

Abstract
Leptin is a product of the obese (OB) gene secreted by adipocytes in proportion to fat mass. It decreases food intake and increases energy expenditure by affecting the balance between orexigenic and anorexigenic hypothalamic pathways. Low leptin levels are responsible for the compensatory increase in appetite and body weight and decreased energy expenditure (EE) following caloric deprivation. The anorexia-cachexia syndrome is a complication of many chronic conditions including cancer, chronic obstructive pulmonary disease, congestive heart failure, chronic kidney disease, and aging, where the decrease in body weight and food intake is not followed by a compensatory increase in appetite or decreased EE. Crosstalk between leptin and inflammatory signaling known to be activated in these conditions may be responsible for this paradox. This manuscript will review the evidence and potential mechanisms mediating changes in the leptin pathway in the setting of anorexia and cachexia associated with chronic diseases.
1. Introdiction
Leptin was discovered in 1994 by Friedman and colleagues after cloning an obese (OB) gene responsible for obesity in ob/ob mice [1]. It is a 167 amino acid peptide produced by adipocytes and it is a member of the adipocytokine family. Leptin has been noted to play a major role in body mass regulation by acting in the central nervous system to both stimulate energy expenditure and decrease food intake [2–4]. Named after the Greek word leptos, meaning lean, leptin was the first adipocyte-secreted hormone discovered, proving the active role of adipocytes in metabolic signaling.
Leptin crosses the blood-brain barrier in a process that is highly regulated [5–8] and its receptors are found both centrally, in the hypothalamus, and peripherally, in pancreatic islets, liver, kidney, lung, skeletal muscle, and bone marrow [9]. Besides its key role on body weight regulation, leptin affects various metabolic pathways, including growth hormone (GH) signaling [10], insulin sensitivity, and lipogenesis [11]. While leptin levels are directly related to adiposity, there are several other factors resulting in individual variability. Leptin secretion is regulated by insulin, glucocorticoids, and catecholamines [3, 12, 13]. Also, females have significantly higher levels of leptin than men, for any degree of fat mass [14]. Along with adiponectin, leptin assists in peripheral insulin sensitization independent of body weight [15–17]. In leptin-deficient (ob/ob) mice, leptin injections led to dose-dependent reductions in serum glucose levels compared to fed ob/ob controls, before any significant change in body weight occurred [1, 18, 19].
Inactivating mutations of leptin or leptin receptor gene result in the body's false perception of starvation and subsequent hyperphagia, decreased energy expenditure, and severe obesity [20, 21]. In the absence of these mutations and presence of diet-induced obesity, increased adipose tissue results in increased leptin levels. A 10% increase in body weight leads to a 300% increase in serum leptin concentrations [22]. These elevated leptin levels should lead to decreased food intake and increased energy expenditure via physical activity and thermogenesis. Although obese humans have high plasma leptin concentrations, these high levels do not result in the predicted decrease in appetite. This was initially thought to be due to leptin resistance. However, an increasing body of evidence now suggests central leptin insufficiency as a mechanism. The conventional idea of leptin resistance was thought to be due to inhibition of leptin signaling pathways in leptin-responsive neurons, defects downstream of leptin receptor, and blood-brain barrier (BBB) transport limitation for leptin [23–27]. Decreased leptin transfer via the BBB does not, however, compromise intracellular signaling in the hypothalamus. Furthermore, centrally administered leptin effectively decreases the rate of fat accumulation, hyperglycemia, insulin resistance, hyperinsulinemia, and progression to metabolic syndrome in obese rodents [28–30]. The validity of the concept of leptin resistance has, thus, been questioned.
Central leptin insufficiency due to dietary and lifestyle changes for extended periods of time has been shown to result in increased fat accrual, decreased energy expenditure, hyperinsulinemia, hyperglycemia, neuroendocrine disorders, osteoporosis, and impaired memory [6, 31, 32]. Leptin permeability across the BBB is modulated by various endogenous factors, including adiposity, daily mealtimes, intrinsic circadian rhythms governing ingestion behavior, and aging [5–8]. Transport of leptin to the brain is reduced by fasting and increased by pretreatment with glucose [33, 34]. Leptin binding proteins in the blood can affect leptin levels available for transport to the brain. For example, hepatic C-reactive protein (CRP), which is increased in obesity, binds leptin and limits leptin receptor binding and transport across the BBB [35, 36]. Central leptin gene therapy in obese mice resulted in multiple benefits, including normoinsulinemia, euglycemia, elimination of fatty liver, increased energy expenditure, and more than doubling of lifespan [32, 37, 38].
Cytokines, such as IL-6 and TNF-α, are increased in obesity and correlate with insulin resistance [39]. After three weeks of a very low-calorie diet, IL-6 levels decrease in adipose tissue as well as in serum. Furthermore, IL-6 knockout mice develop obesity at the young age of 6 months [40]. Unlike leptin, IL-6 has higher CSF concentrations than serum in some obese, but otherwise healthy, men [41]. The presence of increased cytokines, in addition to the hyperleptinemia and central leptin insufficiency, seen in obesity appears to play a key role in the pathophysiology of metabolic syndrome.
Taken together, the data suggest that the leptin system may be more efficient in signaling a decrease in fat mass and lack of nutrients (low leptin state) and triggering a compensatory increase in food intake and a decrease in energy expenditure than as a satiety signal when its serum levels are elevated. Moreover, recent evidence suggests that the neurobiology of leptin signaling in obesity appears to involve central leptin insufficiency, as opposed to the previously postulated notion of leptin resistance. Interestingly, central leptin administration and gene therapy has successfully improved energy homeostasis as well as prevented diet-induced obesity and metabolic syndrome in mice.
