oligonucleotides | ||
| A1A2 | gtaatacgactcactatagggcagtcgacgcgtgcccggtccgac | — |
| A3 | gtcggaccgggc | — |
| A1 | gtaatacgactcactatagggc | — |
| A2 | agtcgacgcgtgcccggtccgac | — |
| A4 | tcgacgcgtgcccggtccgacct | |
| (3) Oligonucleotides used for cap-switch based cDNA amplification | ||
| CDS | aagcagtggtatcaacgcagagtac(t)30 | — |
| riboCS | taacaacgcagagtacgcrgrgrg | — |
| CS | taacaacgcagagtacgcgg | — |
| Primers used for RT–PCR experiments | ||
| (1) LTR specific primers | ||
| LTRfor1 | gtcttgtgaccctgacacatcc | — |
| LTRfor2 | cctccatatgctgaacgctg | — |
| LTRfor3 | ggggcaacccacccctac | — |
| (2) Unique genomic primers specific for LTR 3′ flanking regions | ||
| gLTR55 | taagtggatataattactaagtccagg | AC068381 |
| gLTR38 | ccaacatctgtctcttccctg | AP002754 |
| gLTR22 | gaccatttgcatggacaaatc | AL451165 |
| gLTR9 | ccatcccttccatgccttag | AC069420 |
| gLTR56 | agctttgtggattgtaatttgg | AC072054 |
| gLTR4 | ctcagtaaagatgaaggtatgacaag | AL139421 |
| gLTR57 | gaggcagaggttgcagtgagcc | AC002400 |
| gLTR16 | ataaaggagaaatcttccatgaag | AC026424 |
| gLTR17 | tgtgacggtataatggcctct | AC008648 |
| gLTR58 | ggttatgaataaagttccctcgg | AC027750 |
| gLTR59 | agaatagagcgaacagacacag | AL352982 |
| gLTR24 | aggttattgatacattgcatcgac | AC023201 |
| gLTR12 | caataacagtcattctcactggag | AC118278 |
| gLTR27 | gagttgggatgtggtcttagg | AL353588 |
| gLTR60 | ctcatgctaaactgtctgattatgc | AC105049 |
| gLTR37 | ttgtgcaaactgtctacagcca | BC001407 |
| gLTR18 | aacatacaggttgaggccagg | AC016577 |
| gLTR47 | ttgtagctgaccaacagcctgc | AC068213 |
| gLTR43 | ttaggccagggtctcactgag | U47924 |
| (3) HERV-K (HML-2) proviral gene gag specific primer | ||
| Gag rev | aatggcccaatcattccata | — |
aGenBank accession numbers of corresponding LTRs from non-redundant and high throughout genome sequence databases.
Of course, GREM is not free of limitations. First, it cannot be applied to the analysis of non-polyadenylated transcripts, which form a significant portion of the human transcriptome (43). Therefore, the use of GREM is restricted to RNA polymerase II-transcribed repeats. Also, successful application of this method partly depends on the sequence divergence among repetitive elements under comparison. If this divergence is high, oligonucleotide primers designed from the group consensus sequence may fail to prime PCR with the group members diverged too far from the consensus. However, to improve the priming, degenerated nucleotide primers may be utilized. Alternatively, large groups of repeats could be subdivided into more sequence similar subgroups. Finally, although the stage of ExoIII digestion may seem to complicate the method, this is a common procedure making GREM a one-tube approach. The GREM technique can be similarly applied to any other group of human or non-human repeats.
