

Abstract
Background
Genetic determinants of peripheral arterial disease (PAD) remain largely unknown. To identify genetic variants associated with the ankle-brachial index (ABI), a noninvasive measure of PAD, we conducted a meta-analysis of genome-wide association study data from 21 population-based cohorts.
Methods and Results
Continuous ABI and PAD (ABI≤0.9) phenotypes adjusted for age and sex were examined. Each study conducted genotyping and imputed data to the ~2.5 million SNPs in HapMap. Linear and logistic regression models were used to test each SNP for association with ABI and PAD using additive genetic models. Study-specific data were combined using fixed-effects inverse variance weighted meta-analyses. There were a total of 41,692 participants of European ancestry (~60% women, mean ABI 1.02 to 1.19), including 3,409 participants with PAD and with GWAS data available. In the discovery meta-analysis, rs10757269 on chromosome 9 near CDKN2B had the strongest association with ABI (β= −0.006, p=2.46x10−8). We sought replication of the 6 strongest SNP associations in 5 population-based studies and 3 clinical samples (n=16,717). The association for rs10757269 strengthened in the combined discovery and replication analysis (p=2.65x10−9). No other SNP associations for ABI or PAD achieved genome-wide significance. However, two previously reported candidate genes for PAD and one SNP associated with coronary artery disease (CAD) were associated with ABI : DAB21P (rs13290547, p=3.6x10−5); CYBA (rs3794624, p=6.3x10−5); and rs1122608 (LDLR, p=0.0026).
Conclusions
GWAS in more than 40,000 individuals identified one genome-wide significant association on chromosome 9p21 with ABI. Two candidate genes for PAD and 1 SNP for CAD are associated with ABI.
Keywords: cohort study, genetic association, genome-wide association study, meta-analysis, peripheral vascular disease
Peripheral arterial disease (PAD) affects approximately 27 million people in Europe and North America (1) and is associated with increased risk for myocardial infarction, stroke, and mortality.(2–6) Measurement of ankle and arm blood pressures with a Doppler device and calculation of the ankle-brachial index (ABI) is a simple and reliable method to detect PAD. An ABI≤0.90 is indicative of definite PAD.(7) In previous work, the Ankle Brachial Index Collaboration demonstrated a reverse J shaped relationship of ABI with mortality and coronary events with a low risk ABI ranging from 1.11 to 1.40.(8)
Little is known about genetic susceptibility to PAD but familial aggregation and heritability estimates suggest a significant genetic component.(9–13) A study of 112 biological candidate genes identified only two single nucleotide polymorphisms (SNPs) in NOS3 significantly associated with ABI.(14) The candidate gene approach to identify novel genetic variants for PAD has been limited by modest study sample size, relatively small number of genes examined, and lack of replication in independent samples.(13)
Genome-wide association studies (GWAS) have successfully led to the discovery of novel genetic variants for several common diseases including coronary artery disease (CAD).(15) The association between genetic variants on chromosome 9p21 and CAD has demonstrated replication (16;17), persistent association across race/ethnicity (18), and association with other vascular diseases.(19–21) Notably, GWAS of subclinical atherosclerosis phenotypes such as intima-medial thickness or ABI are sparse. Therefore, we conducted a meta-analysis of GWAS findings for ABI within an international consortium of 21 population-based cohort studies that included 41,692 participants of European ancestry among whom 3,409 participants had PAD (ABI ≤0.90). We conducted replication analyses of our strongest findings in over 16,000 individuals from population-based cohort studies and clinically based samples of PAD. We hypothesized that this approach would lead to the unbiased identification of genetic variants associated with ABI. Further, we hypothesized that some genetic variants for ABI would be identical to those reported to be associated with CAD and/or its risk factors given shared underlying biologic pathways, while some genetic variants would be uniquely associated with PAD.
Methods
Discovery Studies
Our analyses were conducted within the international Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium (22) and included four of the five original CHARGE cohorts: Atherosclerosis Risk in Communities Study (ARIC, n=7,630), the Cardiovascular Health Study (CHS, n=3,193), the Framingham Heart Study (FHS, n=3,572) and the Rotterdam Study (RS-I, n=5,169 and RS-II n=1,642). Ten additional population-based cohorts joined the collaboration for analysis of ABI phenotypes: the Family Heart Study (FamHS, n=1,736), Genetic Epidemiology Network of Arteriopathy Study (GENOA, n=991), Gutenberg Heart Study (GHS, n=3,122), Health, Aging, and Body Composition (Health ABC, n=1,564), the Invecchiare in Chianti Study (InCHIANTI, n=1,130), Cooperative Health Research in the Region of Augsburg (KORA F3, n=1,581 and KORA F4, n=1,407), Netherlands Study of Anxiety and Depression (NESDA, n=1,612), Nijmegen Biomedical Study (NBS, n=544), Study of Health in Pomerania (SHIP, n=543). A further 6 studies derived from population isolates were also available for the analyses: Amish Study (Amish, n=1,183), Croatia-Vis (n=897), Croatia-Korcula (n=851), Croatia-Split (n=499), Erasmus Rucphen Family Study (ERF, n=2,133), and the Orkney Complex Disease Study (ORCADES, n=693). For all studies participating in the meta-analyses, each participant self-identified as European or European American and provided written informed consent and the Institutional Review Board at the parent institution for each respective cohort approved the study protocols. More detailed study-specific information is provided in the Supplementary Methods.
Ankle-brachial index Phenotypes
Ankle and brachial blood pressure measurements for each participating study were obtained from the baseline examination or the first examination the measurement was obtained. Details on the ABI protocol used and the calculation performed in each study are provided in Supplementary Method Table 1. To calculate the ABI for each leg, the systolic blood pressure at each ankle was divided by the systolic blood pressure in the arm. If the systolic blood pressure was measured in both arms, the higher arm reading was used in the ABI calculation. If replicate readings were obtained, the mean of the two measurements for each limb was used to calculate the ABI with the exception of InCHIANTI which used the higher of the two readings of each measurement set to calculate the ABI. The lower of the ABIs from the two legs was used for analysis. In ARIC and FamHS, the ABI was measured in only one leg chosen at random. Participants with an ABI >1.40 were excluded since this high ABI may represent medial sclerosis, fibrocalcific disease secondary to diabetes mellitus, or other causes of non-compressible vessels.
To maximize the sample size and the power to detect genetic variants with modest effects and to examine the entire range of ABI values given the recent evidence of increased CVD risk associated with ABI values up to 1.1(8), we examined the continuous range of ABI <1.40. As a secondary analysis to provide a clinical phenotype, we defined PAD as ABI ≤0.90 and conducted a case (ABI≤0.9)/control (ABI >0.90 and < 1.40) comparison analysis.
Genotyping and Imputation
Different genotyping platforms were used by the 21 studies (Supplementary Methods Table 2). Each study imputed the genotype “dosage” (0–2) for the expected number of alleles for ~2.5 million Phase II HapMap CEU SNPs for each participant using currently available imputation methods. (23) CHS used BIMBAM (available at http://stephenslab.uchicago.edu/software.html) (24), GHS, InCHIANTI, NESDA and SHIP used IMPUTE (25) and all other cohorts used MACH (http://www.sph.umich.edu/csg/abecasis/MaCH/).