The arcuate nucleus (ARC), ventromedial (VMH), dorsomedial (DMH), and lateral (LH) hypothalamic nuclei are important regions regulating food intake and energy expenditure. Disrupting lesions in the ARC, VMH, and DMH of rats resulted in hyperphagia and obesity [42]. Moreover, lesions in the LH resulted in decreased food intake [43]. Binding of leptin to its hypothalamic receptors activates a signaling cascade in the ARC that results in inhibition of orexigenic pathways as indicated by decreased mRNA expression of neuropeptide Y (NPY) and agouti-related peptide (AgRP), and stimulation of anorexigenic pathways as suggested by increases in the mRNA levels of alpha-melanocyte-stimulating hormone (α-MSH) and cocaine and amphetamine-regulated transcript (CART) [44, 45]. Activation of POMC/CART-expressing neurons by leptin results in release of α-MSH, which subsequently binds to melanocotin receptors (MCRs) and leads to anorexia and increased energy expenditure. At the same time, leptin inhibits NPY/AgRP neurons, which stimulate orexigenic responses and directly inhibits POMC neuron expression as indicated by POMC mRNA expression [46, 47]. Interestingly, there is no feedback from POMC neurons to NPY/AgRP neurons, revealing that the default function of the circuit is to promote food intake [48, 49]. Loss of function mutations of the MC-4R, the most important melanocortin receptor (MCR), is the most common genetic etiology of obesity and accounts for 3–5% of severe human obesity [50, 51], highlighting the relevance of this pathway in humans. Leptin modifies postsynaptic action of orexigenic and anorectic signals via the JAK2 (Janus kinase 2)-STAT3 (signal transducer and activator of transcription 3) and PI3K-PDE3B (phosphatidylinositol-3 kinase-phosophodiesterase 3B-cAMP) pathways [52].
Pinto et al. hypothesized that leptin may cause rewiring of the ARC neural circuit when they found that the NPY/AgRP and POMC neurons in ob/ob and wild-type mice differed. Treatment with leptin normalized synaptic density within six hours, even before leptin levels affected food intake. These findings indicate that leptin may also function via neural plasticity in the hypothalamus [53]. Another anorectic hypothalamic pathway has been recently characterized involving the protein nesfatin-1 and it appears to be leptin-independent. Nesfatin-1 targets magnocellular and parvocellular oxytocin neurons as well as nesfatin-1 neurons to stimulate oxytocin release. Oxytocin then activates POMC neurons in the nucleus of the tractus solitaries (NTS) and induces melanocortin-dependent anorexia in leptin-resistant Zucker-fatty rats. Injecting nesfatin-1 was shown to activate the PVN and result in leptin-independent melanocortin-mediated anorexia [54].
In summary, the discovery of ob/ob mice and leptin has provided evidence that there is hormonal communication between adipose tissue and the hypothalamus, regulating food intake and energy metabolism. Leptin controls feeding via the ARC melanocortin system, by altering gene transcription and neural plasticity. The ARC then integrates all the information it receives and accordingly alters feeding and metabolism through hormonal and neural pathways. The hyperleptinemic state of obesity has been associated with leptin resistance or, more likely, central leptin deficiency.
Convincing evidence suggest that one of the main roles of leptin is to signal a state of nutrient deficiency and fat loss. Low leptin levels in this setting will trigger a centrally mediated, compensatory response leading to increased appetite and food intake, decreased energy expenditure, and, ultimately, weight regain. However, this mechanism does not appear to be preserved in most chronic diseases in spite of the weight loss seen. This manuscript will review the evidence and potential mechanisms mediating changes in this pathway in the setting of anorexia and cachexia associated with chronic diseases.
2. Anorexia-Cachexia Syndrome
Anorexia, defined as the loss of desire to eat, despite caloric deprivation, is frequently seen in patients with advanced chronic illness [55]. Cachexia, a term derived from Greek kakos, meaning bad, and hexis, meaning condition, describes a progressive loss of adipose tissue and lean body mass. Increased proteolysis, decreased protein synthesis, and accelerated lipolysis due to high energy demands result in a dramatic decline in lean body mass and fat mass and increase in mortality in this setting [56, 57]. Caloric restriction per se induces a less severe degree of weight loss and a different metabolic pattern characterized by decreased energy expenditure and preservation of lean mass at the expense of fat loss. This suggests that anorexia alone does not cause the extreme weight loss seen in cachexia. Moreover, nutritional support does not reverse cachexia [58]. Clinically, ACS presents with weight loss, decreased appetite, early satiety, muscle atrophy, and weakness. This process has been observed in various illnesses, including cancer, chronic heart disease, pulmonary disease, chronic kidney disease, and aging.
Anorexia-cachexia syndrome appears to be multifactorial, often associated with the underlying disease process, and related to both peripheral and central neurohormonal signals regulating both appetite and energy expenditure. Inflammatory cytokines, such as tumor necrosis factor- (TNF-) α, interleukin- (IL-) 1, IL-6, and interferon- (IFN-) γ have been postulated to play a key pathogenic role in the decreased food intake and increased energy expenditure seen in most chronic conditions associated with the anorexia and cachexia syndrome (ACS) [59]. Increased cytokines in the hypothalamus enhance serotoninergic tone through tryptophan, resulting in activation of POMC neurons and subsequent anorexia [60]. IL-1 inhibition in tumor-bearing animals has been shown to improve appetite and promote weight gain [61]. The somatomedin pathway, including GH and insulin-like growth factor-1 (IGF-1), stimulates skeletal muscle protein synthesis and is inhibited by inflammatory cytokines [62, 63]. In spite of the devastating effect that ACS has in patients, its pathophysiology is only partially understood and there are no approved treatments for this condition.
3. Crosstalk between Leptin Signaling and Inflammation
Leptin receptors belong to the class I cytokine receptor family and have similar structure to the signal-transducing subunits of the IL-6 receptors [64]. Leptin levels decrease with fasting and increase during the postprandial phase afterwards. These changes are directly correlated with changes in hypothalamic interleukin- (IL-) 1β mRNA levels, suggesting that leptin has a proinflammatory role centrally [65]. This link between proinflammatory cytokines and leptin has been illustrated well in animal models. Fasted hamsters treated with the cytokines tumor necrosis factor- (TNF-) α and IL-1 showed increased levels of both circulating leptin and leptin mRNA in adipose tissue. These increases in leptin were associated with a decline in food intake [66]. This is also supported by experiments where peripheral leptin administration caused hypothalamic inflammation and central injection of IL-1 receptor (IL-1r) antagonist inhibited the suppression of food intake caused by central or peripheral injection of leptin [67]. Mice lacking the main IL-1 receptor responsible for IL-1 actions showed no reduction in food intake in response to leptin [68]. Increased inflammatory mediators have been shown to increase hypothalamic POMC mRNA expression [69]. Administering melanocortin receptor antagonist centrally results in blockade of inflammatory anorexia [70].
