


Abstract
Many bacterial genes are controlled by metabolite sensing motifs known as riboswitches, normally located in the 5′ un-translated region of their mRNAs. Small molecular metabolites bind to the aptamer domain of riboswitches with amazing specificity, modulating gene regulation in a feedback loop as a result of induced conformational changes in the expression platform. Here, we report the results of molecular dynamics simulation studies of the S-adenosylmethionine (SAM)-II riboswitch that is involved in regulating translation in sulfur metabolic pathways in bacteria. We show that the ensemble of conformations of the unbound form of the SAM-II riboswitch is a loose pseudoknot structure that periodically visits conformations similar to the bound form, and the pseudoknot structure is only fully formed upon binding the metabolite, SAM. The rate of forming contacts in the unbound form that are similar to that in the bound form is fast. Ligand binding to SAM-II alters the curvature and base-pairing of the expression platform that could affect the interaction of the latter with the ribosome.
INTRODUCTION
Antibiotic resistance is viewed as one of the most pressing public health problems around the world. According to the US Food and Drug Administration, the need for new antibiotics is imperative to the successful treatment of new resistant bacterial infections (1). Several major diseases caused by microbes, such as Tuberculosis, are becoming increasingly more difficult to treat with commonly used drugs. The excess use of antibiotics and evolutionary adaptations of bacteria have exacerbated this problem. The need to develop new antimicrobial drugs is of the utmost importance, and a greater understanding of bacterial gene regulation is a key to solving this problem.
One possible drug target for the new class of antibiotics is the recently discovered metabolite sensing domain of bacterial mRNA (2–5). About 4% of all bacterial genes are regulated at the post-transcriptional level by small metabolites binding to mRNAs (6–9). These transcribed mRNAs typically have a ligand-binding ‘aptamer’ domain and an ‘expression platform’ located in the 5′ un-translated region (UTR). The binding of cognate metabolites, which are generally synthesized by the proteins of the respective mRNAs, to the aptamer triggers conformational changes in the expression platforms resulting in initiation or termination of either transcription or translation. The structured 5′ UTR, known as a ‘riboswitch’, effectively functions as an environmentally sensitive switch capable of sensing local cellular concentrations of metabolites or other ligands in a feedback loop. At the level of translation, riboswitches act by exposing or obscuring the Shine–Dalgarno (SD) sequence and the initiation codon, which are vital elements in ribosomal binding and processing of mRNAs (10). When the bases in the expression platform are hidden, the ribosome cannot bind to the mRNA or initiate translation (11,12). When they are exposed, the ribosome can bind and initiate or resume translation.
Many riboswitches that regulate a host of biosynthetic pathways have been discovered (13). The metabolites they bind include glycine (14), lysine (15–17), adenine (18,19), guanine (20,21), 7-aminomethyl-7-deazaguanine (22,23), thiamine pyrophosphate (8,24–27), flavin mononucleotide (28,29), vitamin B12 (30,31), glucosamine-6-phosphate (32–35), and S-adenosylmethionine (SAM) (36,37). SAM is a co-factor involved in the ubiquitous methionine biosynthesis and related sulfur metabolic pathways in all bacteria (36). Interestingly, the overall fold of the SAM sensing riboswitch is different in different types of bacteria. Five different folds of SAM riboswitches have been discovered to date, vis-à-vis, SAM-I (36,38), SAM-II (39,40), SAM-III (41), SAM-IV (42) and SAM-V (43,44). Of these riboswitches, SAM-I and SAM-IV share similar binding pockets and significant structural elements (42), and SAM-V appears to share similar core structure to SAM-II (13). SAM-II riboswitch is found mainly in proteobacteria and is very specific for SAM (10). SAM-II riboswitch binds SAH (S-adenosylhomocysteine) and other similar byproducts of SAM with much lower affinity (40). The specificity of SAM-II for SAM and the fact that the X-ray crystal structure of SAM-II is one of the only riboswitches containing all of the sequence of the ligand-binding domain and the regulatory expression platform make SAM-II an excellent model system to study (10).
Furthermore, the crystal structure of the SAM-II metabolite-bound riboswitch, solved by Gilbert et al. (10), is a classic H-type pseudoknot shown in Figure 1 (45), a motif that is widespread in RNA biology. The regulatory expression platform (and the SD sequence) is part of the purine-rich region (Figure 1; orange). L1 (green in Figure 1A) interacts with the major groove of the P2a/b helix in a triple helical structure that is characteristic of pseudoknot structures. The expression platform that is in the 3′-end of P2a/b follows the curvature of the helix and forms base-pairing with the 5′-end of P2a/b in the crystal structure of the metabolite-bound riboswitch. The nature of the unbound state of the SAM-II riboswitch at the atomistic detail is not known. However, the response of different regions of the SAM-II riboswitch to ligand binding has been extensively studied using chemical and inline probing experiments (10,39). Ligand-dependent stabilization of L1 and the P2a/b helix were observed in the chemical probing experiments. On the other hand, the P1 helix was predominately unchanged upon ligand binding.
Figure 1.