Statistical Analysis
We devised a GWAS analysis plan for the ABI and PAD phenotypes that each study independently implemented. Sex-specific and age-adjusted residuals of ABI were created from linear regression models and used as phenotypes in the analysis. No transformation of the ABI measure was performed prior to analysis. In FHS, residuals were also obtained separately in the original and offspring cohorts. Multi-site studies (ARIC, CHS, FamHS) additionally adjusted for field study site. Each SNP was tested for association with ABI in additive genetic models using linear regression. The Amish Study, FamHS, FHS, and GENOA cohorts used linear mixed effects (LME) models to account for familial correlations. CROATIA-Vis, CROATIA-Korcula, CROATIA-Split, ERF, and ORCADES used the “mmscore” function of the GenABEL package for R statistical software for the association test under an additive model. This score test for family-based association takes into account pedigree structure and allows unbiased estimations of SNP allelic effect when relatedness is present between examinees. Logistic regression adjusting for age and sex was used to test each SNP for association with the PAD phenotype. The FamHS, FHS, and GENOA cohorts used generalized estimating equations (GEE) clustering on family to account for family correlations.
A genome-wide meta-analysis using a fixed effects approach with inverse-variance weighting, was then conducted in METAL(26) [www.sph.umich.edu/csg/abecasis/metal] for 2,669,158 SNPs in the meta-analysis excluding the population isolates (2,670,732 SNPs including the population isolates) that met imputation and quality control criteria (Supplementary Methods Table 2). Prior to meta-analysis genomic control was applied to each study. The association of ABI per each additional risk allele was quantified by the regression slope (β), its standard error [SE(β)], and the corresponding p-value. We calculated a meta-analysis odds ratio (OR) for each of the most significant SNP associations for PAD. The meta-analysis OR estimates the increase in odds of PAD for each additional copy of the risk allele of the SNP. SNP associations were considered to be significant on a genome-wide level at p<5 x108.(27;28) Standardized gene and SNP annotations were created using a PERL script.(29) We also tested for heterogeneity of study-specific regression parameters using Cochran’s Q statistic. Due to concerns about heterogeneity, we conducted analyses of non-isolate studies and of the full group of studies. We selected SNPs for replication using results from the meta-analysis excluding the population isolates because the available replication samples did not include isolates. We excluded SNP association results if the total meta-analysis sample was less than 20,000 and if the average minor allele frequency of the SNP was <5%.
Replication
We sought to replicate independent SNP associations for ABI that attained genome-wide significance (1 region) and SNPs with suggestive associations (5 regions, p<10−5) and bioinformatics data supporting the signal. The bioinformatic analyses are described in detail in the Supplementary Material. In addition, we sought to replicate one SNP associated with both ABI and PAD at p<10−4. The replication studies included 5 population-based studies and 3 clinically-based studies including a total of over 16,000 participants: the Bruneck Study (n=786), the Copenhagen City Heart Study (CCHS, n=5,330), the Multi-Ethnic Study of Atherosclerosis (MESA, n=2,611), the National Health and Nutrition Examination Surveys (NHANES 1999–2002, n=2,335), Prevention of Renal and Vascular End-stage disease (PREVEND, n=3,691) cohort, Cardiovascular Disease in Intermittent Claudication (CAVASIC, n=443) Study, Genetic Determinants of Peripheral Arterial Disease (GenePAD, n=850), and the Linz Peripheral Arterial Disease (LIPAD, n=671) Study. Each collaborating study was provided with a SNP list and a detailed analysis plan. MESA and PREVEND used in silico genotyping (Supplementary Methods Table 2) and the remaining studies genotyped the SNPs using Taqman assays or Sequenom. Relative excess heterozygosity (REH) analysis demonstrated that all genotyped SNPs were compatible with Hardy-Weinberg equilibrium at the nominal 5% test-level (Supplementary Methods Table 3).(30)
Examination of candidate genes associated with peripheral artery disease and coronary artery disease/myocardial infarction
We selected candidate genes for ABI and/or PAD from the published literature using PubMed search terms “((ankle-brachial index) OR (peripheral arterial disease)) AND polymorphism”. Association studies with at least 100 cases and 100 controls were included regardless of whether the original study results were positive or negative. Using the discovery meta-analysis results for ABI, we then identified the most strongly associated SNP based on p-values within the gene region ±100 kb upstream or downstream of the candidate gene. Due to the high correlation of imputed genotypes, the effective number of loci were calculated for each gene region (31) using the genotype scores from the KORA F4 Study (see Supplementary Methods). Bonferroni correction of p-values was then applied in each region using the effective number of loci. Subsequently, false discovery rates (FDR) were calculated using these corrected p-values, accounting for the number of gene regions examined (see Supplementary Methods). Lastly, we examined the association with ABI of 30 SNPs strongly associated with CAD in recent GWAS.(32–34) Our ABI discovery meta-analysis did not include 2 of the 30 SNPs (rs17465637 and rs3798220) and we were unable to identify proxy SNPs available in our data. Using the p-values for the 28 SNPs in our discovery meta-analysis, we then calculated the FDR for each CAD SNP accounting for the 28 regions examined.
Results
Study Sample
The study sample included 41,692 participants of European ancestry (56% women, 6,256 from population isolates) with ABI data and genome-wide genotyping. Participant characteristics at the time of ABI measurement for each cohort are provided in Supplementary Table 4. Across the studies the mean age ranged from 41.8 years to 73.8 years, the mean ABI ranged from 1.02 to 1.19, and 8.2% (n=3,409) had PAD (ABI<0.9). Characteristics of the replication samples were similar to the discovery set (Supplementary Table 5).
ABI-SNP associations
We conducted a meta-analysis with (n=41,692) and without (n=35,434) the population isolates (Supplementary Figures 1 and 2, QQ-plots and Manhattan plots, study specific lambdas ranged from 0.997 to 1.044). Our primary meta-analysis excluded studies from population isolates because of concern for study heterogeneity and the lack of availability of replication samples from isolates. The strongest SNP association for ABI was rs10757269 on chromosome 9 near CDKN2B (β= −0.006, p=2.46 x 10−8, p for heterogeneity=0.23, Table 1; meta-analysis results including the population isolates, Supplementary Table 7). Among the 96 SNP associations for ABI with p<10−5, 79 were located in the chromosome 9p21 region (Supplementary Table 6). The ABI SNP rs10757269 is in strong LD with several SNPs in the region previously reported to be associated with CAD or myocardial infarction (r2>0.8) but this ABI SNP is not in LD with SNPs previously associated with the type 2 diabetes mellitus (Figure 1). We repeated the meta-analysis to examine the association between ABI and rs10757269 first adjusting for CAD and then excluding individuals with CAD among the non-isolate studies. The association remained but was no longer genome-wide significant (adjusting for CAD: p=5.56 x 10−6; excluding CAD: p=3.79 x 10−5). Next, we sought to replicate the association between rs10757269 and ABI in both population-based and clinically-based samples (n=16,717). The magnitude and direction of the association in the replication studies was similar to the discovery set (β= −0.0035, p=0.0176) providing evidence of replication. In the combined stage 2 discovery plus replication meta-analysis the ABI-rs10757269 association became stronger (p= 2.65 x 10−9). The study-specific estimates of effect for the discovery studies, population isolates, replication studies and overall discovery plus replication meta-analyses are presented in Figure 2. Two studies among the population isolates (the Amish Study and Croatia-Split) had effect estimates in the opposite direction to the other studies. None of the other SNP associations for ABI achieved genome-wide significance. The significance of the associations for the additional SNPs chosen for replication diminished in the discovery plus replication meta-analysis (Table 1, Supplementary Table 7).