Leptin levels are significantly lower in patients with inflammatory states such as cancer [71] despite correction for body fat. These low levels of leptin, however, are not associated with greater appetite or lower energy expenditure, as might be expected. Disturbances in the feedback mechanism in the hypothalamus and/or release of pro-inflammatory cytokines, such as IL-1, IL-6, and TNF-α, are thought to be responsible for cachexia in this setting. These circulating cytokines result in insulin resistance, lipolysis, and loss of skeletal muscle mass [72]. IL-1 influences size, duration, and frequency of meals in rats via hypothalamic signaling [73]. Cytokines also suppress gastric production of the orexigenic peptide ghrelin that decreases production of inflammatory cytokines TNF-α, IL-6, and IL-1β.
Nuclear factor-κB (NF-κB), a transcription factor for inflammation-related proteins, is activated in the hypothalamus of animal models of infection-associated anorexia. These models are created by administration of bacterial and viral products such as lipopolysaccharide (LPS) and HIV-1 transactivator protein (Tat). In vitro, NF-κB activation stimulated POMC transcription, showing the connection of NF-κB in feeding regulation. Hypothalamic injection of LPS and Tat showed reductions in food intake and body weight, while inhibition of NF-κB and melanocortin cancelled these effects. Moreover, hypothalamic NF- κB is activated by leptin and is involved in leptin-stimulated POMC transcription, showing that it may serve as a downstream signaling pathway of leptin [74]. Paradoxically, inflammation is also thought to play a role in obesity [75]. Obesity in both human [76] and animal models [77] has been associated with increased inflammatory markers, including TNF-α and IL-6. In rats and mice with diet-induced obesity, inflammation of both peripheral tissues and the hypothalamus was noted [78–81]. Blocking hypothalamic inflammation signaling via pharmacological approach or gene therapy led to a reduction in food intake and lower body weight in these animals [79, 82].
In summary, the evidence suggests that the central effects of leptin in suppressing appetite and increasing energy expenditure via activation of POMC neurons is at least partially dependent upon inflammation. Moreover, inflammation may influence the same pathways affecting appetite and body weight independently of leptin.
4. Leptin in Cancer Cachexia
Cancer anorexia-cachexia syndrome (Cancer-ACS) is found in 80% of patients with advanced cancer. It has been shown to decrease performance status, quality of life, response to therapy, and survival [56, 57]. Cancer-ACS may account for up to 20% of cancer deaths [58]. Although the tumor itself is primarily responsible, treatments such as chemotherapy and radiation, and associated conditions such as depression, pain, gastrointestinal obstruction, and taste alterations can also contribute to weight loss [83]. Cancer-ACS appears to be multifactorial, involving tumor-host interactions that result in catabolism overwhelming anabolism.
Leptin is thought to play a major role in the pathophysiology of cancer-ACS. Animal studies have shown that circulating leptin levels are decreased in the setting of tumor-induced cachexia, as expected given the decrease in fat mass seen in this setting [84]. However, mRNA levels of NPY were decreased and for POMC were increased in the ARC, unlike what is seen in caloric restriction where low leptin levels cause activation of NPY and suppression of POMC pathways. Levels of phosphorylated signal transducer and activator of transcription-3 (P-Stat3), a central molecule activated via the leptin receptor signaling pathway, are upregulated in subsets of α-MSH and NPY positive neurons that are not responsive to leptin. This pathway appears to be induced by the cytokine macrophage inhibitory cytokine-1 (MIC-1) via activation of the transforming growth factor- (TGF-) β receptor II, suggesting a potential alternative pathway through which MIC-1 could regulate appetite independently of leptin. This is also supported by the fact that MIC-1 infusion can induce anorexia and weight loss in leptin-deficient mice [85].
Leptin levels are significantly decreased in cancer cachexia patients compared to both cancer noncachexia and healthy controls [86, 87]. Proinflammatory cytokines, such as TNF-α, IL-1, and IL-6, have been proposed to cause cachexia in spite of low circulating leptin due to increased expression of the hypothalamic leptin receptor [88]. This dysregulation of the normal feedback loop in cancer cachexia may explain why a decrease in leptin does not increase appetite or lower energy expenditure in patients with cancer cachexia. Interestingly, leptin was found to be directly associated with appetite and insulin resistance [87], suggesting that these patients are resistant to the orexigenic effects of hypoleptinemia. Leptin also has been postulated as an early marker of disease progression in advanced ovarian cancer [89]. Moreover, in these patients, there was a clear correlation between disease stage and performance status with markers of inflammation, such as IL-6 whereas low leptin levels were more closely associated with tumor stage and IL-6 levels than BMI.
Given the large role of proinflammatory cytokines in cancer cachexia, the use of anticytokine antibodies and cytokine receptor antagonists has been investigated as potential therapies. Unfortunately, despite promising experimental data, clinical trials have not been conclusive. In the Yoshida AH-130 model, anti-TNF therapy partially reversed metabolic abnormalities in cachexia [90]. Clinical trials, however, showed transient improvement at best. In a double-blinded, placebo-controlled, randomized study, pentoxyphylline, which inhibits TNFα transcription, failed to show any benefit on cancer cachexia [91]. In small, unrandomized clinical trials, thalidomide, a TNF-α inhibitor, has been shown to improve some cancer-ACS symptoms [92]. Anti-inflammatory cytokines, IL-12 and IL-15, have shown some improvement in cancer-ACS in tumor-bearing animals [93, 94]. Mantovani et al. performed a phase II clinical trial with cyclooxygenase-2 (COX-2) inhibitors in patients with cancer-ACS showing a significant increase in LBM, decrease in TNF-α, and improvement in overall performance status [95]. Whether these interventions that blocked inflammation had an effect on leptin or its pathway is not known given that leptin levels or changes in its downstream mediators were not reported in these studies.