The classic H-type pseudoknot structure of the SAM-II riboswitch (PDB id 2qwy). L1 (green) is localized in the groove of the P2b helix, and the expression platform which follows the curvature of the P2a/b helix is part of the purine rich sequence shown in orange (A). SAM (cyan) is bound in the groove formed by L1 and the 5′-end of P2b, and the adenine moiety of SAM mimics a base on the 5′-end of P2b by intercalating and stacking between U21 and G22 (B). The secondary structure of the SAM-II riboswitch is shown (C), with the base pairing interactions represented using commonly used notations. The other non-standard interactions observed in the X-ray structure that form the remaining tertiary interactions are not shown. The base-pairing interactions between L1, P2b and SAM in the metabolite bound X-ray crystal structure of the SAM-II riboswitch are shown in (B). The distances between N3 of U10 and O4 of U44 (black), O4 of U11 and O4 of U21 (red), and O4 of U12 and N6 of A46 (blue) are used to monitor the interaction of L1 with the major groove of the helix.
SAM binds in the groove between L1 and the 5′-end of the P2 helix of the SAM-II riboswitch. The adenine group of SAM appears (Figure 1) to mimic a base on the 5′-end of P2 by intercalating and stacking between U21 and G22 and interacting with its Hoogsteen base-pairing face to an unpaired U44 on the 3′-end of P2b, stabilizing the helix (Figures 1B). U44 then interacts with U10 on L1, stabilizing the triple helix. The positively charged sulfur moiety of SAM interacts with the two carbonyl oxygen groups on U21 from P2b and U11 from L1, further stabilizing the triple helix between P2a/b and L1, in a site created by the triple base U11•(U21-A45). This interaction site seems to be one of the key components in the specificity of the SAM-II riboswitch for SAM, because SAH, which lacks the single positive charge, binds to SAM-II with an affinity reduced by at least three orders of magnitude (40).
Atomic structures of the unbound state of riboswitches have been solved for only a handful of systems, and these structures include X-ray crystal structures of the glmS (33) and lysine (46,47) riboswitches and NMR structure of the preQ1 (48) riboswitch. NMR studies have also been carried out on the free and bound forms of the purine-sensing riboswitch (49–52). Most of the X-ray crystal structures of riboswitches that undergo large conformational changes have only been solved in their metabolite bound form. Therefore, questions do arise about the nature of their unbound states. For example, how do riboswitches switch conformations from the off to the on state, or vice versa? Direct observations at this level of detail are not always easy to achieve with current experimental techniques. In this regard, all-atom molecular dynamics (MD) simulation has proven invaluable as a complimentary technique to experiments in understanding biomolecular structures, dynamics and function (53–55). This simulation method and related techniques have been used to study other types of riboswitches and have provided very useful information about ligand-induced conformational changes (56–59). We have therefore used MD simulations to probe the dynamics of the SAM-II riboswitch in the bound and unbound forms in order to understand the role of SAM in stabilizing the off state of the riboswitch and the nature of the expression platform in the bound and unbound forms.
RESULTS AND DISCUSSIONS
In order to study the atomistic basis for the switching mechanism between the on and off states of the SAM-II riboswitch, we have carried out two very long all-atom MD simulations (200 ns each) on the bound and unbound forms in explicit water and ions. The snapshots of the first 50 ns of each simulation were discarded in order to make sure that the systems were well equilibrated. The trajectories of the last 150 ns of each simulation were analyzed.
Conformational switching of the pseudoknot structure of the SAM-II riboswitch
There are three major hydrogen bonding and electrostatic interactions between SAM and the RNA (Figure 1B) that are intricately involved in stabilizing the pseudoknot structure. The extended structure of SAM also spans three of the interactions made by L1 and the major groove of P2b, as shown in Figure 1B. We have monitored the three interactions formed by L1 and the major groove of the P2b helix in the presence and absence of SAM in order to probe the role of SAM in preserving these interactions and the integrity of the SAM-II pseudoknot structure. Two of the three interactions L1 makes with the major groove of the P2b helix that are between N3 of U10 and O4 of U44 and between O4 of U11 and O4 of U21 directly involve SAM and are well-formed in the complex (off state) throughout the simulation, as shown in Figure 2 (left column). The distance between O4 of U11 and O4 of U21 is used to monitor the second interaction, because the proximity of these two atoms is essential to the formation of the interaction site for the positively charged sulfur group in SAM. The third interaction between O4 of U12 and N6 of A46, also shown in Figure 2 (left column), is quite stable throughout the simulation with some breathing motions taking place. The third interaction does not form any hydrogen bonding interactions with SAM. SAM locks the first two interactions in place and stabilizes the pseudoknot structure. The third interaction seems to naturally follow.
Figure 2.

Distances between N3 of U10 and O4 of U44 (black), O4 of U11 and O4 of U21 (red) and O4 of U12 and N6 of A46 (blue) in the bound form (left column) and unbound form (right column) of the SAM-II riboswitch during the simulations. The colors are consistent with the corresponding interactions shown in Figure 1B.
We have studied these same interactions in the unbound form of the SAM-II riboswitch, also shown in Figure 2 (right column). The interactions behave quite differently in the unbound form of the riboswitch. The interactions are not as stable as they are in the bound form. They form and break, alternating between different states. As a result, L1 moves in and out of the major groove of the P2a/b helix. One of the states of the unbound form is close to the conformational state of the bound form, with some of the key interactions formed, and the other states can be viewed as a more relaxed and less compact ensemble of conformations. These results are consistent with chemical probing experiments of the SAM-II riboswitch (10). In the chemical probing experiment, the reactivity of a particular domain is directly correlated with its flexibility. It was shown that L1 becomes less reactive upon binding SAM, suggesting that L1 is flexible in the unbound form of the riboswitch and localized in the bound form. Distributions of the first two interactions that are made by L1 with the major groove of P2b in the simulation of the unbound form of the riboswitch can be separated into a small population that is close to the bound form and a more relaxed and open ensemble of conformations (Supplementary Figure S1). More contacts similar to that of the bound state are formed between U10 and U44 than between U11 and U21, and more contacts are formed between U11 and U21 than between U12 and A46 (Supplementary Figure S1). Switching between the different conformational states of the riboswitch in the unbound form is estimated to be very fast and in the nanosecond timescale (Supplementary Figure S2). Therefore, the limiting step of the on–off switch will most likely be the binding and dissociation of SAM that is expected to be much longer than the nanosecond time scale, based on binding studies of other riboswitches (60,61).