It should be mentioned here that the GREM protocol could be markedly simplified under the following conditions: (i) if the transcription initiation point within an RE under study is already unambiguously mapped, thus making it possible to correctly design PCR primers and (ii) if the 3′-terminal part of a repetitive element, that remains in the repeat-driven transcript, is long enough to design PCR primers for exclusive amplification of the sequences containing REs of interest. In this case, the same pool of cDNA-derived tags can be obtained by (i) amplifying total double-stranded cDNA; (ii) digesting it by AluI; (iii) ligating a suppression adapter (such as A1A2) and (iv) performing a two-stage amplification, first with CS and A1 primers, and then (nested stage) with ‘LTRfor2-like’ and A2 primers. There would be no need for complex additional procedures such as isolation of genomic REs' flanks, their hybridization to cDNA-derived products and selective amplification of the product. Actually, it is very difficult to find an example of an RE family with a known uniform transcriptional start site, even among human REs that are thought to be better investigated than the others. Computational approaches are of little help, since their predictions are probabilistic. Moreover, multiple alternative transcriptional start sites may exist, as shown previously, for example, for L1 retrotransposons (35,36) and for HERV-K (HML-2) endogenous retroviruses studied here (7). In principle, the very 3′-terminal sequence of REs might be used for primer design in order to amplify all alternative transcripts, but in many cases this sequence will be insufficient to give a proper primer set for selective amplification of RE-containing cDNAs. For example, a few hundred base pairs long HS LTR 3′-terminal sequence resides also within so called SVA retrotransposons that are far more abundant in human DNA than HS LTRs (17,44).
Concluding remarks
Here, we described the technique termed GREM developed for genome-wide isolation and quantitative analysis of any kind of promoter active repetitive elements. This technique enabled us to make the first attempt to identify genomic repeat-associated promoter activity in a genome-wide study. We were able to both build the first genome-wide map of promoter active human-specific endogenous retroviruses and individual solitary LTRs, and we were able to quantitatively characterize promoter activities of particular elements. A detailed GREM data analysis and GREM profile comparisons for different human tissues will be a further extension of this work.
Acknowledgments
The authors thank Drs Sergey Dmitriev (Belozersky Institute of Physico-Chemical Biology, Moscow), Yuri Lebedev, Tatyana Vinogradova and Lev Nikolayev (Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Moscow) for fruitful discussion, Dr Boris Glotov (Institute of Molecular Genetics, Moscow, Russia) for his valuable comments on the manuscript and Dr Nadezhda Skaptsova (Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Moscow) for synthesis of oligonucleotides. The work was supported by Russian Foundation for Basic Research grants 05-04-48682-a and 2006.20034, by the grant MK-2833.2004.4 of the President of the Russian Federation and by the Molecular and Cellular Biology Program of the Presidium of the Russian Academy of Sciences.
Conflict of interest statement. None declared.
REFERENCES
- 1.Weiner, A.M., Deininger, P.L., Efstratiadis, A. 1986. Nonviral retroposons: genes, pseudogenes, and transposable elements generated by the reverse flow of genetic information Annu. Rev. Biochem. 55631–661 [DOI] [PubMed] [Google Scholar]
- 2.Mouse genome sequencing consortium. 2002. Initial sequencing and comparative analysis of the mouse genome Nature 420520–562 [DOI] [PubMed] [Google Scholar]
- 3.International human genome sequencing consortium. 2001. Initial sequencing and analysis of the human genome Nature 409860–921 [DOI] [PubMed] [Google Scholar]
- 4.Buzdin, A.A. 2004. Retroelements and formation of chimeric retrogenes Cell. Mol. Life. Sci. 612046–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kidwell, M.G. and Lisch, D. 1997. Transposable elements as sources of variation in animals and plants Proc. Natl Acad. Sci. USA 947704–7711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van de Lagemaat, L.N., Landry, J.R., Mager, D.L., Medstrand, P. 2003. Transposable elements in mammals promote regulatory variation and diversification of genes with specialized functions Trends Genet. 19530–536 [DOI] [PubMed] [Google Scholar]
