

With great interest we read the paper of Khalil and Läer on the concepts of physiologically based pharmacokinetic (PBPK) modeling and its applications to pediatric drug development [1]. The authors clearly described the methodology, applications, and limitations of PBPK models in pediatric drug development [1]. In essence, PBPK models hold the promise to design and perform a pediatric study based on “a well-educated guess” to paraphrase what the authors refer to as “confirmatory” instead of “exploratory” approach. The approach described by the authors hereby mainly reflects what we would like to describe as a “top-down (from model to clinical observations)” concept: based on the available knowledge on developmental anatomy and physiology, a PBPK model is developed, undergoes validation, and will subsequently facilitate pediatric studies [1]. We would like to further challenge research groups active in the field of PBPK modeling not to get too disconnected from the “in vivo” world of pediatric developmental pharmacology and also consider what we would like to describe as a “bottom-up (from clinical observations to model)” concept: from compound specific observations to mechanism-based models [2–5]. It is our strong opinion that active comparison between PBPK and mechanism based models using the same in vivo datasets can be helpful to further improve clinical care but also provides guidance for more focused studies on aspects of developmental physiology.
During the last 2-3 decades, hundreds of compound specific clinical pharmacology studies have been conducted to investigate the impact of ontogeny on clinical pharmacology in pediatric populations.
As clinical pharmacologists and neonatologists with specific interest in developmental pharmacology, we would like to raise awareness for the relevance of “rich data sets” that contain both clinical characteristics and concentration-time (pharmacokinetics) or concentration-effect (pharmacodynamics) profiles [6]. The description of a compound specific pattern is beyond compound specific relevance (“bottom-up”) [1–5]. The maturational patterns described and the extent of the impact of covariates can subsequently be applied to predict in vivo concentration-time profiles for compounds that undergo similar routes of elimination. Through improved predictability, such maturational mechanism-based models can serve to improve clinical care and feasibility of clinical studies in neonates. The same in vivo observations can also be used as a “bottom-up” approach to learn more about the maturational patterns, and to guide research on gaps in the knowledge on developmental anatomy and physiology.
The concept of a comparative approach using both PBPK and mechanism-based models for drugs cleared by renal elimination is illustrated in Figure 1. In the left panel of Figure 1, a mechanism-based approach (bottom-up: description based on observations, subsequent validation) is considered, in the right panel, a PBPK-based approach (top-down: development of a PBPK model based on renal maturational physiology data, that is, ontogeny of glomerular filtration rate [GFR] is applied). Based on mechanism-based models, we described covariates of amikacin and vancomycin clearance in 531 neonates and documented that size, postmenstrual age, growth restriction, and coadministration of ibuprofen explained 85% of the interindividual variability in clearance [7]. The same amikacin dataset has more recently been used to validate a PBPK model (left panel, Figure 1) that aimed to describe maturational GFR in (pre)term neonates [8].
Figure 1.
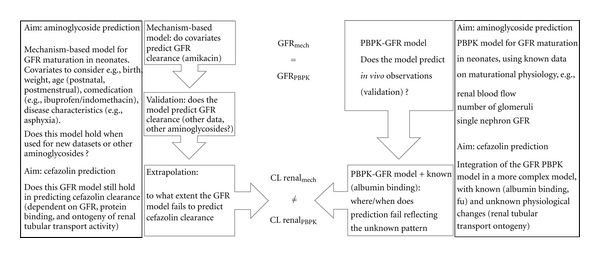
Integration of in vivo datasets analyzed by mechanism-based and PBPK predictive models results in a switch from “explorative, hypothesis-driven” to “confirmative” approach in the field of developmental physiology. This is illustrated for renal drug clearance in neonates, which reflects glomerular filtration rate (e.g., aminoglycosides) or a more complex pattern of known (glomerular filtration rate, protein binding) and still unknown (renal tubular transport ontogeny) (e.g., cefazolin) maturational processes.