These results suggest that the unbound form of the riboswitch periodically forms contacts similar to that of the bound form that are necessary to stabilize the ensemble of conformations of the bound form. The unbound form of the SAM-II riboswitch partially samples the conformational space of the bound form, and upon binding of SAM, the classic pseudoknot conformation is fully formed after ‘zipping’ up L1 with the help of the additional RNA–ligand interactions. The simulation results therefore suggest that the recognition mechanism of the SAM-II riboswitch is due to a combination of conformational changes of the unbound form to conformations similar to that of the bound form and subsequent rearrangement of the structure, as exemplified by the conformational selection (62–64) and induced fit (65) models of ligand binding, respectively. Interestingly, this interplay between induced fit and conformational selection has been previously observed in the guanine-sensing riboswitch (49).
We have shown that the binding of SAM to the SAM-II riboswitch localizes L1 in the major groove of the P2b helix, and our results provide atomistic description of earlier experimental observation (10) of the flexibility of L1 in the unbound form. What functional role does L1 have, since it behaves so differently in the bound and unbound forms of the SAM-II riboswitch and is a central feature in stabilizing the classic H-type pseudoknot? For example, how does the motion of L1 correlate with other parts of the riboswitch? In order to answer these questions, we have calculated the cross-correlation coefficients of each residue with all of the other residues of the SAM-II riboswitch in the simulation of the unbound form, as shown in Figure 3. Figure 3 shows the normalized correlation matrix plot of both positive (red) and negative (blue) correlated motions between each residue in the unbound form of the SAM-II riboswitch. Several positive correlations will naturally show up between different residues, if they are on the same domain or directly interacting with each other. For example, the diagonal shows up as positive correlated motions because it represents the correlation between each individual residue to itself. However, the interesting correlated motions are the ones that are long-ranged and do not involve direct interactions between the residues in question. For example, in Figure 3 we clearly see strong negative correlated motions between L1 and the purine-rich sequence of the expression platform. We also see correlated motions between binding site residues, especially residues U21 and G22, and the expression platform. These results therefore suggest and provide evidence that binding and dissociation of the ligand, coupled with the ensuing motions of L1 in and out of the major groove of the P2b helix, are tied to the motions of the expression platform. The correlation of the motions between the binding site residues, L1, and the expression platform is interesting, because the mechanism of action of the riboswitch is to control the dynamics and conformational changes in the expression platform through binding and dissociation of the ligand.
Figure 3.

Correlated motions of the SAM-II riboswitch calculated using the simulation of the unbound form. The color bar shows the relationship between the cross-correlation coefficients and the intensity of the colors. Red and blue represent strong positive and strong negative correlations, respectively. The residue numbers of the L1 region of the SAM-II riboswitch that is in the major groove of the P2a/b helix are highlighted with a green line, and the residue numbers representing the purine-rich region of the expression platform are highlighted with an orange line. The positions of the two residues (U21 and G22) in the binding site, sandwiching the adenine moiety of SAM in the bound form are shown with arrows. The region showing correlated motions between L1 and the expression platform is labeled as (A), and the region showing correlated motions between the binding site residues and the expression platform is labeled as (B).
Ligand-induced curvature and base-pairing of the expression platform
The expression platform is a key component of bacterial mRNA structure and directs the ribosome to the proper location in order to initiate protein synthesis. The ultimate goal of conformational switching in the SAM-II riboswitch is to initiate or terminate translation due to induced conformational changes in the expression platform that expose or conceal the ribosomal recognition sequence. The AUG start codon, which is not part of the crystal structure, would normally be found downstream, immediately after the 3′-end of the riboswitch. As we have already seen above, the motions of the binding site, L1 and the expression platform are correlated. A hypothesis based on occlusion of the recognition SD sequence in repressing translation was proposed as a result of the pseudoknot structure of the SAM-II riboswitch and chemical probing experiments (10). It was assumed that stabilization of the pseudoknot structure by SAM locks the expression platform in an ensemble of conformations that prevent the bases of the recognition sequence from ultimately interacting with the ribosome. One main observation of the expression platform in the bound form (as shown in Figure 1) is that it is an integral part of the helical structure formed by the P2a/b helix, following the curvature of the helix, in the bound structure. Since the metabolite-bound form of the pseudoknot structure of SAM-II represses translation, it is obvious that this particular conformation would not allow ribosomal assembly or translation to proceed. It is therefore assumed that a different ensemble of conformations of the expression platform would serve as a proper recognition conformation or a proper conformation to initiate translation. We have therefore studied the curvature of the expression platform in the bound and unbound forms of the riboswitch, as shown in Figure 4. In order to calculate the effective curvature, we fitted the phosphorus atoms of six of the last seven phosphate groups to an effective circle.
Figure 4.

