

Abstract
Septins belong to the GTPase superclass of conserved proteins and have been identified to play a role in diverse aspects of cell biology, from cytokinesis to the maintenance of cellular morphology. At least 14 septins have been identified in humans. With their complex patterns in gene expressions and interaction, it has been reported that alterations in septin expression are observed in human diseases. Although much is not known about the role of human septins in oral carcinogenesis, circumstantial evidence does indicate that it may play a major role. This review intends to summarize the basis of septin biology, with the focus being on the evidence for septin involvement in human oral cancer.
Keywords: Cancer, oral, septin
INTRODUCTION
The final stage in mitosis is the physical separation of two daughter cells, referred to as cytokinesis. During cytokinesis, a contractile ring consisting of actin, myosin, and other proteins is formed. This drives the constriction of the plasma membrane, resulting in two daughter cells connected by a cytoplasmic bridge. This intracellular bridge contains the mid-body, which gets resolved during the final stage of abscission. Failure of complete cytokinesis has been known to promote tumorigenesis by leading to tetraploidy and ensuing chromosomal instability.[1]
Septins were first identified as essential genes for yeast cell division in the 1970s. Yeast cells divide asymmetrically producing daughter cells from the surface of the mother cell in the form of buds. The bud neck controls the exchange of material between mother and daughter cells by means of a diffusion barrier that is controlled by septin genes. The diffusion barrier restricts certain products of aging, such as the extrachromosomal ribosomal DNA circles (ERCs–circular episomes formed by recombination in the rDNA locus and containing an autonomous replicating sequence, accumulate as they progress through successive rounds of DNA replication) to the mother cell so that even as the mother yeast cell ages, the bud is born with a full lifespan potential.[2,3] Mutations in yeast septin genes lead to incomplete cytokinesis[1] and hence probably promote tumorigenesis.
Since its initial identification and description in yeast, a number of septin genes have also been identified in humans. These genes are widely distributed in the genome and show considerable sequence conservation; certain aspects of genomic architecture and gene control are remarkably similar in humans and yeasts.[3] In humans, septin genes encode a family of GTPase-binding proteins with a common, conserved central core domain, a P loop-based GTP-binding domain in the N-terminal half, bordered by a polybasic domain and more often a coiled-coil region. They play a major role in cytokinesis, vesicle trafficking, polarity determination, membrane diffusion, barrier formation, as well as in microtubule and actin dynamics. Septins have the ability to form hetero-oligomeric complexes and have been noted to function as dynamic protein scaffolds. It is observed that there are complex patterns and interactions of human septin genes, and alterations in their expression are seen in human neoplastic processes.[4]
This article reviews the human septin genes, mutations of these genes, and its potential role in initiation of oral carcinogenesis.
FUNCTIONS OF SEPTINS
Some of the known major functions of human septins have been outlined in Table 1. Recent insights on septin interaction with the microtubular cytoskeleton have added to our existing understanding of their role in cytokinesis. By influencing microtubule dynamics, forming protein scaffolds, and through actin-based cytoskeletal interactions, septins may possibly act as modulators of multiple cellular events, such as cytokinesis, spindle assembly, chromosome congression, anaphase chromosome movement, chromosome attachment to spindle microtubules, nuclear orientation, vesicle trafficking along microtubules, and cell abscission. Septins play a role in positioning actin and other cytoskeletal structures relative to each other in order to facilitate cell division.[5]
Table 1.
Functions of septins and cancers involved in humans

CARCINOGENESIS AND SEPTINS
It is logical to assume that the major role played by septins in cytokinesis, membrane dynamics,[3] apoptosis,[4] association with actin and tubulin,[1,4] epithelial cell phenotype, and cell polarity, as well as their association with the Rho signaling pathway,[3] will reflect fairly on the processes that may lead to neoplasia. Septins also play a vital role in context-specific spatial cues that organize the spatial arrangement of other proteins.[3] Several hallmark observations, including those of alteration in apoptosis (isoform of SEPT4),[6] phenotypic changes of epithelial cell (mutants of SEPT9),[7] and interaction with microtubules, have been made.[5] The first example of septin–carcinogenesis association was with SEPT9 interacting with the MLL gene in leukemias.[2] Since then, several cancers, including those of ovary and breast and gliomas, have been associated with various septins.[5]
ORAL CARCINOGENESIS
Recent discoveries have exponentially increased our understanding of the neoplastic physiology and the biochemical and molecular mechanisms that control the sequential events and biological processes that lead to head and neck squamous cell cancers (HNSCC). It is known that neoplastic cells are derived from the clonal expansion and aberrant growth of a single stem cell or few cells that have acquired self-renewal capacity owing to mutation(s).[8] During oral oncogenesis, numerous mutations and dysregulation of molecular networks occur.[8] One among the numerous such dysregulations is the alteration of septin genes and their products.[7] As genes of the septin family control a lot of vital pathways that are dysregulated, it can be safely hypothesized that septin might have hitherto undescribed mechanisms by which carcinogenesis is influenced. Though little evidence regarding the role of septin in HNSCC can be found in literature, there is plenty of circumstantial evidence on the role of various septins. Table 2 lists a few mutations reported in HNSCC that share their loci with genes for septins.