In summary, cancer anorexia-cachexia syndrome is a major predictive factor of mortality. In both animal and human models, circulating leptin levels decrease in the setting of cancer-ACS. However, this decrease in leptin is not associated with a compensatory increase in appetite and food intake. Animal studies suggest that hypothalamic inflammation may account for the lack of response of leptin targets to the effects of hypoleptinemia. Although cytokines appear to play a major role in the development of cancer-ACS via central and peripheral effects, preliminary studies targeting this pathway have not shown convincing evidence of a beneficial effect.
5. Leptin in Chronic Heart Failure-Induced Cachexia
The incidence of chronic heart failure is steadily rising and carries a poor prognosis. Cardiac cachexia is defined as nonedematous weight loss of >6% of previous normal weight observed over a period of >6 months and is associated with poor prognosis [96]. CHF patients with cardiac cachexia have been noted to have a mortality of 50% at 18 months, versus 17% in noncachectic patients [97]. Various factors can contribute to the weight loss seen in CHF-induced cachexia, including malnutrition from medications, metabolic disturbances (i.e., hyponatremia, renal failure), and hepatic congestion; malabsorption from severe heart failure; or increased nutritional requirements. The basal metabolic rate in these patients is increased by 20% [98]. As in other chronic conditions, inflammation has been implicated as a key aspect of CHF-induced cachexia [99].
Some controversy exists regarding leptin levels in cachectic versus noncachectic CHF patients. While many studies show lower leptin levels in cachectic patients [10, 100, 101], as it would be expected with their decreased fat tissue, other studies report normal levels [102]. These differences may exist due to sex distribution and presence of cachexia in selected subjects. When fat tissue was normalized, leptin levels for both cachectic and noncachectic CHF patients were elevated in comparison to non-CHF controls [10]. Several groups have hypothesized that leptin has a cardioprotective role and that this increase in its levels in the setting of CHF may represent a compensatory response rather than simply a marker of fat atrophy [103–105]. This could also explain the fact that circulating leptin levels directly correlate with NYHA class and overall prognosis in this setting. It is also possible that this increase in leptin is at least partially responsible for the weight loss and anorexia in CHF patients since an increase in leptin would lead to activation of the melanocortin system that in turn would cause an increase in energy expenditure and a decrease in food intake [106]. This hypothesis remains to be tested. It is also postulated that hyperleptinemia in these patients may be a result of insulin resistance [107, 108]. Regardless of the reason, this hyperleptinemia in both cachectic and noncachectic CHF patients suggests that leptin-mediated decrease in appetite and food intake is not particularly important in the development of CHF-induced cachexia.
Leptin may also contribute to CHF-cachexia and obesity-related cardiomyopathy by various cardiovascular mechanisms, including increasing sympathetic activity and producing vasodilation by an endothelium-dependent mechanism peripherally. It also promotes inflammation, calcification, proliferation, and thrombosis in the vasculature [109]. Animal models, however, do not show any increase in blood pressure, despite this increase in sympathetic activity [110, 111]. It is hypothesized that this may be due to leptin's vasodilatory effects via unclear mechanisms [109, 112].
The presence of proinflammatory cytokines in this setting suggests that inflammation plays an important role in the pathogenesis of CHF. Increasing levels of TNF-α, IL-1, and IL-6, lead to activation of the renin-angiotensin-aldosterone-system, improving renal and organ perfusion early on. However, TNF-α also induces apoptosis and activates protein breakdown in various tissues, including striate muscle. It also contributes to endothelial dysfunction and subsequent decreased blood flow to skeletal muscle, which results in decreased exercise endurance and nutrient supply [113]. Plasma TNF-α receptor levels in CHF patients have been associated with poor- long and short-term prognosis [114, 115]. Importantly, TNF-α increases the expression of leptin [66, 116]. No current therapy is approved to target cardiac cachexia. Standard treatment of CHF including ACE-inhibitors and beta-blockers has been shown to increase weight in CHF patients [96]. However, it appears that increases in leptin in noncachectic patients with CHF are primarily driven by body weight increases [117].
Although CHF is associated with elevated leptin levels, these are closely related to the amount of fat tissue; hence, levels are lower in cachectic individuals compared to noncachectic, CHF controls. This elevation in leptin may be due in part to insulin resistance but a direct cardioprotective effect of leptin also has been proposed. Taken together, the data suggests that leptin's role in this setting is not entirely related to body weight regulation and that the decreased appetite and increased energy expenditure seen in CHF-induced cachexia are more likely due to other factors. There is a scarcity of therapeutic data on patients with CHF-cachexia, so little is known about leptin responses to treatment at this time.
6. Leptin in Pulmonary Cachexia
Chronic obstructive pulmonary disease (COPD), including chronic bronchitis, emphysema, asthmatic bronchitis, and bronchiolitis obliterans, is a leading cause of morbidity and mortality worldwide. Cachexia has been reported in 20–40% of COPD patients and is associated with negative prognosis [118]. Although the increased work of breathing may be partly responsible for increased energy expenditure, this alone does not explain the reported weight loss [119, 120]. It is hypothesized that many different pathways are involved in the pathophysiology of COPD cachexia, including anorexia, nutritional deficiency, hypoxia, increased metabolic rate, inactivity, sympathetic upregulation, inflammation, and anabolic hormone deficiency.
Circulating levels of TNF-α and TNF-α production by peripheral monocytes are increased in patients with pulmonary cachexia [121, 122]. In both animal and human models, endotoxin or cytokine (TNF-α or IL-1) administration produces a dose-dependence increase in serum leptin levels [66, 123, 124]. However, COPD patients were reported to have lower leptin levels compared to healthy controls and these levels correlated well with BMI and percentage body fat [125]. Moreover, there is a loss of circadian variation in leptin levels that may represent alterations in the autonomic nervous system tone. Conversely, serum TNF-α levels were significantly higher in COPD patients compared to healthy controls and did not correlate with leptin levels [126], suggesting that in pulmonary cachexia, leptin levels are physiologically regulated and are independent of inflammatory markers, such as TNF-α.