Probability distributions, p(κ), of the effective curvature, κ, of the expression platform in the bound (black) and unbound (red) forms of the of the SAM-II riboswitch. Ensembles of structures of the bound state (right inset) with a curved expression platform and unbound state (left inset) with a less curved expression platform are shown as insets. The structures represent a 5 ns segment of the simulation that matches the conformations at the peaks of the distributions.
Figure 4 shows distributions of the effective curvature of the expression platform during the entire simulations with and without the presence of SAM. It can be seen that the expression platform of the bound form is more curved and has a narrower distribution than that of the unbound form of the riboswitch. The bound riboswitch exhibits a stable, tightly curved binding platform; however, the unbound riboswitch shows variation in curvature with an overall unwinding of the platform. The distribution of the effective curvature of the unbound riboswitch partly overlaps with that of the bound form, providing further evidence that the unbound form of the riboswitch partially samples the conformational space close to that of the bound form. The results suggest that in the absence of SAM, the platform uncurls and exposes the binding platform for possible ribosomal assembly or initiation of translation. As the binding platform straightens out, the bases of P2a/b become exposed and readily accessible, as shown in Figure 5A.
Figure 5.

Probability distributions of distances, p(r), between the bases of three base-pairs in the purine-rich expression platform of the P2a/b helix. The distances are measured between N3 of U20 and N1 of A46, N6 of A19 and N3 of A47, and O2 of U18 and N6 of A48 (A) from the simulations of the bound (black) and unbound (red) forms of the SAM-II riboswitch. The base-pairing interactions are shown in the X-ray crystal structure of the SAM-II riboswitch (B). Probability distributions of distances, p(r), between the bases of three base-pairs around the center of the P1 helix are also shown (C). The distances are measured between N1 of G3 and N3 of C29, N3 of C4 and N1 of G28, and N1 of G5 and N3 of C27 from the simulations of the metabolite bound (black) and unbound (red) forms of the SAM-II riboswitch.
Figure 5 shows the distribution of the interactions between the three most upstream bases of the purine-rich sequence (Figures 1 and 5B; orange) on the P2a/b helix that contains the expression platform on the 3′-end and the bases on the 5′-end of the same helix in the bound and unbound forms of the SAM-riboswitch. It can be seen that the interactions between these bases and their pairing partners on the opposite strand, as depicted in the X-ray crystal structure (Figure 5B), are very stable and well formed in the ligand bound form of the SAM-II riboswitch. However, in the unbound form of the riboswitch, these same interactions are loosely formed and the distributions are much broader than that of the bound form. Unlike the P2a/b helix, bases pairing in the P1 helix are unaffected by metabolite binding (Figure 5C), also in agreement with chemical and inline probing experiments (10,39). Figure 5C shows distributions of the interactions between the bases of three base pairs around the center of the P1 helix in the metabolite-bound and unbound forms of the SAM-II riboswitch. The distribution of the base pairing distances in the unbound form is almost identical to that of the bound form.
These results therefore suggest pairing of the bases in the expression platform upon binding of the metabolite, concealing the bases from direct interaction with cellular components in the off state. On the other hand, the bases are hardly paired in the unbound form, and these loosely paired bases would be pivotal for the assembly of the ribosome. Alternatively, the expression platform of the metabolite-bound pseudoknot structure of the SAM-II riboswitch could still bind the ribosome, but blocks translation until after dissociation of the metabolite and the subsequent conformational changes in the expression platform. Previously, Marzi et al. (66) reported a series of cryo-electron microscopy images suggestive of the fact that folded 5′ UTR of mRNAs may still bind the ribosome, but blocks initiation of translation until the mRNA unfolds. The simulation results suggest that the changes in the curvature and base-pairing of the expression platform partly control the interactions of the riboswitch with the ribosome.
Fully understanding how such conformational changes accompany ligand binding and how they direct initiation or termination of translation (or transcription, as in the case of other riboswitches) would provide a valuable insight into bacterial gene regulation. The ability to modulate gene regulation in bacteria holds tremendous promise in designing new classes of antibiotics. The metabolite sensing riboswitches have so far only been discovered in prokaryotes, fungi and plants (13,67,68), therefore, targeting this gene regulation mechanism as a therapeutic strategy could potentially have little or no side-effect to a human host.
CONCLUSIONS
Most of the X-ray crystal structures of riboswitches solved to date that undergo large conformational changes are in the metabolite-bound form. Therefore, there is limited structural information about their unbound states and the mechanism of the on–off switch. Our MD simulation studies have revealed the nature of the ligand-induced conformational changes of the SAM-II riboswitch at atomistic detail and agree very well with previous chemical and inline probing experiments. The classic H-type pseudoknot structure of the SAM-II riboswitch is only fully formed in the metabolite-bound structure. In the unbound structure, the SAM-II riboswitch alternates between a loosely formed pseudoknot structure and an ensemble of conformations that are close to that of the bound form. Ligand binding stabilizes L1 in the major groove of the P2a/b helix, causing the helix to partially wrap around L1 and induces curvature and stabilizes base-pairing in the expression platform. The loose and flexible nature of the unbound form of the SAM-II riboswitch would make it difficult to investigate the unbound form of riboswitches using X-ray crystallography. However, other experimental techniques, such as NMR, SAXS and chemical probing, could provide valuable insight into the nature of the unbound states of riboswitches. Similarly, theoretical and computational methods can complement experiments in further understanding and providing another perspective of the switching mechanism of riboswitches in bacterial gene regulation at atomistic detail, as we have done in these studies.