Table 2.
Genetic abnormalities in head and neck squamous cell carcinoma reported from loci of septins
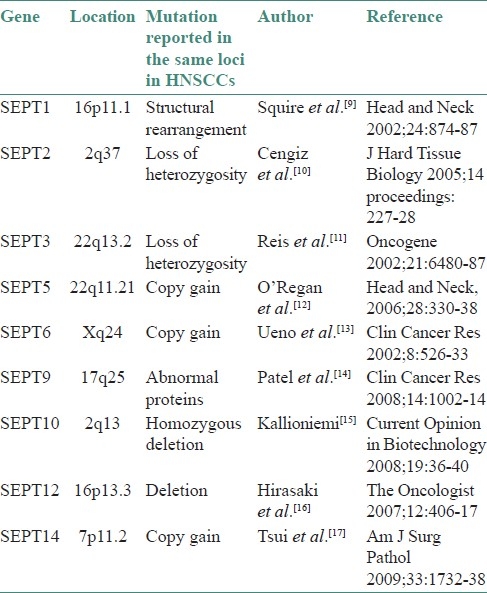
The following discussion focuses on individual septins and their probable role/influence on oral carcinogenesis.
Septin 1
The septin 1 locus was observed to be involved in HNSC carcinogenesis. A study was done to determine whether genes located in specific loci are subject to alterations in gene expression. Spectral karyotyping (to visualize the numerical and structural chromosomal changes in metaphase preparations) identified the involvement of 16p11.1–q11.1 region in HNSC carcinogenesis.[9] This loci codes for septin 1, which functions as a filament-forming cytoskeletal GTPase with a possible role in cytokinesis. Furthermore, the normal protein SEPT1 is known to localize to spindle poles of HeLa cells throughout mitosis and to the midbody during telophase.[5] An abnormality in this protein could play a role in the development of HNSCC.
Septin 2
Chromosomal region 2q22-37.3 is highly populated with several candidate tumor suppressor genes, including ING5, CASP8, CASP10, PPP1R7, and BOK.[10] The septin 2 gene (SEPT2) is also present within this region at position 2q37. This gene codes for a filament-forming cytoskeletal GTPase, which is required for normal organization of the actin cytoskeleton. It also plays a role in the biogenesis of polarized columnar-shaped epithelium by maintaining polyglutamylated microtubules, facilitating efficient vesicle transport, and by impeding MAP4 binding to tubulin. During mitosis it forms a scaffold at the mid-plane of the mitotic spindle, which is required to maintain CENPE (centromere protein E) localization at the kinetochores and consequently achieve chromosome congression. During anaphase, this protein may be required for chromosome segregation and spindle elongation.[5] Considering the vital role of this protein in orchestrating cell division, it can be assumed that this gene could be involved or mutated during HNSC tumorigenesis as the region has been proved to harbor numerous loss of heterozygosity as well as deletions.
Septin 3
The DIA1 gene, located in 22q13.2-13.31, and the microsatellite marker D22S274 are both mapped to 22q13.31 and are spaced 2.5 cM apart. The concomitant loss of both sequences in several HNSCC cases suggests the involvement of a large region of deletion associated with prognostic factors in tongue and floor-of-the-mouth carcinomas. This region contains CpG islands and poly-A sequences, indicating that this region may contain a variety of genes.[11] The septin 3 gene (SEPT3) has been localized here.[5]
SEPT3 localizes predominantly to presynaptic terminals, colocalizing with synaptophysin and dynamin I. SEPT3 is specifically enriched in synaptosomes and in peripheral membrane extract and is not found in soluble form or membrane extracts, suggesting that SEPT3 is involved in synaptic vesicle recycling. It has been also associated with dynamic microtubule-based vesicle transport.[5] Microtubule assembly is an integral part of the cell and is dysregulated in oncogenesis.[8] Hence, it is possible that this gene product, when mutated, could have a role in HNSCC oncogenesis.