Table 1.
Meta-analysis results: ABI-SNP associations with p<10−5 in the primary discovery analysis with population isolates excluded.
| SNP | Chr | Physical Position | Closest Gene | Risk/Non-risk Allele | Risk Allele frequency | Meta-analysis | N | Beta | SE | P value | P het |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs10757269 | 9 | 22062264 | CDKN2B | G/A | 0.49 | ABI Discovery | 35036 | −0.0056 | 0.001 | 2.46E-08 | 0.23 |
| ABI Replication | 16672 | −0.0035 | 0.0015 | 1.76E-02 | 0.67 | ||||||
| ABI Combined | 51708 | −0.0049 | 0.0008 | 2.65E-09 | 0.38 | ||||||
| PAD† Discovery | 34555 | 0.0849 | 0.0296 | 4.15E-03 | 0.32 | ||||||
| rs4659996 | 1 | 238912747 | GREM2 | A/G | 0.48 | ABI Discovery | 28087 | −0.006 | 0.0012 | 4.44E-07 | 0.34 |
| ABI Replication | 16658 | −0.0018 | 0.0016 | 2.67E-01 | 0.65 | ||||||
| ABI Combined | 44745 | −0.0045 | 0.001 | 2.12E-06 | 0.32 | ||||||
| PAD Discovery | 27574 | 0.0725 | 0.0319 | 2.31E-02 | 0.52 | ||||||
| rs7003385 | 8 | 41705907 | ANK1‡ | T/C | 0.67 | ABI Discovery | 35375 | −0.0053 | 0.0011 | 5.24E-07 | 0.49 |
| ABI Replication | 16690 | −0.002 | 0.0016 | 2.20E-01 | 0.52 | ||||||
| ABI Combined | 52065 | −0.0043 | 0.0009 | 1.11E-06 | 0.43 | ||||||
| PAD Discovery | 34903 | 0.0838 | 0.0314 | 7.57E-03 | 0.24 | ||||||
| rs819750 | 1 | 99469651 | LPPR4‡ | G/T | 0.12 | ABI Discovery | 35278 | −0.007 | 0.0015 | 3.64E-06 | 0.51 |
| ABI Replication | 16660 | 0.0022 | 0.0023 | 3.22E-01 | 0.99 | ||||||
| ABI Combined | 51938 | −0.0041 | 0.0013 | 1.01E-03 | 0.31 | ||||||
| PAD Discovery | 34780 | 0.1068 | 0.0437 | 1.45E-02 | 0.06 | ||||||
| rs9485528 | 6 | 102221473 | GRIK2‡ | A/G | 0.17 | ABI Discovery | 35339 | −0.0061 | 0.0013 | 4.63E-06 | 0.78 |
| ABI Replication | 16679 | 0.0002 | 0.002 | 9.24E-01 | 0.63 | ||||||
| ABI Combined | 52018 | −0.0041 | 0.0011 | 1.77E-04 | 0.48 | ||||||
| PAD Discovery | 34850 | 0.1172 | 0.0380 | 2.02E-03 | 0.80 | ||||||
| rs722453 | 7 | 84037497 | SEMA3A | G/A | 0.42 | ABI Discovery | 26200 | −0.0054 | 0.0012 | 6.43E-06 | 0.69 |
| ABI Replication | 6300 | −0.0046 | 0.0025 | 5.74E-02 | 0.08 | ||||||
| ABI Combined | 32500 | −0.0052 | 0.0011 | 1.02E-06 | 0.59 | ||||||
| PAD Discovery | 25706 | 0.0575 | 0.0318 | 7.05E-02 | 0.63 | ||||||
| rs16824978 | 2 | 211380306 | CPS1 | T/C | 0.25 | ABI Discovery | 34950 | −0.0054 | 0.0012 | 7.77E-06 | 0.37 |
| ABI Replication | 14340 | 0.0000 | 0.0019 | 9.94E-01 | 0.22 | ||||||
| ABI Combined | 49290 | −0.0039 | 0.001 | 1.48E-04 | 0.11 | ||||||
| PAD Discovery | 34518 | 0.0760 | 0.0343 | 2.65E-02 | 0.39 |
Phet= p value for heterogeneity
SNP is located within the gene; rs819750 is within 60kb of the gene
PAD discovery: ABI≤0.9 vs. ABI>0.9
Figure 1.
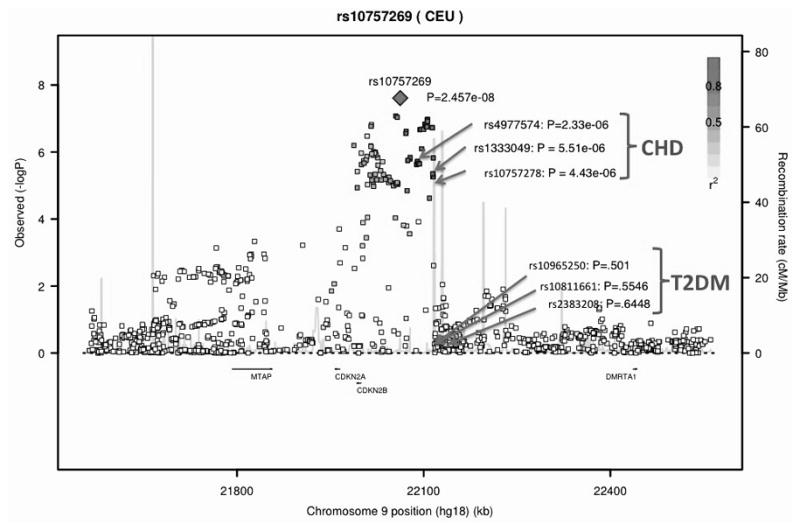
Genomic context of the genome-wide significant signal at chromosome 9p21 plotted against the −log10 p values. r2 between the top signal (rs10757269) and each SNP shown in red. SNPs previously reported from GWAS to be associated with coronary artery disease (CAD, green arrows) and type 2 diabetes (T2DM, orange arrows) and p-value for association with ankle-brachial index shown. Chromosome positions are based on build hg18.
Figure 2.

Ankle-brachial index-chromosome 9p21 (rs10757269) association: study-specific estimates of effect for the discovery studies, population isolates, replication studies and overall discovery and replication meta-analyses.
PAD-SNP Associations
None of the SNP associations for the PAD phenotype (defined by an ABI≤0.9) achieved genome-wide significance (Table 2, for meta-analysis results including population isolates Supplementary Table 8). The strongest association was found for rs6584389 on chromosome 10 near the PAX2 gene (odds ratio 1.17, 95% confidence interval 1.10, 1.25, p=2.34 x 10−6). Of note, the chromosome 9 SNP rs10757269 association with PAD was in a direction consistent with the ABI association but did not achieve statistical significance (Table 1, β=0.0849, p=0.004, increasing the odds of PAD).