Our simulations show that the rate of sampling the different conformational states of the unbound form of the SAM-II riboswitch, including conformational states similar to that of the bound form, is on the nanosecond timescale. The results therefore suggest that the rate of dissociation of SAM will determine whether the mechanism of action of the SAM-II riboswitch is thermodynamically or kinetically driven. Since the lifetime of prokaryotic mRNAs is very short (minutes) (69), designing new drugs that can compete with SAM and bind to the SAM-II riboswitch with a very slow dissociation rate would therefore be a good strategy in shutting down the translational machinery of the SAM-II riboswitch.
Computational methods
MD simulations
The bound crystal structure of the SAM-II riboswitch (PDB id:2qwy; chain A) solved by Gilbert et al. (10) was the starting point for all simulations. All MD simulations were carried out using the Amber 10 suite of programs (70). In order to obtain the unbound form of the riboswitch, the structure was edited to remove the ligand. All of the crystallographic water molecules and ions were preserved. The modified nucleic acid parameters (parmbsc0) (71) of the Cornell et al. (72) Amber force field were used. These parameters along with the structure of the unbound riboswitch were loaded into the LEAP module in Amber. The system was placed in an explicit TIP3P (73) truncated octahedron water box consisting of 11 257 water molecules, and 50 Na+ counter ions were added to bring the system to neutrality. After minimizing the system, a 10 ns equilibration was carried out using the PMEMD module. The Langevin thermostat was used during the equilibration in order to bring the system from 100K to 300K using a collision frequency of 1.0 /ps. Following the 10 ns equilibration, a 200 ns long MD simulation was carried out. Particle mesh Ewald (74) method was used to treat long range electrostatics. A 2 fs time-step was used to solve Newton’s Equations of Motion at a constant pressure of 1 bar and a constant temperature of 300K using the NTP ensemble. All bonds involving hydrogen atoms were restrained using the SHAKE algorithm (75), and a 9 Å cutoff was applied to all long ranged non-bonded interactions.
Starting with the SAM ligand previously removed from the crystal structure of the riboswitch, the GaussView program (76) was used to add hydrogens to the ligand and the structure was quantum mechanically optimized using the Gaussian program at the HF/6-31-g(d) level of theory. Force field parameters were obtained using the Antechamber module in Amber, and partial charges were generated with the RESP (77) method from the electrostatic potential calculated using the Gaussian program, also at the HF/6-31g(d) level of theory. These force field parameters and charges were loaded into LEAP along with the default nucleic acid parameters in order to prepare the SAM-II riboswitch complex. The complex was also solvated in a truncated octahedron TIP3P water box consisting of 11 254 water molecules. The positive charge of the ligand resulted in the addition of only 49 Na+ counter ions to the system in order to bring it to neutrality. Equilibration and dynamics were carried out from this point on under the same conditions and for the same length of time (200 ns) as for the free riboswitch.
Cross-correlation coefficients
The residue–residue cross-correlation coefficients were calculated using the trajectories of the entire simulation of the unbound form of the SAM-II riboswitch. The cross-correlation coefficient 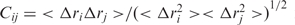 for the displacement of any two atoms i and j, where Δri is the displacement from the average position of i. The cross-correlation coefficients, Cij, is therefore a 2D matrix with the value of each coefficient between +1.0 and −1.0, where +1.0 signifies perfect positive correlated motions, and −1.0 signifies perfect negative correlated motions. The cross-correlation coefficients were averaged over each residue, such that there was only one Cij per residue. The cross-correlation coefficients are then plotted using a color-coded 2D matrix from red (+1) to blue (−1).
for the displacement of any two atoms i and j, where Δri is the displacement from the average position of i. The cross-correlation coefficients, Cij, is therefore a 2D matrix with the value of each coefficient between +1.0 and −1.0, where +1.0 signifies perfect positive correlated motions, and −1.0 signifies perfect negative correlated motions. The cross-correlation coefficients were averaged over each residue, such that there was only one Cij per residue. The cross-correlation coefficients are then plotted using a color-coded 2D matrix from red (+1) to blue (−1).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by Georgia State's; IBM System p5 supercomputer, acquired through a partnership of the Southeastern Universities Research Association and IBM supporting the SURAgrid initiative. D.H. is a Georgia Cancer Coalition (GCC) scholar. This work was supported in part by Research Initiation Grant from Georgia State University, the Department of Chemistry at Georgia State University, and the Georgia Cancer Coalition. Funding for open access charge: Department of Chemistry, Georgia State University.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Drs T. Shen and W.D. Wilson for helpful discussions during the preparation of this manuscript. They would also like to thank Dr Urmi Doshi for reading the manuscript and providing helpful suggestions.