Septin 5
The septin 5 gene (SEPT5) codes for a filament-forming cytoskeletal GTPase with a possible role in cytokinesis and platelet secretion.[5] The protein has been mapped to locus 22q11.21. The protein TBX1 encoded at 22q11.2 has been shown to be highly mutated in HNSCC in young individuals.[12] It is likely that SEPT5 could also mutated in the process of oral carcinogenesis.
Septin 6
Septin 6 is a filament-forming cytoskeletal GTPase required for normal organization of the actin cytoskeleton and cytokinesis. Its gene (SEPT6) has been mapped to the X chromosome. A study on oropharyngeal carcinoma established the link between mutation of this particular loci and HNSCC, especially esophageal carcinomas.[13] Circumstantial evidence points to involvement of SEPT6 in HNSCC, but this association warrants further investigation.
Septin 9
The first confirmed instance of septin–carcinogenesis association was seen with SEPT9 in leukemias.[2] A proteomic analysis of HNSCC using laser capture microdissection on a novel proteomic platform found an upregulation of several proteins involved in cell cycle progression, particularly those in G2-M transition and mitosis. Septin 9 was detected only in tumor samples and not in normal tissue, reflecting their active role in proliferation. Methylation of the promoter region of a gene generally results in silencing of the locus. The silencing is achieved by condensing the chromatin that, in turn, limits the transcription machinery's access to the locus.[18]
SEPT9 interacts with HIF-1a to prevent its ubiquitination and degradation, which can allow TGF-β feedback activation.[18] SEPT9 is normally associated with microtubules in interphase cell and is localized to the mitotic spindle during mitosis. An isoform, SEPT9_v1 has been shown to be indicative of poor prognosis in HNSCC.[8,18] The high degree of expression and association of septin 9 in HNSCC tumor tissues, coupled with the known functions of SEPT9, indicate the high degree of probability of SEPT9 being involved in HNSC carcinogenesis.
Septin 10
Homozygous deletion of 2q13 has been associated with carcinogenesis and lymphomas.[15] The region codes for the pro-apoptotic BIM gene as well as the septin 10 gene (SEPT10). This regional association between apoptotic genes and SEPT10 requires further investigations.
Septin 12
The exact function of the septin 12 gene (SEPT12) and its products has not been elucidated. SEPT12 is located on 16p13.3. Recently, in esophageal carcinoma, a novel loss (indicating homozygous deletion) was recognized at 16p13.3, which contains several genes, including ZNF434, ZNF174, ZNF597, FLJ14154, LOC390671, and CLUAP1. As the loci is frequently mutated, there exists a high degree of probability that SEPT12 could be involved in HNSCC.[16]
Septin 14
The septin 14 gene (SEPT14) shares its locus with CHRNB1, a microsatellite marker. This region has been demonstrated to be involved in tongue carcinoma.[17] Further studies are necessary to elucidate the involvement of SEPT14 in head and neck carcinogenesis.
MALIGNANT TRANSFORMATION OF LEUKOPLAKIA
A pioneer study in oral systems biology, using functional proteomics, was done to elucidate the mechanistic aspects and potential involvement of proteins in premalignant to malignant conversions. The conversion to malignancy from potentially malignant states involves a huge number of genes and complex interactions. In the study, leukoplakia tissues exhibited considerable levels of septin 2 expression along with other differentially expressed proteins involved in malignant transformation. Dysregulated protein genes in leukoplakia and oral SCC that were involved in common pathways in the malignant transformation process were analyzed using the new software, Pathway Studio®. Proteins with their SWISS-PROT numbers were loaded into the software and analyzed using Pathway Studio® with the ResNet® 2.5 database. About 85 protein genes with a high confidence index of interactions were included in the pathway layout graph. Septin 2 was one of the top 85 proteins that had this high confidence index, indicating that septin has a major role to play in the malignant transformation of oral leukoplakia. Functional genome (Gene) ontology analysis has also proved the role of such proteins in malignant transformation.[19]
ASSOCIATED PROTEINS AND LOCATION OF SEPTIN