Table 2.
Meta-analysis results: SNP associations for PAD (ABI ≤0.9 vs ABI >0.9) with p<10−5 with population isolates excluded.
| SNP | Chr | Physical Position | Closest Gene | Risk/Non- risk Allele | Risk Allele frequency | N | OR | 95% Confidence Interval | P value | P het |
|---|---|---|---|---|---|---|---|---|---|---|
| rs6584389 | 10 | 102459392 | PAX2 | C/A | 0.50 | 24474 | 1.17 | (1.10, 1.25) | 2.34E-06 | 0.37 |
|
|
||||||||||
| rs9998941 | 4 | 162544312 | FSTL5* | A/G | 0.23 | 34670 | 1.18 | (1.10, 1.27) | 2.34E-06 | 0.61 |
|
|
||||||||||
| rs11751656 | 6 | 42751046 | UBR2* | G/A | 0.07 | 27470 | 1.61 | (1.32, 1.96) | 2.46E-06 | 0.75 |
|
|
||||||||||
| rs4535726 | 8 | 68938371 | DEPDC2 | T/C | 0.20 | 34915 | 1.18 | (1.10, 1.26) | 3.79E-06 | 0.01 |
|
|
||||||||||
| rs2090205 | 17 | 73897869 | PGS1* | A/C | 0.24 | 34912 | 1.16 | (1.09, 1.24) | 5.01E-06 | 0.17 |
|
|
||||||||||
| rs11933540 | 4 | 25729099 | RBPJ | C/T | 0.30 | 34830 | 1.15 | (1.08, 1.23) | 9.86E-06 | 0.08 |
Phet= p value for heterogeneity
SNP is located within the gene
Overlap in SNP Associations for ABI and PAD
While the directions of effect for the ABI SNPs in Table 1 were consistent with the PAD association result (lower ABI, increased odds of PAD), there was little overlap in the top associations for the two phenotypes. Only three regions marked by SNPs in/near IDE (10q23-q25 ), DAB21P (9q33.2), and GRAMD1C (3q13.31) in addition to the chromosome 9p21 region showed association with both ABI and PAD at the p<10−4 level (Supplementary Table 9). SNP rs7100623 in IDE demonstrated the strongest novel association with both ABI (β= −0.005, p=1.89 x 10−5) and PAD (β= 0.139, p=8.39 x 10−5) at p<10−4; however the association p-value was not significant in the replication stage and diminished in the combined discovery plus replication meta-analysis.
Examination of PAD Candidate Genes
Among the 55 candidate genes or regions previously tested for association with ABI and/or PAD, eight regions showed nominally significant p-values (p<0.05) after correction for the number of effective loci for each gene region. After accounting for the number of regions examined using a false discovery rate (FDR<0.10), we found evidence of association between ABI and CYBA (rs3794624, uncorrected p=6.3 x 10−5, corrected p=0.0036, FDR=0.0665) and DAB21P (rs13290547, uncorrected p=3.6 x 10−5, corrected p=0.0035, FDR=0.0665) in addition to the chromosome 9p21 locus (rs1333049) reported to be associated with ABI (Table 3) (35). We found no evidence of association between ABI and any of the other candidate genes previously tested for association with ABI or PAD (Supplementary Table 10).
Table 3.
Literature-reported candidate genes for peripheral artery disease (PAD) and coronary artery disease (CAD) and their association with ankle-brachial index (ABI) in the CHARGE GWAS discovery sample (population isolates excluded) with FDR <0.10†.
| SNP | Chr | Physical Position | Closest Gene | Risk/Non-risk Allele | Risk Allele frequency | N | Beta | SE | P value* | # of effective loci ‡ | P value corrected‡ | False discovery rate‡ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PAD genes |
|
|||||||||||
| rs10757269 | 9 | 22,062,264 | CDKN2B | G/A | 0.51 | 35036 | −0.006 | 0.001 | 2.50E-08 | 69 | 1.70E-06 | 9.32E-05
|
| rs3794624 | 16 | 87,244,575 | CYBA | G/A | 0.34 | 31035 | −0.005 | 0.001 | 6.30E-05 | 58 | 3.60E-03 | 0.0665
|
| rs13290547 | 9 | 123,527,316 | DAB2IP | T/C | 0.06 | 32135 | −0.009 | 0.002 | 3.60E-05 | 97 | 3.50E-03 | 0.0665
|
| CAD genes |
|
|||||||||||
| rs4977574 | 9 | 22,088,574 | CDKN2B | G/A | 0.49 | 35411 | −0.0047 | 0.001 | 2.33E-06 | - | - | 6.52E-05
|
| rs1122608 | 19 | 11,024,601 | LDLR | G/T | 0.74 | 35384 | −0.0035 | 0.001 | 2.56E-03 | - | - | 0.036 |
P value from Discovery GWAS of ABI
Candidate genes for PAD were selected for testing with ABI if an association study with at least 100 cases and 100 controls was available in the literature independent of whether the study was positive or negative. Genes for CAD were only considered for testing with ABI if they were identified by recent GWAS to be genome-wide significantly associated with CAD. The table shows only the genes which showed an experiment-wise significant association with ABI after correction for multiple testing. The entire list of genes can be seen in Supplementary tables 10 and 11 for PAD and CAD genes, respectively.
Due to the high correlation of imputed genotype scores, the effective number of loci was calculated for each PAD gene region (31) using the genotype scores from the KORA F4 Study. Bonferroni correction of p-values was then applied in each region using this number. Furthermore, the corrected P value thresholds of significance for 28 CAD loci (tested in Suppl Table 11, α=0.05/28, 1.85 x 10−3) and 55 PAD loci (tested in Suppl Table 10, α=0.05/effective number of loci) were calculated. We also calculated a false discovery rate (FDR) using the corrected p-values accounting for the number of gene regions examined. An FDR <0.10 defined evidence of a significant association.
Examination of Coronary Artery Disease/Myocardial Infarction Candidate Genes
Among the 30 SNPs previously reported by GWAS to be associated with CAD or myocardial infarction, 28 SNPs were available in our discovery meta-analysis of ABI and 2 of these SNPs demonstrated an association (FDR <0.10) with ABI including rs4977574 near CDKN2B (p=2.33 x 10−6) and rs1122608 in LDLR (p=0.0026) (Table 3, Supplementary Table 11).
Discussion
Our GWAS meta-analysis for ABI conducted in more than 40,000 adults of European ancestry has several notable findings. First, we identified and replicated one genome-wide significant association between a SNP in the chromosome 9p21 region and ABI. No other ABI-SNP associations achieved genome-wide significance. Second, in our discovery sample over 3000 adults had PAD (ABI≤0.9); however, none of the SNP associations were significant. Third, the directions of effect were consistent across the two phenotypes for the most significant ABI SNPs (lower ABI, increased odds of PAD): however, we observed minimal overlap in the top SNP associations for ABI and PAD. Finally the effect size for the 9p21 SNP was modest. The association itself is, however, intriguing and may provide insights into the biologic mechanisms contributing to generalized atherosclerosis.