In addition to the effects of chronic inflammation, it appears that hypoxia plays a major role in COPD-cachexia. Leptin-deficient animals show CO2 retention and respiratory depression; leptin administration to these animals increases minute ventilation and improves lung mechanics suggesting that an increase in leptin levels in patients with lung disease may represent a compensatory response to hypoxia [127–130]. Consistent with this hypothesis, elevated leptin has been described in hypoxic patients compared to BMI-matched controls [131]. It has been shown that expression of the human leptin gene is induced by hypoxia through the hypoxia-inducible factor-1 (HIF-1) pathway. The introduction of noninvasive positive airway pressure ventilation (NIPPV) in patients with severe COPD serves to both decrease energy expenditure and hypoxia. Moreover, body weight significantly improved (increased 12%) in patients with severe COPD and cachexia placed on NIPPV for 1 year [132]. This elevation in leptin was reversed by improving hypoxia, although this was associated with a decrease in fat accumulation in some studies that may have accounted at least partially for the changes in leptin.
In summary, pulmonary cachexia likely involves multiple pathways, including hypoxia and inflammation. Circulating leptin levels are decreased in patients with pulmonary cachexia suggesting that the physiologic regulation of leptin is maintained despite weight loss. Hypoxia also appears to play a role in leptin expression via the HIF-1 pathway that is reversible by correction of hypoxia. Furthermore, correction of hypoxia is associated with weight gain in spite of a decrease in leptin levels. These findings are consistent with the hypothesis that leptin plays a role in regulating the respiratory drive besides its usual role as a metabolic sensor.
7. Leptin in CKD Cachexia
Chronic kidney disease (CKD) is a common illness associated with a state of chronic inflammation and, oftentimes, cachexia. CKD-associated cachexia is linked to higher morbidity and mortality [133]. Uremic anorexia appears to be multifactorial. High plasma levels of insulin, leptin, and uremic toxins induce MC4-R stimulation to increase energy expenditure and decrease food intake [134]. Leptin levels are significantly elevated in CKD and ESRD patients and are associated with markers of poor nutritional status, such as low serum albumin and hypercatabolism as well as decline in renal function [135]. Serum leptin concentrations have been shown to inversely associate with survival in some studies [136]. In others, leptin was shown to correlate with fat mass rather than independently affecting food intake or mortality [137]. The hyperleptinemia seen in dialysis patients may be due to poor renal clearance, overproduction, or both. It has been postulated that uremic patients may have an acquired leptin receptor disorder resulting in central insensitivity or resistance, similar to obese individuals. Leptin reduces hypothalamic NPY levels and increases sympathetic activity with hyperinsulinemia, resulting in appetite suppression [138]. Supporting this hypothesis is the observation of increased sympathetic activity, via elevated dopamine, norepinephrine, and serotonin levels, found in uremic patients [139]. Elevated serum acute phase reactants, including C-reactive protein (CRP) and several cytokines, most prominently IL-6 and TNF-α, are found in CKD patients and may be associated with reduced appetite in dialysis patients [140]. Increased inflammation in renal failure is multifactorial, and possible factors include decreased renal clearance and increased production of proinflammatory cytokines [141, 142]. The mediators of inflammation act on the central nervous system to alter both appetite and metabolic rate [143, 144].
The administration of AgRP to mice with CKD resulted in increased food intake, normalization of basal metabolic rate, and increases in total body weight and lean body mass, independent of caloric or protein intake. Also, studies in db/db mice show that the lack of leptin receptor is protective against CKD-induced cachexia. Furthermore, manipulation of leptin's downstream mediators in the hypothalamus, MC4 by either gene deletion or by using antagonists of its receptor, confirms the relevance of this pathway in mediating anorexia and weigh loss in the setting of CKD [145].
A cross-sectional study of 217 hemodialysis patients followed for 31 months showed that those in the lowest tertile of ghrelin levels were the oldest and had the highest BMI, and highest CRP and leptin levels. These patients all had increased mortality risk, despite adjustment for age, gender, and dialysis history. Moreover, those in this group with protein-energy wasting had the highest all-cause and cardiovascular mortality risk (hazards ratios 3.34 and 3.54, resp.) [151]. In the setting of CKD, there is the opportunity to manipulate leptin levels not only by administering recombinant leptin but also by removing leptin from circulation using super-flux polysulfone dialyzers. van Tellingen et al. tried such approach and although leptin levels were significantly reduced, no other parameters such as appetite or body composition were examined [152, 153]. Therefore, the effectiveness of this intervention remains unknown.
Taken together, the evidence shows that CKD and ESRD-induced cachexia are associated with poor prognosis. Elevated levels of inflammatory mediators and leptin are likely results of decreased renal clearance and disease-related inflammation. Activation of the melanocortin system by leptin is key in the pathophysiology of CKD cachexia. Further studies to explore the efficacy of therapeutic options, including polysulfone dialyzers to lower leptin levels, are needed to determine the role of leptin in this setting.
8. Leptin in Aging
Weight loss in the geriatric population is a strong predictor of morbidity and mortality [154]. Normal aging involves a decline in appetite, decrease in lean body mass, increase in fat mass, and decrease in energy expenditure [155]. Aging has significant effects on energy homeostasis and dysregulation of adipokines, including leptin. These effects appear to be mediated at least in part by a decrease in the tone of the orexigenic AgRP/NPY pathway, an increase of the anorexigenic CART/POMC pathway, and failure of these pathways in responding to caloric restriction.
Elevated circulating leptin levels with decreased hypothalamic leptin responsiveness have been found in both animal and human models of aging [146]. Using a rodent model of aging, Wolden-Hanson showed that caloric restriction in aged Brown Norway rats failed to induce a compensatory increase in appetite after refeeding, unlike what is seen in young animals [147]. Leptin levels are increased in aged animals paralleling changes in fat mass but fail to decrease in response to fasting, suggesting that hyperleptinemia may contribute to this energy balance dysregulation and play a causative role in the poor tolerance of aged individuals to catabolic conditions. Also, leptin resistance has been proposed as one of the alterations seen in the elderly [156]. Hence, hyperleptinemia may be a compensatory mechanism to overcome the impaired leptin action in the brain. Uptake of leptin in the hypothalamus is significantly lower in old animals [157]. In aged rats, leptin administration does not suppress appetite, hypothalamic NPY expression, circulating leptin levels, or ob mRNA levels in white adipose tissue to the same extent as in young animals [148]. Although expression of the leptin receptor was not investigated in this report, others have shown a decrease in expression of the long form of this receptor during aging [157]. Downstream of leptin, abnormalities in other hypothalamic neuropeptides have been reported as well. Transcript mRNA expression of the orexigenic peptides NPY and AgRP decreases and fails to increase in response to caloric restriction [149, 158]; while CART mRNA expression increases with aging. Although response to exogenous AgRP appears to be maintained, response to NPY administration was significantly blunted in aged animals [159].