Immunohistological data indicates that the septin-binding protein anillin is widely expressed in diverse human tissues, including nonproliferative (e.g., neurons, terminally differentiated epithelial cells, and quiescent stromal cells) and proliferative cellular compartments. The presence of anillin mRNA in large quantities and subsequent protein expression is not a mere marker of proliferation but has further unexplored and unexplained functions. The correlation of anillin and Ki67 mRNA expression in neoplastic tissue indicates that cell cycle–dependent factors govern anillin expression. However, anillin is also expressed in nondividing cells in many tissues. Alternatively, in neoplasia, amplification of the anillin locus is a possibility. Other proteins associated with cytokinesis, including kinetochore proteins (CENP-F, CENP-A, and Nek2 kinase), aurora kinases, citron kinase, polo-like kinases, myosin, and myosin II, are also altered in neoplasia.[20]
Mammalian septins have been identified in the cell cortex, contractile ring, and midbody of mitotic cells (SEPT2, SEPT4, SEPT6, SEPT7, and SEPT9) and in the cell cortex, actin stress fibers (SEPT2, SEPT4, SEPT6, SEPT7, and SEPT9), and microtubules (SEPT9) of interphase cells. In the mammalian nervous system, septins are observed on the cytoplasmic side of presynaptic membranes (SEPT7), synaptic vesicles (SEPT5 and SEPT6), and in the end terminal of astroglia (SEPT4 and SEPT7). Cytokinesis is perturbed by microinjection of anti-septin antibodies (against SEPT2 and SEPT9) or transfection of siRNAs (against SEPT2, SEPT7, and SEPT9).[7] Depletion of SEPT2 or SEPT7 protein by RNA interference also causes disorganization of actin stress fibers, leading to flat cell morphology in interphase cells. Although SEPT5 is highly expressed in mature nervous systems, no brain abnormality is seen in SEPT5-null mice, probably because of compensation by redundant septin species.[21]
FUTURE DIRECTIONS IN SEPTIN-ORAL CARCINOGENESIS
HNSCCs are potential candidates for studies on septin interactions owing to their relatively large size and variations in the degree of dysplasia within these tumors, which can be used to demonstrate different septin expression patterns.
Originally identified as a key player in cytokinesis, it is clear that even in yeast septins have multiple functions. The complex distribution of mammalian septins could be associated with an increased range of cellular functions. Data from multiple sources indicate that human septins can interact with other septins as well as with components of the cytoskeleton such as actin and tubulin. In addition, several nondividing cells such as neurons exhibit upregulation of several of these proteins. Present evidence indicates that septin is probably a late player in carcinogenesis and is associated with poor clinical outcomes. However, this entity does not, by itself, form a clone of neoplastic cells. With its ability to interact with actin–myosin microtubular assembly and communication, vesicle mediate transportation, and apoptosis and given its key role in cytokinesis, septin could progressively alter the neoplastic clone, and this alteration could be responsible for poor clinical outcomes in neoplasia.
A large proportion of publications on septin-related diseases are studies in which alterations in septin expression have been documented by proteomic, immunochemical, or gene expression analyses. The data emanating is correlative in nature and in only very few situations are there sufficient data to suggest that septins have a pathogenetic role in disease. Such correlation does not indicate a direct causation of Neoplasia but it reflects that septins play a vital septins play a vital role in the prognosis of the disease.
In the years to come, septin biology in neoplasia will require new perspectives and approaches with regard to protein levels, stoichiometry and the nature and distribution of protein complexes, and will ultimately require a much higher-resolution analysis, utilizing new reagents and new molecular biological approaches. Currently, the role of septins in neoplastic processes warrant further investigations, especially in cases with poor clinical outcomes, to underline the pathways and mechanism via which septins interact with the neoplastic clone. As different molecular techniques have different sensitivities and specificities, accurate estimation of these proteins has to remain the first priority. The stoichiometry and temporal spatial expression of these proteins need to be investigated along with their interaction with key neoplastic processes. Soon, biotechnological manipulation of septin complexes in tumors may provide new insights regarding therapeutic modalities, suggesting that there are grounds for optimism about the development of a cure for cancer.