Chromosome 9p21 locus and atherosclerosis susceptibility
Common genetic variants in the 9p21 locus are strongly associated with myocardial infarction and CAD (17;33;36) and confer risk for other atherosclerotic diseases including stroke (19), cerebral and abdominal aortic aneurysm (20;21), and clinically diagnosed PAD; however, the relation with PAD was diminished when coronary artery disease cases were excluded.(20) SNP associations at the 9p21 locus with subclinical measures of atherosclerosis have been conflicting. Initially no association was observed with carotid intima-medial thickness or flow mediated dilation in young or older adults (37;38); however more recent reports demonstrate an association with the development and progression of carotid atherosclerosis (39) and with the suggestion of a stronger effect in men.(40) To further investigate the ABI-9p21 SNP association noted in this study, we conducted the meta-analysis after adjusting for CAD and after exclusion of individuals with CAD. Not surprisingly, the association persisted but was no longer genome-wide significant. Both CAD and PAD are manifestations of underlying atherosclerosis and nearly two-thirds of individuals with PAD have coexisting coronary or cerebrovascular disease.(41) One previous report conducted in three studies of older adults identified an association between a variant at 9p21 and lower ABI as well as an increased risk for PAD.(35) The primary affect of the chromosome 9p21 region may be on the atherosclerotic process itself, and there are likely to be many other factors both genetic and environmental that determine whether it manifests as CAD, PAD, or another clinical atherosclerotic phenotype. The primary biologic mechanism underlying the association with ABI is unknown but appears to be independent of two major PAD risk factors, diabetes and smoking, as the ABI SNP in the 9p21 region we identified is not in linkage disequilibrium with the SNPs in the region associated with diabetes risk (42;43) or smoking related behaviors.(44) The mechanism may be related to modulation of platelet reactivity (45), atheroma formation, plaque instability, thrombosis, or biologic processes not yet identified.(46) The SNP associated with ABI is nearest to CDKN2B, a well recognized tumor-suppressor gene that encodes a cyclin-dependent kinase inhibitor and is involved in regulation of the cell cycle. CDKN2B is abundantly expressed in human atherosclerotic lesions (47) and animal models suggest that altered CDKN2A/B expression results in abnormal regulation of vascular cell proliferation.(48) Functional studies reveal a long non-coding RNA at this locus named ANRIL, and a mouse model has confirmed the essential role of ANRIL in regulation of CDKN2B expression through a cis-acting mechanism.(49;50) ANRIL is implicated in proliferation and senescence.
PAD Candidate Genes
We performed a literature search to identify all candidate gene regions previously investigated for association with PAD and/or ABI, irrespective of whether the association was reported to be positive or negative. This approach revealed two further associated gene regions: DAB2IP and CYBA. DAB2IP rs13290547 was not only associated with ABI but also with PAD (p=3.62 x 10−5 and 2.2 x 10−5, respectively) (Supplementary Table 10). The DAB2IP gene encodes an inhibitor that is involved in the regulation of cell survival and proliferation. One variant in the DAB2IP gene (rs70254486) has recently been detected in a GWAS of abdominal aortic aneurysm.(51) That study also detected an association with PAD as a secondary endpoint in 3,690 cases versus 12,271 controls (p=3.9x10−5). The same SNP showed an association with CVD within a meta-analysis of case-control studies.(52) The CYBA gene is involved in NADPH oxidase regulation, which contributes to oxidative stress and plays a key role in the pathophysiology of coronary disease. Only one report investigated a SNP (rs4673) in this gene for association with PAD among 324 cases and 295 controls, but did not find an association.(53) Our study found an association of rs3794624 (r2=0.5 with rs4673), with continuous ABI, which may indicate that the earlier study likely lacked power to find this association. None of the other gene regions had sufficient evidence for association with continuous ABI in our meta-analysis. Another very wide-reaching approach designed to systematically examine a large number of genes related to intermediate phenotypes of atherosclerosis such as blood pressure regulation, lipoprotein metabolism, inflammation, oxidative stress, vascular wall biology, obesity and diabetes found only eNOS to be significantly associated with ABI.(14) This gene could not be confirmed by our candidate gene examination.
Coronary candidate genes
Besides the chromosome 9 locus, one other SNP reported to be associated with coronary disease in recent GWAS, also showed an association with ABI in our study; rs1122608 in LDLR. The LDLR gene plays an important role in cholesterol homeostasis and mutations at this gene have been shown to influence LDL cholesterol levels and the subsequent risk for coronary disease.(54) The association of LDLR gene with ABI in our study is a confirmation of the shared biologic pathways underlying both subclinical and clinically apparent disease.
Strengths/limitations
Our meta-analysis represents the largest collaborative effort to date to identify genome-wide SNP associations for variation in ABI and PAD (ABI ≤0.90) and our findings suggest the absence of common variants with large effects on ABI. Use of ABI as our primary phenotype has major advantages of detecting asymptomatic PAD as the ABI is an objective measurement whereas clinical PAD requires subjective symptoms of exertional leg discomfort and mobility of the individual. However, several limitations of our meta-analysis merit comment. The blood pressure measurement protocol and ABI calculation was heterogeneous across participating studies. While protocols were standardized within each study, the studies were not designed to be fully standardized and comparable across studies (Supplementary Methods Table 1). This phenotype heterogeneity may have impacted our ability to detect associations. Furthermore, for many studies information about a previous revascularization intervention was not available. This lack of data may have resulted in the misclassification of some of the most affected persons by placing them into an ABI range of unaffected individuals and consequently reducing our power to detect true associations. Our sample was restricted to individuals of European ancestry and thus our findings cannot yet be generalized to individuals of other race/ethnic groups. Furthermore, some PAD susceptibility variants may be race/ethnic specific and can only be uncovered through the study of non-Europeans. For example, African Americans have a higher prevalence of PAD that cannot be attributed to traditional or novel risk factors.(55) This observation raises the hypothesis that polymorphisms unique to African Americans may partially be responsible for the higher prevalence of PAD.(55) We did not evaluate gene by environment interactions which may be especially relevant for cigarette smoking, a strong risk factor for PAD (56) and a factor known to interact with other genes to modulate atherosclerosis. (57)
Conclusions
In conclusion, a common variant near the CDKN2B gene in the chromosome 9p21 locus is associated with a lower ABI. PAD represents a diffuse form of atherosclerosis associated with increased risk for death and incident CVD events. Thus, the identification of genetic variants associated with ABI may provide an important opportunity not only to unravel the biologic basis of PAD but also to improve our understanding of the causes of the variation in degree of atherosclerosis from one arterial bed to another. Additional studies are warranted to identify the causal variants in the 9p21 locus and to characterize their functional significance. The search for genes influencing predilection to PAD remains elusive and alternative approaches are warranted.