REFERENCES
- 1.Lewis R. The rise of antibiotic-resistant infections. FDA Consum. 1995;29:11–15. [PubMed] [Google Scholar]
- 2.Barrick JE, Breaker RR. The power of riboswitches. Sci. Am. 2007;296:50–57. doi: 10.1038/scientificamerican0107-50. [DOI] [PubMed] [Google Scholar]
- 3.Winkler WC, Breaker RR. Genetic control by metabolite-binding riboswitches. Chembiochem. 2003;4:1024–1032. doi: 10.1002/cbic.200300685. [DOI] [PubMed] [Google Scholar]
- 4.Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 5.Blount KF, Breaker RR. Riboswitches as antibacterial drug targets. Nat. Biotechnol. 2006;24:1558–1564. doi: 10.1038/nbt1268. [DOI] [PubMed] [Google Scholar]
- 6.Nudler E, Mironov AS. The riboswitch control of bacterial metabolism. Trends Biochem. Sci. 2004;29:11–17. doi: 10.1016/j.tibs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Soukup JK, Soukup GA. Riboswitches exert genetic control through metabolite-induced conformational change. Curr. Opin. Struct.Biol. 2004;14:344–349. doi: 10.1016/j.sbi.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Thore S, Leibundgut M, Ban N. Structure of the eukaryotic thiamine pyrophosphate riboswitch with its regulatory ligand. Science. 2006;312:1208–1211. doi: 10.1126/science.1128451. [DOI] [PubMed] [Google Scholar]
- 9.Coppins RL, Hall KB, Groisman EA. The intricate world of riboswitches. Curr. Opin. Microbiol. 2007;10:176–181. doi: 10.1016/j.mib.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilbert SD, Rambo RP, Van Tyne D, Batey RT. Structure of the SAM-II riboswitch bound to S-adenosylmethionine. Nat. Struct. Mol. Biol. 2008;15:177–182. doi: 10.1038/nsmb.1371. [DOI] [PubMed] [Google Scholar]
- 11.Serganov A. The long and the short of riboswitches. Curr. Opin. Struct. Biol. 2009;19:251–259. doi: 10.1016/j.sbi.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montange RK, Batey RT. Riboswitches: emerging themes in RNA structure and function. Annu. Rev. Biophys. 2008;37:117–133. doi: 10.1146/annurev.biophys.37.032807.130000. [DOI] [PubMed] [Google Scholar]
- 13.Roth A, Breaker RR. The structural and functional diversity of metabolite-binding riboswitches. Annu. Rev. Biochem. 2009;78:305–334. doi: 10.1146/annurev.biochem.78.070507.135656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–279. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 15.Grundy FJ, Lehman SC, Henkin TM. The L box regulon: lysine sensing by leader RNAs of bacterial lysine biosynthesis genes. Proc. Natl Acad. Sci. USA. 2003;100:12057–12062. doi: 10.1073/pnas.2133705100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Regulation of lysine biosynthesis and transport genes in bacteria: yet another RNA riboswitch? Nucleic Acids Res. 2003;31:6748–6757. doi: 10.1093/nar/gkg900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sudarsan N, Wickiser JK, Nakamura S, Ebert MS, Breaker RR. An mRNA structure in bacteria that controls gene expression by binding lysine. Genes Dev. 2003;17:2688–2697. doi: 10.1101/gad.1140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johansen LE, Nygaard P, Lassen C, Agerso Y, Saxild HH. Definition of a second Bacillus subtilis pur regulon comprising the pur and xpt-pbuX operons plus pbuG, nupG (yxjA), and pbuE (ydhL) J. Bacteriol. 2003;185:5200–5209. doi: 10.1128/JB.185.17.5200-5209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandal M, Breaker RR. Adenine riboswitches and gene activation by disruption of a transcription terminator. Nat. Struct. Mol. Biol. 2004;11:29–35. doi: 10.1038/nsmb710. [DOI] [PubMed] [Google Scholar]
- 20.Batey RT, Gilbert SD, Montange RK. Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature. 2004;432:411–415. doi: 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- 21.Serganov A, Yuan YR, Pikovskaya O, Polonskaia A, Malinina L, Phan AT, Hobartner C, Micura R, Breaker RR, Patel DJ. Structural basis for discriminative regulation of gene expression by adenine- and guanine-sensing mRNAs. Chem. Biol. 2004;11:1729–1741. doi: 10.1016/j.chembiol.2004.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roth A, Winkler WC, Regulski EE, Lee BW, Lim J, Jona I, Barrick JE, Ritwik A, Kim JN, Welz R, et al. A riboswitch selective for the queuosine precursor preQ1 contains an unusually small aptamer domain. Nat. Struct. Mol. Biol. 2007;14:308–317. doi: 10.1038/nsmb1224. [DOI] [PubMed] [Google Scholar]
- 23.Klein DJ, Edwards TE, Ferre-D'A;mare AR. Cocrystal structure of a class I preQ1 riboswitch reveals a pseudoknot recognizing an essential hypermodified nucleobase. Nat. Struct. Mol. Biol. 2009;16:343–344. doi: 10.1038/nsmb.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mironov AS, Gusarov I, Rafikov R, Lopez LE, Shatalin K, Kreneva RA, Perumov DA, Nudler E. Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell. 2002;111:747–756. doi: 10.1016/s0092-8674(02)01134-0. [DOI] [PubMed] [Google Scholar]
- 25.Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J. Biol. Chem. 2002;277:48949–48959. doi: 10.1074/jbc.M208965200. [DOI] [PubMed] [Google Scholar]