- 7.Kovalskaya, E., Buzdin, A., Gogvadze, E., Vinogradova, T., Sverdlov, E. 2005. Functional human endogenous retroviral LTR transcription start sites are located between the R and U5 regions Virology 346373–378 [DOI] [PubMed] [Google Scholar]
- 8.Jasinska, A. and Krzyzosiak, W.J. 2004. Repetitive sequences that shape the human transcriptome FEBS Lett. 567136–141 [DOI] [PubMed] [Google Scholar]
- 9.Kazazian, H.H., Jr. 2004. Mobile elements: drivers of genome evolution Science 3031626–1632 [DOI] [PubMed] [Google Scholar]
- 10.Matz, M.V. and Lukyanov, S.A. 1998. Different strategies of differential display: areas of application Nucleic Acids Res. 265537–5543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Badge, R.M., Alisch, R.S., Moran, J.V. 2003. ATLAS: a system to selectively identify human-specific L1 insertions Am. J. Hum. Genet. 72823–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diatchenko, L., Lukyanov, S., Lau, Y.F., Siebert, P.D. 1999. Suppression subtractive hybridization: a versatile method for identifying differentially expressed genes Methods Enzymol. 303349–380 [DOI] [PubMed] [Google Scholar]
- 13.Akopyants, N.S., Fradkov, A., Diatchenko, L., Hill, J.E., Siebert, P.D., Lukyanov, S.A., Sverdlov, E.D., Berg, D.E. 1998. PCR-based subtractive hybridization and differences in gene content among strains of Helicobacter pylori Proc. Natl Acad. Sci. USA 9513108–13113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chalaya, T., Gogvadze, E., Buzdin, A., Kovalskaya, E., Sverdlov, E.D. 2004. Improving specificity of DNA hybridization-based methods Nucleic Acids Res. 32e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Velculescu, V.E., Vogelstein, B., Kinzler, K.W. 2000. Analysing uncharted transcriptomes with SAGE Trends Genet. 16423–425 [DOI] [PubMed] [Google Scholar]
- 16.Matz, M., Shagin, D., Bogdanova, E., Britanova, O., Lukyanov, S., Diatchenko, L., Chenchik, A. 1999. Amplification of cDNA ends based on template-switching effect and step-out PCR Nucleic Acids Res. 271558–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buzdin, A., Ustyugova, S., Khodosevich, K., Mamedov, I., Lebedev, Y., Hunsmann, G., Sverdlov, E. 2003. Human-specific subfamilies of HERV-K (HML-2) long terminal repeats: three master genes were active simultaneously during branching of hominoid lineages (small star, filled) Genomics 81149–156 [DOI] [PubMed] [Google Scholar]
- 18.Medstrand, P. and Mager, D.L. 1998. Human-specific integrations of the HERV-K endogenous retrovirus family J. Virol. 729782–9787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vinogradova, T.V., Leppik, L.P., Nikolaev, L.G., Akopov, S.B., Kleiman, A.M., Senyuta, N.B., Sverdlov, E.D. 2001. Solitary human endogenous retroviruses-K LTRs retain transcriptional activity in vivo, the mode of which is different in different cell types Virology 29083–90 [DOI] [PubMed] [Google Scholar]
- 20.Turner, G., Barbulescu, M., Su, M., Jensen-Seaman, M.I., Kidd, K.K., Lenz, J. 2001. Insertional polymorphisms of full-length endogenous retroviruses in humans Curr. Biol. 111531–1535 [DOI] [PubMed] [Google Scholar]
- 21.Belshaw, R., Dawson, A.L., Woolven-Allen, J., Redding, J., Burt, A., Tristem, M. 2005. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): implications for present-day activity J. Virol. 7912507–12514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dewannieux, M., Blaise, S., Heidmann, T. 2005. Identification of a functional envelope protein from the HERV-K family of human endogenous retroviruses J. Virol. 7915573–15577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayer, J., Stuhr, T., Reus, K., Maldener, E., Kitova, M., Asmus, F., Meese, E. 2005. Haplotype analysis of the human endogenous retrovirus locus HERV-K(HML-2.HOM) and its evolutionary implications J. Mol. Evol. 61706–715 [DOI] [PubMed] [Google Scholar]