Supplementary Material
Acknowledgments
Funding Sources: The Amish Study was supported by grants R01 088119, R01 AR046838, U01 HL72515, and R01 AG18728, the University of Maryland General Clinical Research Center, Grant M01 RR 16500, Mid-Atlantic Nutrition Obesity Research Center Grant P30 DK072488, General Clinical Research Centers Program, National Center for Research Resources (NCRR) and the Baltimore Veterans Administration Geriatric Research and Education Clinical Center (GRECC). Dr. Montasser was supported by AG000219. ARIC is carried out as a collaborative study supported by NHLBI contracts(HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C), R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402; and NIH contract HHSN268200625226C and infrastructure support UL1RR025005. CHS research was supported by NHLBI contracts N01-HC-85239, N01-HC-85079 through N01-HC-85086; N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133 and NHLBI grants HL080295, HL075366, HL087652, HL105756 with additional contribution from NINDS. Additional support was provided through AG-023629, AG-15928, AG-20098, and AG-027058 from the NIA. DNA handling and genotyping was supported in part by National Center for Research Resources grant M01-RR00425 and National Institute of Diabetes and Digestive and Kidney Diseases grant DK063491. The Family Heart Study GWAS was funded by grant HL08770002 and the work was supported by NHLBI contract numbers R01HL08770003I, and R01DK06833603 and R01DK07568101 from NIDDK. This work was partially supported by the National Heart, Lung and Blood Institute’s Framingham Heart Study (Contract No. N01-HC-25195) and its contract with Affymetrix, Inc for genotyping services (Contract No. N02-HL-6-4278). A portion of this research utilized the Linux Cluster for Genetic Analysis (LinGA-II) funded by the Robert Dawson Evans Endowment of the Department of Medicine at Boston University School of Medicine and Boston Medical Center. “Genetic Epidemiology Network of Arteriopathy (GENOA) study is supported by the NIH, grant number 5R01HL087660.” The Gutenberg Heart Study is funded through Rheinland-Pfalz (“Stiftung Rheinland Pfalz für Innovation”, contract number AZ 961-386261/733), the research programs “Wissen schafft Zukunft” and “Schwerpunkt Vaskuläre Prävention” of the Johannes Gutenberg-University of Mainz and its contract with Boehringer Ingelheim and PHILIPS Medical Systems including an unrestricted grant for the Gutenberg Heart Study. This research was also supported by the National Genome Network “NGFNplus” (contract number project A3 01GS0833 and 01GS0831) by the Federal Ministry of Education and Research, Germany. Health ABC was supported by NIA contracts N01AG62101, N01AG62103, and N01AG62106. The GWAS was funded by NIA grant 1R01AG032098-01A1 to Wake Forest University Health Sciences and genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C. The InCHIANTI study baseline (1998–2000) was supported as a “targeted project”(ICS110.1/RF97.71) by the Italian Ministry of Health and in part by the NIA (Contracts: 263 MD 9164 and 263 MD 821336); the InCHIANTI Follow-up 1 (2001–2003) was funded by NIA (Contracts: N.1-AG-1-1 and N.1-AG-1-2111); the InCHIANTI Follow-ups 2 and 3 studies (2004–2010) were funded by NIA ( Contract: N01-AG-5-0002); supported in part by the NIA Intramural research program. A portion of the support was through a R&D contract with MedStar Health Research Institute. KORA F3 and KORA F4 was partially funded by the “Genomics of Lipid-associated Disorders - GOLD” of the “Austrian Genome Research Programme GEN-AU” and by the Austrian Heart Fund to F. Kronenberg and by the Austrian National Bank (Project-Nr. 13662) to Barbara Kollerits. The MONICA/KORA Augsburg cohort study was financed by the Helmholtz Zentrum München and the German National Genome Research Net NGFN2 and NGFNplus (H.-E. Wichmann 01GS0823). NESDA was supported by the Geestkracht program of ZonMW [grant 10-000-1002]; matching funds from universities and mental health care institutes involved in NESDA (GGZ Buitenamstel-Geestgronden, Rivierduinen, University Medical Center Groningen, GGZ 25 Lentis, GGZ Friesland, GGZ Drenthe). Genotyping was funded by the Genetic Association Information Network (GAIN) of the Foundation for the US NIH, and analysis was supported by grants from GAIN and the NIMH (MH081802). NBS support was obtained from RUNMC. The measurement of ABI was supported by Grant 2003B057 of the Netherlands Heart Foundation. This work was sponsored by the Stichting Nationale Computerfaciliteiten (National Computing Facilities Foundation, NCF) for the use of supercomputer facilities, with financial support from the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (Netherlands Organization for Scientific Research, NWO). The Rotterdam GWA study was funded by the Netherlands Organisation of Scientific Research NWO Investments (nr. 175.010.2005.011, 911-03-012), the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Consortium for Healthy Aging (NCHA) project nr. 050-060-810. Dr Jacqueline Witteman is supported by Netherlands Organization for Scientific Research (NOW) grant (vici, 918-76-619). Abbas Dehghan is supported by Erasmus University Rotterdam (EUR) fellowship. The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. SHIP is funded by the Federal Ministry of Education and Research (grants no. 01ZZ9603, 01ZZ0103, and 01ZZ0403), the Ministry of Cultural Affairs as well as the Social Ministry of the Federal State of Mecklenburg-West Pomerania. Genome-wide data have been supported by the Federal Ministry of Education and Research (grant no. 03ZIK012) and a joint grant from Siemens Healthcare, Erlangen, Germany and the Federal State of Mecklenburg- West Pomerania. CROATIA Studies (Croatia-Vis, Croatia-Korcula, Croatia-Split) were supported by grants from the Medical Research Council UK and Ministry of Science, Education and Sport of the Republic of Croatia (No. 108-1080315-0302) and CROATIA-Vis by the European Union framework program 6 European Special Populations Research Network (EUROSPAN) project (contract LSHG-CT-2006-018947). The genotyping for the ERF study was supported by EUROSPAN (European Special Populations Research Network) and the European Commission FP6 STRP grant (018947; LSHG-CT-2006-01947). The ERF study was further supported by grants from the Netherlands Organisation for Scientific Research, Erasmus MC, the Centre for Medical Systems Biology (CMSB) and the Netherlands Brain Foundation (HersenStichting Nederland). ORCADES was supported by the Chief Scientist Office of the Scottish Government, the Royal Society and the European Union framework program 6 EUROSPAN project (contract no. LSHG-CT-2006-018947). DNA extractions were performed at the Wellcome Trust Clinical Research Facility in Edinburgh. The Bruneck Study was supported by the Pustertaler Verein zur Prävention von Herz- und Hirngefaesserkrankungen, Gesundheitsbezirk Bruneck, and the Assessorat fuer Gesundheit, Province of Bolzano, Italy. The Copenhagen City Heart Study was supported by a Specific Targeted Research Project grant from the European Union, Sixth Framework Programme Priority (FP-2005-LIFESCIHEALTH-6) contract 037631, the Danish Medical Research Council (Copenhagen), the Research Fund at Rigshospitalet, Copenhagen University Hospital (Copenhagen), Chief Physician Johan Boserup and Lise Boserup’s Fund (Copenhagen), Ingeborg and Leo Dannin’s Grant (Copenhagen), and Henry Hansen’s and Wife’s Grant (Copenhagen). Genotyping was supported by a grant from the Austrian Heart Fund. National Health and Nutrition Examination Surveys (NHANES) are supported by the Centers for Disease Control and Prevention. The Vanderbilt University Center for Human Genetics Research, Computational Genomics Core provided computational and/or analytical support for this work. MESA and the MESA SHARe project are conducted and supported by the NHLBI in collaboration with MESA investigators. Support is provided by grants and contracts N01 HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169 and RR-024156. “Funding for SHARe genotyping was provided by NHLBI Contract N02-HL-6-4278.” The PREVEND study is supported by the Dutch Kidney Foundation (Grant E033), EU project grant GENECURE (FP-6 LSHM CT 2006 037697), and NWO VENI (grant number 916.76.170). The CAVASIC study was partially funded by the “Genomics of Lipid-associated Disorders - GOLD” of the “Austrian Genome Research Programme GEN-AU” and by the Austrian Heart Fund and the Austrian National Bank (Project-Nr. 13662). GenePAD was supported by grants RO1 HL-75774, 1K12 HL087746, 1P50HL083800 as well as grant M01 RR 00070 (General Clinical Research Center, Stanford University School of Medicine) and the Stanford Cardiovascular Institute. The LIPAD project was supported in part by a grant from the Upper Austrian Government. Genotyping was supported by a grant from the Austrian Heart Fund.