- 26.Serganov A, Polonskaia A, Phan AT, Breaker RR, Patel DJ. Structural basis for gene regulation by a thiamine pyrophosphate-sensing riboswitch. Nature. 2006;441:1167–1171. doi: 10.1038/nature04740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards TE, Ferre-D'A;mare AR. Crystal structures of the thi-box riboswitch bound to thiamine pyrophosphate analogs reveal adaptive RNA-small molecule recognition. Structure. 2006;14:1459–1468. doi: 10.1016/j.str.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Serganov A, Huang L, Patel DJ. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature. 2009;458:233–237. doi: 10.1038/nature07642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winkler WC, Cohen-Chalamish S, Breaker RR. An mRNA structure that controls gene expression by binding FMN. Proc. Natl Acad. Sci. USA. 2002;99:15908–15913. doi: 10.1073/pnas.212628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borovok I, Gorovitz B, Schreiber R, Aharonowitz Y, Cohen G. Coenzyme B12 controls transcription of the Streptomyces class Ia ribonucleotide reductase nrdABS operon via a riboswitch mechanism. J. Bacteriol. 2006;188:2512–2520. doi: 10.1128/JB.188.7.2512-2520.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warner DF, Savvi S, Mizrahi V, Dawes SS. A riboswitch regulates expression of the coenzyme B12-independent methionine synthase in Mycobacterium tuberculosis: implications for differential methionine synthase function in strains H37Rv and CDC1551. J. Bacteriol. 2007;189:3655–3659. doi: 10.1128/JB.00040-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jansen JA, McCarthy TJ, Soukup GA, Soukup JK. Backbone and nucleobase contacts to glucosamine-6-phosphate in the glmS ribozyme. Nat. Struct. Mol. Biol. 2006;13:517–523. doi: 10.1038/nsmb1094. [DOI] [PubMed] [Google Scholar]
- 33.Klein DJ, Ferre-D'A;mare AR. Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science. 2006;313:1752–1756. doi: 10.1126/science.1129666. [DOI] [PubMed] [Google Scholar]
- 34.Winkler WC, Nahvi A, Roth A, Collins JA, Breaker RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature. 2004;428:281–286. doi: 10.1038/nature02362. [DOI] [PubMed] [Google Scholar]
- 35.Klein DJ, Been MD, Ferre-D'A;mare AR. Essential role of an active-site guanine in glmS ribozyme catalysis. J. Am. Chem. Soc. 2007;129:14858–14859. doi: 10.1021/ja0768441. [DOI] [PubMed] [Google Scholar]
- 36.Grundy FJ, Henkin TM. The S box regulon: a new global transcription termination control system for methionine and cysteine biosynthesis genes in gram-positive bacteria. Mol. Microbiol. 1998;30:737–749. doi: 10.1046/j.1365-2958.1998.01105.x. [DOI] [PubMed] [Google Scholar]
- 37.Epshtein V, Mironov AS, Nudler E. The riboswitch-mediated control of sulfur metabolism in bacteria. Proc. Natl Acad. Sci. USA. 2003;100:5052–5056. doi: 10.1073/pnas.0531307100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winkler WC, Nahvi A, Sudarsan N, Barrick JE, Breaker RR. An mRNA structure that controls gene expression by binding S-adenosylmethionine. Nat. Struct. Biol. 2003;10:701–707. doi: 10.1038/nsb967. [DOI] [PubMed] [Google Scholar]
- 39.Corbino KA, Barrick JE, Lim J, Welz R, Tucker BJ, Puskarz I, Mandal M, Rudnick ND, Breaker RR. Evidence for a second class of S-adenosylmethionine riboswitches and other regulatory RNA motifs in alpha-proteobacteria. Genome Biol. 2005;6:R70. doi: 10.1186/gb-2005-6-8-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim J, Winkler WC, Nakamura S, Scott V, Breaker RR. Molecular-recognition characteristics of SAM-binding riboswitches. Angew. Chem. Int. Ed. Engl. 2006;45:964–968. doi: 10.1002/anie.200503198. [DOI] [PubMed] [Google Scholar]
- 41.Fuchs RT, Grundy FJ, Henkin TM. The S(MK) box is a new SAM-binding RNA for translational regulation of SAM synthetase. Nat. Struct. Mol. Biol. 2006;13:226–233. doi: 10.1038/nsmb1059. [DOI] [PubMed] [Google Scholar]
- 42.Weinberg Z, Regulski EE, Hammond MC, Barrick JE, Yao Z, Ruzzo WL, Breaker RR. The aptamer core of SAM-IV riboswitches mimics the ligand-binding site of SAM-I riboswitches. RNA. 2008;14:822–828. doi: 10.1261/rna.988608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang JX, Breaker RR. Riboswitches that sense S-adenosylmethionine and S-adenosylhomocysteine. Biochem. Cell Biol. 2008;86:157–168. doi: 10.1139/O08-008. [DOI] [PubMed] [Google Scholar]
- 44.Meyer MM, Ames TD, Smith DP, Weinberg Z, Schwalbach MS, Giovannoni SJ, Breaker RR. Identification of candidate structured RNAs in the marine organism ‘Candidatus Pelagibacter ubique'. BMC Genomics. 2009;10:268. doi: 10.1186/1471-2164-10-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hilbers CW, Michiels PJ, Heus HA. New developments in structure determination of pseudoknots. Biopolymers. 1998;48:137–153. doi: 10.1002/(SICI)1097-0282(1998)48:2<137::AID-BIP4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 46.Garst AD, Heroux A, Rambo RP, Batey RT. Crystal structure of the lysine riboswitch regulatory mRNA element. J. Biol. Chem. 2008;283:22347–22351. doi: 10.1074/jbc.C800120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serganov A, Huang L, Patel DJ. Structural insights into amino acid binding and gene control by a lysine riboswitch. Nature. 2008;455:1263–1267. doi: 10.1038/nature07326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang M, Peterson R, Feigon J. Structural Insights into riboswitch control of the biosynthesis of queuosine, a modified nucleotide found in the anticodon of tRNA. Mol. Cell. 2009;33:784–790. doi: 10.1016/j.molcel.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 49.Noeske J, Buck J, Furtig B, Nasiri HR, Schwalbe H, Wohnert J. Interplay of ‘induced fit’ and preorganization in the ligand induced folding of the aptamer domain of the guanine binding riboswitch. Nucleic Acids Res. 2007;35:572–583. doi: 10.1093/nar/gkl1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noeske J, Richter C, Grundl MA, Nasiri HR, Schwalbe H, Wohnert J. An intermolecular base triple as the basis of ligand specificity and affinity in the guanine- and adenine-sensing riboswitch RNAs. Proc. Natl Acad. Sci. USA. 2005;102:1372–1377. doi: 10.1073/pnas.0406347102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buck J, Furtig B, Noeske J, Wohnert J, Schwalbe H. Time-resolved NMR spectroscopy: ligand-induced refolding of riboswitches. Methods Mol. Biol. 2009;540:161–171. doi: 10.1007/978-1-59745-558-9_12. [DOI] [PubMed] [Google Scholar]