- 24.Frank, O., Giehl, M., Zheng, C., Hehlmann, R., Leib-Mosch, C., Seifarth, W. 2005. Human endogenous retrovirus expression profiles in samples from brains of patients with schizophrenia and bipolar disorders J. Virol. 7910890–10901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hughes, J.F. and Coffin, J.M. 2004. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution Proc. Natl Acad. Sci. USA 1011668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buzdin, A.A., Lebedev Iu, B., Sverdlov, E.D. 2003. Human genome-specific HERV-K intron LTR genes have a non-random orientation relative to the direction of transcription, and, possibly, participated in antisense gene expression regulation Bioorg. Khim. 29103–106 [DOI] [PubMed] [Google Scholar]
- 27.Altschul, S.F., Gish, W., Miller, W., Myers, E.W., Lipman, D.J. 1990. Basic local alignment search tool J. Mol. Biol. 215403–410 [DOI] [PubMed] [Google Scholar]
- 28.Siebert, P.D., Chenchik, A., Kellogg, D.E., Lukyanov, K.A., Lukyanov, S.A. 1995. An improved PCR method for walking in uncloned genomic DNA Nucleic Acids Res. 231087–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lavrentieva, I., Broude, N.E., Lebedev, Y., Gottesman, I.I, Lukyanov, S.A., Smith, C.L., Sverdlov, E.D. 1999. High polymorphism level of genomic sequences flanking insertion sites of human endogenous retroviral long terminal repeats FEBS Lett. 443341–347 [DOI] [PubMed] [Google Scholar]
- 30.Buzdin, A., Khodosevich, K., Mamedov, I., Vinogradova, T., Lebedev, Y., Hunsmann, G., Sverdlov, E. 2002. A technique for genome-wide identification of differences in the interspersed repeats integrations between closely related genomes and its application to detection of human-specific integrations of HERV-K LTRs Genomics 79413–422 [DOI] [PubMed] [Google Scholar]
- 31.Buzdin, A., Ustyugova, S., Gogvadze, E., Lebedev, Y., Hunsmann, G., Sverdlov, E. 2003. Genome-wide targeted search for human specific and polymorphic L1 integrations Hum. Genet. 112527–533 [DOI] [PubMed] [Google Scholar]
- 32.Weiner, A.M. 2002. SINEs and LINEs: the art of biting the hand that feeds you Curr. Opin. Cell. Biol. 14343–350 [DOI] [PubMed] [Google Scholar]
- 33.Kazazian, H.H., Jr. 2000. Genetics. L1 retrotransposons shape the mammalian genome Science 2891152–1153 [DOI] [PubMed] [Google Scholar]
- 34.Esnault, C., Maestre, J., Heidmann, T. 2000. Human LINE retrotransposons generate processed pseudogenes Nat. Genet. 24363–367 [DOI] [PubMed] [Google Scholar]
- 35.Lavie, L., Maldener, E., Brouha, B., Meese, E.U., Mayer, J. 2004. The human L1 promoter: variable transcription initiation sites and a major impact of upstream flanking sequence on promoter activity Genome Res. 142253–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Speek, M. 2001. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes Mol. Cell. Biol. 211973–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rebrikov, D.V., Britanova, O.V., Gurskaya, N.G., Lukyanov, K.A., Tarabykin, V.S., Lukyanov, S.A. 2000. Mirror orientation selection (MOS): a method for eliminating false positive clones from libraries generated by suppression subtractive hybridization Nucleic Acids Res. 28e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brosius, J. 2005. Waste not, want not—transcript excess in multicellular eukaryotes Trends Genet. 21287–288 [DOI] [PubMed] [Google Scholar]
- 39.Brosius, J. 1999. RNAs from all categories generate retrosequences that may be exapted as novel genes or regulatory elements Gene 238115–134 [DOI] [PubMed] [Google Scholar]
- 40.Costa, F.F. 2005. Non-coding RNAs: new players in eukaryotic biology Gene 35783–94 [DOI] [PubMed] [Google Scholar]
- 41.Sverdlov, E.D. 2000. Retroviruses and primate evolution Bioessays 22161–171 [DOI] [PubMed] [Google Scholar]
- 42.Ermolaeva, O.D., Lukyanov, S.A., Sverdlov, E.D. 1996. The mathematical model of subtractive hybridization and its practical application Proc. Int. Conf. Intell. Syst. Mol. Biol. 452–58 [PubMed] [Google Scholar]
- 43.Bentwich, I., Avniel, A., Karov, Y., Aharonov, R., Gilad, S., Barad, O., Barzilai, A., Einat, P., Einav, U., Meiri, E., et al. 2005. Identification of hundreds of conserved and nonconserved human microRNAs Nat. Genet. 37766–770 [DOI] [PubMed] [Google Scholar]
- 44.Ostertag, E.M., Goodier, J.L., Zhang, Y., Kazazian, H.H., Jr. 2003. SVA elements are nonautonomous retrotransposons that cause disease in humans Am. J. Hum. Genet. 731444–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]