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Belch JJ, Topol EJ, Agnelli G, Bertrand M, Califf RM, Clement DL, et al. Critical issues in peripheral arterial disease detection and management: a call to action. Arch Intern Med. 2003;163:884–892. doi: 10.1001/archinte.163.8.884. [DOI] [PubMed] [Google Scholar]
- 2.Criqui MH, Langer RD, Fronek A, Feigelson HS, Klauber MR, McCann TJ, et al. Mortality over a period of 10 years in patients with peripheral arterial disease. N Engl J Med. 1992;326:381–386. doi: 10.1056/NEJM199202063260605. [DOI] [PubMed] [Google Scholar]
- 3.Newman AB, Shemanski L, Manolio TA, Cushman M, Mittelmark M, Polak JF, et al. Ankle-arm index as a predictor of cardiovascular disease and mortality in the Cardiovascular Health Study. The Cardiovascular Health Study Group. Arterioscler Thromb Vasc Biol. 1999;19:538–545. doi: 10.1161/01.atv.19.3.538. [DOI] [PubMed] [Google Scholar]
- 4.Murabito JM, Evans JC, Larson MG, Nieto K, Levy D, Wilson PW. The ankle-brachial index in the elderly and risk of stroke, coronary disease, and death: the Framingham Study. Arch Intern Med. 2003;163:1939–1942. doi: 10.1001/archinte.163.16.1939. [DOI] [PubMed] [Google Scholar]
- 5.Weatherley BD, Nelson JJ, Heiss G, Chambless LE, Sharrett AR, Nieto FJ, et al. The association of the ankle-brachial index with incident coronary heart disease: the Atherosclerosis Risk In Communities (ARIC) study, 1987–2001. BMC Cardiovasc Disord. 2007;7:3. doi: 10.1186/1471-2261-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamina C, Meisinger C, Heid IM, Lowel H, Rantner B, Koenig W, et al. Association of ankle-brachial index and plaques in the carotid and femoral arteries with cardiovascular events and total mortality in a population-based study with 13 years of follow-up. Eur Heart J. 2006;27:2580–2587. doi: 10.1093/eurheartj/ehl228. [DOI] [PubMed] [Google Scholar]
- 7.Hirsch AT, Haskal ZJ, Hertzer NR, Bakal CW, Creager MA, Halperin JL, et al. ACC/AHA 2005 Guidelines for the Management of Patients With Peripheral Arterial Disease (Lower Extremity, Renal, Mesenteric, and Abdominal Aortic): A Collaborative Report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease) J Am Coll Cardiol. 2006;47:e1–e192. [Google Scholar]
- 8.Fowkes FG, Murray GD, Butcher I, Heald CL, Lee RJ, Chambless LE, et al. Ankle brachial index combined with Framingham Risk Score to predict cardiovascular events and mortality: a meta-analysis. JAMA. 2008;300:197–208. doi: 10.1001/jama.300.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valentine RJ, Guerra R, Stephan P, Scoggins E, Clagett GP, Cohen J. Family history is a major determinant of subclinical peripheral arterial disease in young adults. J Vasc Surg. 2004;39:351–356. doi: 10.1016/j.jvs.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 10.Kullo IJ, Turner ST, Kardia SL, Mosley TH, Jr, Boerwinkle E, de Andrade M. A genome-wide linkage scan for ankle-brachial index in African American and non-Hispanic white subjects participating in the GENOA study. Atherosclerosis. 2006;187:433–438. doi: 10.1016/j.atherosclerosis.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Murabito JM, Guo CY, Fox CS, D’Agostino RB. Heritability of the ankle-brachial index: the Framingham Offspring study. Am J Epidemiol. 2006;164:963–968. doi: 10.1093/aje/kwj295. [DOI] [PubMed] [Google Scholar]
- 12.Carmelli D, Fabsitz RR, Swan GE, Reed T, Miller B, Wolf PA. Contribution of genetic and environmental influences to ankle-brachial blood pressure index in the NHLBI Twin Study. National Heart, Lung, and Blood Institute. Am J Epidemiol. 2000;151:452–458. doi: 10.1093/oxfordjournals.aje.a010230. [DOI] [PubMed] [Google Scholar]
- 13.Knowles JW, Assimes TL, Li J, Quertermous T, Cooke JP. Genetic susceptibility to peripheral arterial disease: a dark corner in vascular biology. Arterioscler Thromb Vasc Biol. 2007;27:2068–2078. doi: 10.1161/01.ATV.0000282199.66398.8c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kardia SL, Greene MT, Boerwinkle E, Turner ST, Kullo IJ. Investigating the complex genetic architecture of ankle-brachial index, a measure of peripheral arterial disease, in non-Hispanic whites. BMC Med Genomics. 2008;1:16. doi: 10.1186/1755-8794-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–1491. doi: 10.1126/science.1142447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schunkert H, Gotz A, Braund P, McGinnis R, Tregouet DA, Mangino M, et al. Repeated replication and a prospective meta-analysis of the association between chromosome 9p21.3 and coronary artery disease. Circulation. 2008;117:1675–1684. doi: 10.1161/CIRCULATIONAHA.107.730614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen GQ, Li L, Rao S, Abdullah KG, Ban JM, Lee BS, et al. Four SNPs on chromosome 9p21 in a South Korean population implicate a genetic locus that confers high cross-race risk for development of coronary artery disease. Arterioscler Thromb Vasc Biol. 2008;28:360–365. doi: 10.1161/ATVBAHA.107.157248. [DOI] [PubMed] [Google Scholar]
- 19.Gschwendtner A, Bevan S, Cole JW, Plourde A, Matarin M, Ross-Adams H, et al. Sequence variants on chromosome 9p21.3 confer risk for atherosclerotic stroke. Ann Neurol. 2009;65:531–539. doi: 10.1002/ana.21590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helgadottir A, Thorleifsson G, Magnusson KP, Gretarsdottir S, Steinthorsdottir V, Manolescu A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–224. doi: 10.1038/ng.72. [DOI] [PubMed] [Google Scholar]
- 21.Thompson AR, Golledge J, Cooper JA, Hafez H, Norman PE, Humphries SE. Sequence variant on 9p21 is associated with the presence of abdominal aortic aneurysm disease but does not have an impact on aneurysmal expansion. Eur J Hum Genet. 2009;17:391–394. doi: 10.1038/ejhg.2008.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Psaty BM, O’Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Servin B, Stephens M. Imputation-based analysis of association studies: candidate regions and quantitative traits. PLoS Genet. 2007;3:e114. doi: 10.1371/journal.pgen.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 26.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pe’er I, Yelensky R, Altshuler D, Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008;32:381–385. doi: 10.1002/gepi.20303. [DOI] [PubMed] [Google Scholar]