- 52.Ottink OM, Rampersad SM, Tessari M, Zaman GJ, Heus HA, Wijmenga SS. Ligand-induced folding of the guanine-sensing riboswitch is controlled by a combined predetermined induced fit mechanism. RNA. 2007;13:2202–2212. doi: 10.1261/rna.635307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheatham TE., 3rd Simulation and modeling of nucleic acid structure, dynamics and interactions. Curr. Opin. Struct. Biol. 2004;14:360–367. doi: 10.1016/j.sbi.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 54.Adcock SA, McCammon JA. Molecular dynamics: survey of methods for simulating the activity of proteins. Chem. Rev. 2006;106:1589–1615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mackerell AD, Jr, Nilsson L. Molecular dynamics simulations of nucleic acid-protein complexes. Curr. Opin. Struct. Biol. 2008;18:194–199. doi: 10.1016/j.sbi.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang W, Kim J, Jha S, Aboul-Ela F. A mechanism for S-adenosyl methionine assisted formation of a riboswitch conformation: a small molecule with a strong arm. Nucleic Acids Res. 2009;37:6528–6539. doi: 10.1093/nar/gkp664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharma M, Bulusu G, Mitra A. MD simulations of ligand-bound and ligand-free aptamer: molecular level insights into the binding and switching mechanism of the add A-riboswitch. RNA. 2009;15:1673–1692. doi: 10.1261/rna.1675809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Villa A, Wohnert J, Stock G. Molecular dynamics simulation study of the binding of purine bases to the aptamer domain of the guanine sensing riboswitch. Nucleic Acids Res. 2009;37:4774–4786. doi: 10.1093/nar/gkp486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Whitford PC, Schug A, Saunders J, Hennelly SP, Onuchic JN, Sanbonmatsu KY. Nonlocal helix formation is key to understanding S-adenosylmethionine-1 riboswitch function. Biophys. J. 2009;96:L7–L9. doi: 10.1016/j.bpj.2008.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wickiser JK, Cheah MT, Breaker RR, Crothers DM. The kinetics of ligand binding by an adenine-sensing riboswitch. Biochemistry. 2005;44:13404–13414. doi: 10.1021/bi051008u. [DOI] [PubMed] [Google Scholar]
- 61.Wickiser JK, Winkler WC, Breaker RR, Crothers DM. The speed of RNA transcription and metabolite binding kinetics operate an FMN riboswitch. Mol. Cell. 2005;18:49–60. doi: 10.1016/j.molcel.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 62.Bosshard HR. Molecular recognition by induced fit: how fit is the concept? News Physiol. Sci. 2001;16:171–173. doi: 10.1152/physiologyonline.2001.16.4.171. [DOI] [PubMed] [Google Scholar]
- 63.Berger C, Weber-Bornhauser S, Eggenberger J, Hanes J, Pluckthun A, Bosshard HR. Antigen recognition by conformational selection. FEBS Lett. 1999;450:149–153. doi: 10.1016/s0014-5793(99)00458-5. [DOI] [PubMed] [Google Scholar]
- 64.Monod J, Wyman J, Changeux JP. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 65.Koshland DE. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl Acad. Sci. USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marzi S, Myasnikov AG, Serganov A, Ehresmann C, Romby P, Yusupov M, Klaholz BP. Structured mRNAs regulate translation initiation by binding to the platform of the ribosome. Cell. 2007;130:1019–1031. doi: 10.1016/j.cell.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 67.Sudarsan N, Barrick JE, Breaker RR. Metabolite-binding RNA domains are present in the genes of eukaryotes. RNA. 2003;9:644–647. doi: 10.1261/rna.5090103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wachter A, Tunc-Ozdemir M, Grove BC, Green PJ, Shintani DK, Breaker RR. Riboswitch control of gene expression in plants by splicing and alternative 3′ end processing of mRNAs. Plant Cell. 2007;19:3437–3450. doi: 10.1105/tpc.107.053645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Selinger DW, Saxena RM, Cheung KJ, Church GM, Rosenow C. Global RNA half-life analysis in Escherichia coli reveals positional patterns of transcript degradation. Genome Res. 2003;13:216–223. doi: 10.1101/gr.912603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perez A, Marchan I, Svozil D, Sponer J, Cheatham TE, 3rd, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A 2nd generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 73.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 74.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 75.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-integration of cartesian equations of motion of a system with constraints—molecular-dynamics of N-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 76.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, et al. Wallingford CT: Gaussian, Inc.; 2004. in Gaussian 03 Revision E.01 ed. [Google Scholar]
- 77.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charge—The RESP model. J. Phys. Chem. 1993;97:10269–10280. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.