Both approaches should describe GFR ontogeny. However, when these PBPK-GFR models are subsequently used to predict renal elimination clearance of compounds that do not only depend on GFR, but also on protein binding and renal tubular functions (e.g., cefazolin), the PBPK models will fail because data on renal tubular ontogeny are not yet available. In contrast, the use of mechanism-based models to describe the covariates of cefazolin clearance in such a dataset and comparison with the PBPK-GFR model may unveil thresholds and patterns of renal tubular maturation that can subsequently guide researchers to explore ontogeny of renal tubular activity in specific subpopulations, resulting in a similar confirmatory instead of exploratory approach.
In conclusion, similar to PBPK models for clinical care (top-down), mechanism-based models (bottom-up) and comparison between both approaches may further guide and facilitate both clinical and fundamental research on developmental physiology and anatomy [1–5]. Discrepancies serve as indicators for “missing” links in our knowledge on maturational anatomy or physiology (e.g., drug receptor activity, receptor expression) and in this way may also shape fundamental research in the field of developmental physiology. In this way, PBPK models do not only hold the promise (top-down) to be helpful in the clinical design, but may also serve as indicators to perform developmental anatomy/physiology research projects as “confirmatory” instead of “exploratory.” In this way, improved knowledge on developmental pharmacology does not only serve the individual clinician and the patient, but can also improve focused fundamental research on aspects of developmental biology that are currently hardly understood and difficult to explore.
Acknowledgments
K. Allegaert is supported by the Fund for Scientific Research, Flanders (Belgium) (FWO Vlaanderen) by a Fundamental Clinical Investigatorship (1800209N). J. van den Anker is supported in part by NIH Grants (R01HD060543, K24DA027992, R01HD048689, and U54HD071601) and FP7 Grants TINN (223614), TINN2 (260908), and NEUROSIS (223060).
References
- 1.Khalil F, Läer S. Physiologically based pharmacokinetic modeling: methodology, applications, and limitations with a focus on its role in pediatric drug development. Journal of Biomedicine and Biotechnology. 2011;2011:13 pages. doi: 10.1155/2011/907461. Article ID 907461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Cock RF, Piana C, Krekels EH, Danhof M, Allegaert K, Knibbe CAJ. The role of population PK-PD modelling in paediatric clinical research. European Journal of Clinical Pharmacology. 2011;67(supplement 1):S5–S16. doi: 10.1007/s00228-009-0782-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Läer S, Barrett JS, Meibohm B. The in silico child: using simulation to guide pediatric drug development and manage pediatric pharmacotherapy. Journal of Clinical Pharmacology. 2009;49(8):889–904. doi: 10.1177/0091270009337513. [DOI] [PubMed] [Google Scholar]
- 4.Bartelink IH, Rademaker CM, Schobben AF, van den Anker JN. Guidelines on paediatric dosing on the basis of developmental physiology and pharmacokinetic considerations. Clinical Pharmacokinetics. 2006;45(11):1077–1097. doi: 10.2165/00003088-200645110-00003. [DOI] [PubMed] [Google Scholar]
- 5.Johnson TN, Rostami-Hodjegan A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Paediatric Anaesthesia. 2011;21(3):291–301. doi: 10.1111/j.1460-9592.2010.03323.x. [DOI] [PubMed] [Google Scholar]
- 6.Allegaert K, Verbesselt R, Naulaers G, et al. Developmental pharmacology: neonates are not just small adults. Acta Clinica Belgica. 2008;63(1):16–24. doi: 10.1179/acb.2008.003. [DOI] [PubMed] [Google Scholar]
- 7.Allegaert K, Anderson BJ, van den Anker JN, Vanhaesebrouck S, de Zegher F. Renal drug clearance in preterm neonates: relation to prenatal growth. Therapeutic Drug Monitoring. 2007;29(3):284–291. doi: 10.1097/FTD.0b013e31806db3f5. [DOI] [PubMed] [Google Scholar]
- 8.Claassen K, Willmann S, Thelen K, Coboeken K, Allegaert K, Lippert. J. Physiology-based simulations of amikacin pharmacokinetics in preterm neonates. PAGE, 19th meeting Berlin, abstract 1859, 2010, http://www.page-meeting.org/?abstract=1859.