- 29.Johnson AD, O’Donnell CJ. An open access database of genome-wide association results. BMC Med Genet. 2009;10:6. doi: 10.1186/1471-2350-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziegler A, Van Steen K, Wellek S. Investigating Hardy-Weinberg equilibrium in case-control or cohort studies or meta-analysis. Breast Cancer Res Treat. 2011;128:197–201. doi: 10.1007/s10549-010-1295-z. [DOI] [PubMed] [Google Scholar]
- 31.Gao X. Multiple testing corrections for imputed SNPs. Genet Epidemiol. 2011;35:154–158. doi: 10.1002/gepi.20563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43:339–344. doi: 10.1038/ng.782. [DOI] [PubMed] [Google Scholar]
- 33.Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang F, Xu CQ, He Q, Cai JP, Li XC, Wang D, et al. Genome-wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. Nat Genet. 2011;43:345–349. doi: 10.1038/ng.783. [DOI] [PubMed] [Google Scholar]
- 35.Cluett C, McDermott MM, Guralnik J, Ferrucci L, Bandinelli S, Miljkovic I, et al. The 9p21 myocardial infarction risk allele increases risk of peripheral artery disease in older people. Circ Cardiovasc Genet. 2009;2:347–353. doi: 10.1161/CIRCGENETICS.108.825935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 37.Cunnington MS, Mayosi BM, Hall DH, Avery PJ, Farrall M, Vickers MA, et al. Novel genetic variants linked to coronary artery disease by genome-wide association are not associated with carotid artery intima-media thickness or intermediate risk phenotypes. Atherosclerosis. 2009;203:41–44. doi: 10.1016/j.atherosclerosis.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samani NJ, Raitakari OT, Sipila K, Tobin MD, Schunkert H, Juonala M, et al. Coronary artery disease-associated locus on chromosome 9p21 and early markers of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:1679–1683. doi: 10.1161/ATVBAHA.108.170332. [DOI] [PubMed] [Google Scholar]
- 39.Ye S, Willeit J, Kronenberg F, Xu Q, Kiechl S. Association of genetic variation on chromosome 9p21 with susceptibility and progression of atherosclerosis: a population-based, prospective study. J Am Coll Cardiol. 2008;52:378–384. doi: 10.1016/j.jacc.2007.11.087. [DOI] [PubMed] [Google Scholar]
- 40.Lin HF, Tsai PC, Lin RT, Khor GT, Sheu SH, Juo SH. Sex differential genetic effect of chromosome 9p21 on subclinical atherosclerosis. PLoS One. 2010;5:e15124. doi: 10.1371/journal.pone.0015124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhatt DL, Steg PG, Ohman EM, Hirsch AT, Ikeda Y, Mas JL, et al. International prevalence, recognition, and treatment of cardiovascular risk factors in outpatients with atherothrombosis. JAMA. 2006;295:180–189. doi: 10.1001/jama.295.2.180. [DOI] [PubMed] [Google Scholar]
- 42.Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 44.Caporaso N, Gu F, Chatterjee N, Sheng-Chih J, Yu K, Yeager M, et al. Genome-wide and candidate gene association study of cigarette smoking behaviors. PLoS One. 2009;4:e4653. doi: 10.1371/journal.pone.0004653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Musunuru K, Post WS, Herzog W, Shen H, O’Connell JR, McArdle PF, et al. Association of single nucleotide polymorphisms on chromosome 9p21.3 with platelet reactivity: a potential mechanism for increased vascular disease. Circ Cardiovasc Genet. 2010;3:445–453. doi: 10.1161/CIRCGENETICS.109.923508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cunnington MS, Keavney B. Genetic Mechanisms Mediating Atherosclerosis Susceptibility at the Chromosome 9p21 Locus. Curr Atheroscler Rep. 2011;13:193–201. doi: 10.1007/s11883-011-0178-z. [DOI] [PubMed] [Google Scholar]
- 47.Holdt LM, Sass K, Gabel G, Bergert H, Thiery J, Teupser D. Expression of Chr9p21 genes CDKN2B (p15(INK4b)), CDKN2A (p16(INK4a), p14(ARF)) and MTAP in human atherosclerotic plaque. Atherosclerosis. 2011;214:264–270. doi: 10.1016/j.atherosclerosis.2010.06.029. [DOI] [PubMed] [Google Scholar]
- 48.Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–412. doi: 10.1038/nature08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pasmant E, Sabbagh A, Vidaud M, Bieche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 2011;25:444–448. doi: 10.1096/fj.10-172452. [DOI] [PubMed] [Google Scholar]
- 50.Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gretarsdottir S, Baas AF, Thorleifsson G, Holm H, den Heijer M, de Vries JP, et al. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet. 2010;42:692–697. doi: 10.1038/ng.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harrison SC, Holmes MV, Agu O, Humphries SE. Genome wide association studies of abdominal aortic aneurysms-Biological insights and potential translation applications. Atherosclerosis. 2011;217:47–56. doi: 10.1016/j.atherosclerosis.2011.02.045. [DOI] [PubMed] [Google Scholar]
- 53.Renner W, Schallmoser K, Gallippi P, Krauss C, Toplak H, Wascher TC, et al. C242T polymorphism of the p22 phox gene is not associated with peripheral arterial occlusive disease. Atherosclerosis. 2000;152:175–179. doi: 10.1016/s0021-9150(99)00448-7. [DOI] [PubMed] [Google Scholar]
- 54.Linsel-Nitschke P, Gotz A, Erdmann J, Braenne I, Braund P, Hengstenberg C, et al. Lifelong reduction of LDL-cholesterol related to a common variant in the LDL-receptor gene decreases the risk of coronary artery disease--a Mendelian Randomisation study. PLoS One. 2008;3:e2986. doi: 10.1371/journal.pone.0002986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Allison MA, Criqui MH, McClelland RL, Scott JM, McDermott MM, Liu K, et al. The effect of novel cardiovascular risk factors on the ethnic-specific odds for peripheral arterial disease in the Multi-Ethnic Study of Atherosclerosis (MESA) J Am Coll Cardiol. 2006;48:1190–1197. doi: 10.1016/j.jacc.2006.05.049. [DOI] [PubMed] [Google Scholar]
- 56.Conen D, Everett BM, Kurth T, Creager MA, Buring JE, Ridker PM, et al. Smoking, smoking status, and risk for symptomatic peripheral artery disease in women: a cohort study. Ann Intern Med. 2011;154:719–726. doi: 10.1059/0003-4819-154-11-201106070-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Viiri LE, Viiri KM, Ilveskoski E, Huhtala H, Maki M, Tienari PJ, et al. Interactions of functional apolipoprotein E gene promoter polymorphisms with smoking on aortic atherosclerosis. Circ Cardiovasc Genet. 2008;1:107–116. doi: 10.1161/CIRCGENETICS.108.791764. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.