Previous studies have shown protective effects of fat mass on morbidity and mortality in the geriatric population [160]. Lipoatrophy and lipodystrophy in aging have been associated with dysregulation of adipokines and, subsequently, metabolic derangements such as insulin resistance, dyslipidemia, metabolic syndrome, hypertension, and hyperglycemia. Centenarians, models of health and longevity, had been reported to exhibit preserved insulin sensitivity and intact adipokine profiles. Studying this population revealed that poor prognosis was associated with dysregulated adipokines, including leptin levels inappropriately low for fat mass [161]. Leptin concentrations in 19 elderly patients with protein-energy malnutrition were significantly lower than their age-matched controls. However, others have reported an increase in leptin levels in aged individuals even after adjusting for fat mass [162]. It has also been shown that the there are increased levels of IL-6 and CRP in aging [150]. Studies in obese patients have suggested an association between hypothalamic inflammation and decreased leptin action via persistence activation of the melanocortin system; no studies, however, have been performed in the elderly [69, 163]. Functional status, anthropometry, and serum markers of nutrition and inflammation, including leptin and CRP, in seventy elderly patients versus controls revealed that those with the lowest functional status and highest frailty indices displayed features of cachexia. Moreover, they had low leptin levels, appropriate for their low body fat, as well as high CRP and IL-6 levels [164]. This suggests that the mechanism for cachexia in the elderly may involve disrupted hypothalamic feedback of leptin from the effects of proinflammatory cytokines like other chronic inflammatory states.
In summary, weight loss in the geriatric population is associated with higher mortality. In normal aging, fat mass is increased and hyperleptinemia arises. Despite these high levels of circulating leptin, however, there appears to be decreased leptin action and subsequently no decrease in appetite, similar to obese individuals. Moreover, a failed hypothalamic response to caloric restriction appears to be responsible for the poor tolerance of the elderly to catabolic stress. Elderly patients with cachexia tend to have elevated inflammatory markers and low leptin levels, both correlating with worsened prognosis.
9. Conclusion
Leptin, a product of the obese gene secreted by adipose tissue, acts centrally to suppress appetite and increase thermogenesis by activating the POMC neurons in the arcuate nucleus and triggering the release of α-MSH from POMC axon terminals and subsequently activating MC4-R (Figure 1). Moreover, the NPY and AgRP-producing neurons of the arcuate nucleus, which are suppressed by leptin, can antagonize these anorexigenic melanocortin cells. Consequently, low levels of leptin cause an increase in appetite and reduce energy expenditure.
Figure 1.
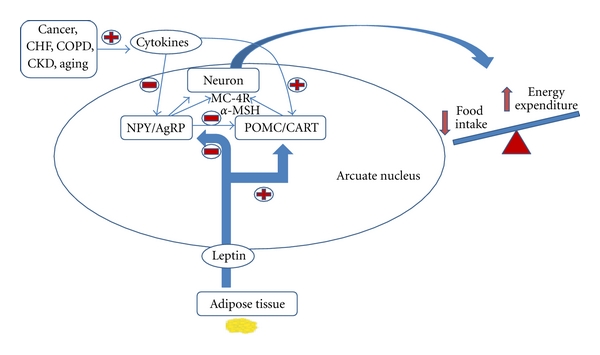
Summrary of the effects of peripheral hormones on hypothalamic regulation of food intake and energy expenditure. NPY = neuropeptide Y; AgRP = Agoui-relaed peptide; POMC = pro-opiomelacortin; CART = cocaine-amphetamine-related peptide; α-MSH = alpha-melanocyte-stimulating hormone; MC-4R = type-4 melanocortin receptor.
Cachexia is a unique process characterized by depletion of adipose tissue and lean body mass found in various chronic diseases often accompanied by anorexia. Anorexia-cachexia can be seen in cancer, CHF, COPD, CKD, and aging (Table 1). All of these conditions are associated with elevated inflammatory markers such as TNF-α, IL-6, IL-2, and IL-1β. These inflammatory markers may regulate hypothalamic feedback mechanisms and are thought to contribute to the development of cachexia. Leptin receptors belong to the class I cytokine family and there is crosstalk between leptin signaling and inflammation. This crosstalk could explain why, despite low levels of leptin in chronic inflammatory processes such as cancer, COPD, and aging, patients do not have the expected increased appetite or lower energy expenditure.
Table 1.
Summary of markers of appetite regulation in various cachectic states.
| Condition | Appetite | Body Weight | Circulating Leptin Levels | POMC/α-MSH hypothalamic levels | NPY/AgRP hypothalamic levels | Circulating inflammatory markers | Hypothalamic inflammatory markers | References |
|---|---|---|---|---|---|---|---|---|
| Cancer cachexia | ↓∗# | ↓∗# | ↓∗# | ↑* | ↓* | ↑∗# | ↑* | [60, 84–87] |
| CHF-induced cachexia | ↓∗# | ↓∗# | ↑∗# | unknown | unknown | ↑∗# | unknown | [10, 99–102, 115] |
| Pulmonary cachexia | ↓∗# | ↓∗# | ↓∗# | unknown | unknown | ↑∗# | unknown | [121, 122, 124–126] |
| CKD cachexia | ↓∗# | ↓∗# | ↑∗# | unknown | unknown | ↑∗# | unknown | [135, 137, 139, 140, 142, 145] |
| Aging cachexia | ↓∗# | ↓∗# | ↓∗# | ↑* | ↓* | ↑∗# | unknown | [146–150] |
#—supported by human model data; *—supported by animal model data.