ACKNOWLEDGEMENT
The authors would like to thank Dr. S. Ramachandran, Principal, Ragas Dental College and Prof. Dr. A. Kanagaraj, Chairman of Jaya Group of Institution, Chennai for their constant support and encouragement.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.Sagona AP, Stenmark H. Cytokinesis and cancer. FEBS Lett. 2010;584:2652–61. doi: 10.1016/j.febslet.2010.03.044. [DOI] [PubMed] [Google Scholar]
- 2.Russell SH, Hall PA. Do septins have a role in cancer? Br J Cancer. 2005;93:499–503. doi: 10.1038/sj.bjc.6602753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caudron F, Barral Y. Septins and the lateral compartmentalization of eukaryotic membranes. Dev Cell. 2009;16:493–506. doi: 10.1016/j.devcel.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Barral Y. The gates of immortality. Scientist. 2010;24:39–43. [Google Scholar]
- 5.Silvermann-Gavrila RV, Silvermann-Gavrila LB. Septins: New microtubule interacting partners. Scientific World Journal. 2008;8:611–20. doi: 10.1100/tsw.2008.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J. 2004;23:1627–35. doi: 10.1038/sj.emboj.7600155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stanbery L, D’Silva NJ, Lee JS, Bradford CR, Carey TE, Prince ME, et al. High SEPT9_v1 Expression is Associated with Poor Clinical Outcomes in Head and Neck Squamous cell Carcinoma. Transl Oncol. 2010;3:239–45. doi: 10.1593/tlo.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45:324–34. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Squire JA, Bayani J, Luk C, Unwin L, Tokunaga J, MacMillan C, et al. Molecular cytogenetic analysis of head and neck squamous cell carcinoma: By comparative genomic hybridization, spectral karyotyping, and expression array analysis. Head Neck. 2002;24:874–87. doi: 10.1002/hed.10122. [DOI] [PubMed] [Google Scholar]
- 10.Cengiz B, Gunduz M, Gunduz E, Ouchida M, Shimizu K, Inoue T, et al. Deletion Mapping of 2q21-37 region of oral cancer. J Hard Tissue Biol. 2005;14:227. [Google Scholar]
- 11.Reis PP, Rogatto SR, Kowalski LP, Nishimoto IN, Montovani JC, Corpus G, et al. Quantitative real-time PCR identifies a critical region of deletion on 22q13 related to prognosis in oral cancer. Oncogene. 2002;21:6480–7. doi: 10.1038/sj.onc.1205864. [DOI] [PubMed] [Google Scholar]
- 12.O’Regan EM, Toner ME, Smyth PC, Finn SP, Timon C, Cahill S, et al. Distinct array Comparative genomic hybridization profiles in oral squamous cell carcinoma occurring in young patients. Head Neck. 2006;28:330–8. doi: 10.1002/hed.20354. [DOI] [PubMed] [Google Scholar]
- 13.Ueno T, Tangoku A, Yoshino S, Abe T, Toshimitsu H, Furuya T, et al. Gain of 5p15 detected by comparative genomic hybridization as an independent marker of poor prognosis in patients with esophageal squamous cell carcinoma. Clin Cancer Res. 2002;8:526–33. [PubMed] [Google Scholar]
- 14.Patel V, Hood BL, Molinolo AA, Lee NH, Conrads TP, Braisted JC, et al. Proteomic analysis of laser-captured paraffin-embedded tissues: A molecular portrait of head and neck cancer progression. Clin Cancer Res. 2008;14:1002–14. doi: 10.1158/1078-0432.CCR-07-1497. [DOI] [PubMed] [Google Scholar]
- 15.Kallioniemi A. CGH microarrays and cancer. Curr Opin Biotechnol. 2008;19:36–40. doi: 10.1016/j.copbio.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Hirasaki S, Noguchi T, Mimori K, Onuki J, Morita K, Inoue H, et al. BAC clones related to prognosis in patients with esophageal squamous carcinoma: An array comparative genomic hybridization study. Oncologist. 2007;12:406–17. doi: 10.1634/theoncologist.12-4-406. [DOI] [PubMed] [Google Scholar]
- 17.Tsui IF, Garnis C, Poh CF. A dynamic oral cancer field – unraveling the underlying biology and its clinical implication. Am J Surg Pathol. 2009;33:1732–8. doi: 10.1097/PAS.0b013e3181b669c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett KL, Karpenko M, Lin MT, Claus R, Arab K, Dyckhoff G, et al. Frequently methylated tumor suppressor genes in head and neck squamous cell carcinoma. Cancer Res. 2008;68:4494–9. doi: 10.1158/0008-5472.CAN-07-6509. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Feng X, Liu X, Jiang L, Zeng X, Ji N, et al. Involvement of potential pathways in malignant transformation from oral leukoplakia to squamous cell carcinoma revealed by proteomic analysis. BMC Genomics. 2009;10:383. doi: 10.1186/1471-2164-10-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall PA, Todd CB, Hyland PL, McDade SS, Grabsch H, Dattani M, et al. The septin-binding protein anillin is overexpressed in diverse human tumors. Clin Cancer Res. 2005;11:6780–6. doi: 10.1158/1078-0432.CCR-05-0997. [DOI] [PubMed] [Google Scholar]
- 21.Kinoshita M. The Septins. Genome Biol. 2003;4:236. doi: 10.1186/gb-2003-4-11-236. [DOI] [PMC free article] [PubMed] [Google Scholar]