Cancer, COPD, and aging-associated cachexia are all associated with low leptin levels, in spite of low appetite and elevated energy expenditure suggesting a state of resistance to the effects of hypoleptinemia. On the contrary, elevated levels have been noted in CKD- and CHF-induced cachexia. In CHF-induced cachexia, the reason for this elevation is unclear but there is no association with weight or fat mass change or with appetite suggesting that leptin may have a different function in this setting and that it likely does not play a major role in the ensuing cachexia. In CKD and ESRD, circulating levels of leptin and inflammatory agents are likely elevated due to poor renal clearance but there is no association with the degree of weight loss or anorexia.
Given the key role that inflammation appears to play in the pathogenesis of leptin-mediated cachexia, therapeutic intervention with anti-inflammatory drugs may prove to be beneficial in restoring sensitivity to the effect of hypoleptinemia in ACS. COX-2 inhibitors have shown some promise in patients with cancer cachexia. In the setting of uremic cachexia, polysulfone dialysers decrease leptin levels but more studies are needed to evaluate the effect of this intervention on appetite and weight parameters. As we gain more insight into the pathophysiology of cachexia, the therapeutic possibilities increase. Further investigations into anti-inflammatory drugs, appetite stimulants, and immunomodulators in these various conditions are warranted.
Acknowledgment
J. Garcia received research support from a M. MERIT grant from the Department of Veterans Affairs (I01-BX000507).
References
- 1.Halaas JL, Gajiwala KS, Maffei M, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269(5223):543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 2.Friedman JM. A tale of two hormones. Nature Medicine. 2010;16(10):1100–1106. doi: 10.1038/nm1010-1100. [DOI] [PubMed] [Google Scholar]
- 3.Friedman JM. Modern science versus the stigma of obesity. Nature Medicine. 2004;10(6):563–569. doi: 10.1038/nm0604-563. [DOI] [PubMed] [Google Scholar]
- 4.Flier JS, Maratos-Flier E. Lasker lauds leptin. Cell. 2010;143(1):9–12. doi: 10.1016/j.cell.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 5.Banks WA. Enhanced leptin transport across the blood-brain barrier by α1-adrenergic agents. Brain Research. 2001;899(1-2):209–217. doi: 10.1016/s0006-8993(01)02242-9. [DOI] [PubMed] [Google Scholar]
- 6.Banks WA. Is obesity a disease of the blood-brain barrier? Physiological, pathological, and evolutionary considerations. Current Pharmaceutical Design. 2003;9(10):801–809. doi: 10.2174/1381612033455350. [DOI] [PubMed] [Google Scholar]
- 7.Banks WA, Farrell CL. Impaired transport of leptin across the blood-brain barrier in obesity is acquired and reversible. American Journal of Physiology. 2003;285(1):E10–E15. doi: 10.1152/ajpendo.00468.2002. [DOI] [PubMed] [Google Scholar]
- 8.Kastin AJ, Pan W. Dynamic regulation of leptin entry into brain by the blood-brain barrier. Regulatory Peptides. 2000;92(1–3):37–43. doi: 10.1016/s0167-0115(00)00147-6. [DOI] [PubMed] [Google Scholar]
- 9.Margetic S, Gazzola C, Pegg GG, Hill RA. Leptin: a review of its peripheral actions and interactions. International Journal of Obesity. 2002;26(11):1407–1433. doi: 10.1038/sj.ijo.0802142. [DOI] [PubMed] [Google Scholar]
- 10.Doehner W, Pflaum CD, Rauchhaus M, et al. Leptin, insulin sensitivity and growth hormone binding protein in chronic heart failure with and without cardiac cachexia. European Journal of Endocrinology. 2001;145(6):727–735. doi: 10.1530/eje.0.1450727. [DOI] [PubMed] [Google Scholar]
- 11.Doehner W, Anker SD. Cardiac cachexia in early literature: a review of research prior to Medline. International Journal of Cardiology. 2002;85(1):7–14. doi: 10.1016/s0167-5273(02)00230-9. [DOI] [PubMed] [Google Scholar]
- 12.Mueller WM, Gregoire FM, Stanhope KL, et al. Evidence that glucose metabolism regulates leptin secretion from cultured rat adipocytes. Endocrinology. 1998;139(2):551–558. doi: 10.1210/endo.139.2.5716. [DOI] [PubMed] [Google Scholar]
- 13.Leroy P, Dessolin S, Villageois P, et al. Expression of ob gene in adipose cells: regulation by insulin. Journal of Biological Chemistry. 1996;271(5):2365–2368. doi: 10.1074/jbc.271.5.2365. [DOI] [PubMed] [Google Scholar]
- 14.Mantzoros CS, Moschos SJ. Leptin: in search of role(s) in human physiology and pathophysiology. Clinical Endocrinology. 1998;49(5):551–567. doi: 10.1046/j.1365-2265.1998.00571.x. [DOI] [PubMed] [Google Scholar]
- 15.Khan A, Narangoda S, Ahren B, Holm C, Sundler F, Efendic S. Long-term leptin treatment of ob/ob mice improves glucose-induced insulin secretion. International Journal of Obesity. 2001;25(6):816–821. doi: 10.1038/sj.ijo.0801628. [DOI] [PubMed] [Google Scholar]
- 16.Koch C, Augustine RA, Steger J, et al. Leptin rapidly improves glucose homeostasis in obese mice by increasing hypothalamic insulin sensitivity. Journal of Neuroscience. 2010;30(48):16180–16187. doi: 10.1523/JNEUROSCI.3202-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JW, Romsos DR. Leptin administration normalizes insulin secretion from islets of Lepob/Lepob mice by food intake-dependent and -independent mechanisms. Experimental Biology and Medicine. 2003;228(2):183–187. doi: 10.1177/153537020322800208. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz MW, Baskin DG, Bukowski TR, et al. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes. 1996;45(4):531–535. doi: 10.2337/diab.45.4.531. [DOI] [PubMed] [Google Scholar]
- 19.Pelleymounter MA, Cullen MJ, Baker MB, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269(5223):540–543. doi: 10.1126/science.7624776. [DOI] [PubMed] [Google Scholar]
- 20.Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387(6636):903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 21.Clément K, Vaisse C, Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392(6674):398–401. doi: 10.1038/32911. [DOI] [PubMed] [Google Scholar]
- 22.Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. New England Journal of Medicine. 1996;334(5):292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- 23.Burguera B, Couce ME, Curran GL, et al. Obesity is associated with a decreased leptin transport across the blood-brain barrier in rats. Diabetes. 2000;49(7):1219–1223. doi: 10.2337/diabetes.49.7.1219. [DOI] [PubMed] [Google Scholar]
- 24.Caro JF, Kolaczynski JW, Nyce MR, et al. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. The Lancet. 1996;348(9021):159–161. doi: 10.1016/s0140-6736(96)03173-x. [DOI] [PubMed] [Google Scholar]
- 25.Kalra SP. Circumventing leptin resistance for weight control. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(8):4279–4281. doi: 10.1073/pnas.091101498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocrine Reviews. 1999;20(1):68–100. doi: 10.1210/edrv.20.1.0357. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D., Jr. Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nature Medicine. 1996;2(5):589–593. doi: 10.1038/nm0596-589. [DOI] [PubMed] [Google Scholar]
- 28.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269(5223):546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 29.Dube MG, Beretta E, Dhillon H, Ueno N, Kalra PS, Kalra SP. Central leptin gene therapy blocks high-fat diet-induced weight gain, hyperleptinemia, and hyperinsulinemia: increase in serum ghrelin levels. Diabetes. 2002;51(6):1729–1736. doi: 10.2337/diabetes.51.6.1729. [DOI] [PubMed] [Google Scholar]
- 30.Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(16):8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baldock PA, Sainsbury A, Allison S, et al. Hypothalamic control of bone formation: distinct actions of leptin and Y2 receptor pathways. Journal of Bone and Mineral Research. 2005;20(10):1851–1857. doi: 10.1359/JBMR.050523. [DOI] [PubMed] [Google Scholar]
- 32.Dhillon H, Kalra SP, Prima V, et al. Central leptin gene therapy suppresses body weight gain, adiposity and serum insulin without affecting food consumption in normal rats: a long-term study. Regulatory Peptides. 2001;99(2-3):69–77. doi: 10.1016/s0167-0115(01)00237-3. [DOI] [PubMed] [Google Scholar]
- 33.Kastin AJ, Akerstrom V. Fasting, but not adrenalectomy, reduces transport of leptin into the brain. Peptides. 2000;21(5):679–682. doi: 10.1016/s0196-9781(00)00195-9. [DOI] [PubMed] [Google Scholar]
- 34.Kastin AJ, Akerstrom V. Glucose and insulin increase the transport of leptin through the blood-brain barrier in normal mice but not in streptozotocin-diabetic mice. Neuroendocrinology. 2001;73(4):237–242. doi: 10.1159/000054640. [DOI] [PubMed] [Google Scholar]
- 35.Chen K, Li F, Li J, et al. Induction of leptin resistance through direct interaction of C-reactive protein with leptin. Nature Medicine. 2006;12(4):425–432. doi: 10.1038/nm1372. [DOI] [PubMed] [Google Scholar]
- 36.Florez H, Castillo-Florez S, Mendez A, et al. C-reactive protein is elevated in obese patients with the metabolic syndrome. Diabetes Research and Clinical Practice. 2006;71(1):92–100. doi: 10.1016/j.diabres.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Beretta E, Dube MG, Kalra PS, Kalra SP. Long-term suppression of weight gain, adiposity, and serum insulin by central leptin gene therapy in prepubertal rats: effects on serum ghrelin and appetite-regulating genes. Pediatric Research. 2002;52(2):189–198. doi: 10.1203/00006450-200208000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Boghossian S, Ueno N, Dube MG, Kalra P, Kalra S. Leptin gene transfer in the hypothalamus enhances longevity in adult monogenic mutant mice in the absence of circulating leptin. Neurobiology of Aging. 2007;28(10):1594–1604. doi: 10.1016/j.neurobiolaging.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 39.Bastard JP, Jardel C, Bruckert E, et al. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. Journal of Clinical Endocrinology and Metabolism. 2000;85(9):3338–3342. doi: 10.1210/jcem.85.9.6839. [DOI] [PubMed] [Google Scholar]
- 40.Wernstedt I, Olsson B, Jernås M, et al. Increased levels of acylation-stimulating protein in interleukin-6- deficient (IL-6−/−) mice. Endocrinology. 2006;147(6):2690–2695. doi: 10.1210/en.2005-1133. [DOI] [PubMed] [Google Scholar]
- 41.Stenlöf K, Wernstedt I, Fjällman T, Wallenius V, Wallenius K, Jansson JO. Interleukin-6 levels in the central nervous system are negatively correlated with fat mass in overweight/obese subjects. Journal of Clinical Endocrinology and Metabolism. 2003;88(9):4379–4383. doi: 10.1210/jc.2002-021733. [DOI] [PubMed] [Google Scholar]
- 42.Bray GA, York DA. Hypothalamic and genetic obesity in experimental animals: an autonomic and endocrine hypothesis. Physiological Reviews. 1979;59(3):719–809. doi: 10.1152/physrev.1979.59.3.719. [DOI] [PubMed] [Google Scholar]
- 43.Anand BK, Brobeck JR. Localization of a "feeding center" in the hypothalamus of the rat. Proceedings of the Society for Experimental Biology and Medicine. 1951;77(2):323–324. doi: 10.3181/00379727-77-18766. [DOI] [PubMed] [Google Scholar]
- 44.Cowley MA, Smart JL, Rubinstein M, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411(6836):480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 45.Ibrahim N, Bosch MA, Smart JL, et al. Hypothalamic proopiomelanocortin neurons are glucose responsive and express KATP channels. Endocrinology. 2003;144(4):1331–1340. doi: 10.1210/en.2002-221033. [DOI] [PubMed] [Google Scholar]
- 46.Horvath TL, Naftolin F, Kalra SP, Leranth C. Neuropeptide-Y innervation of β-endorphin-containing cells in the rat mediobasal hypothalamus: a light and electron microscopic double immunostaining analysis. Endocrinology. 1992;131(5):2461–2467. doi: 10.1210/endo.131.5.1425443. [DOI] [PubMed] [Google Scholar]
- 47.Horvath TL, Garcia-Segura LM, Naftolin F. Control of gonadotropin feedback: the possible role of estrogen-induced hypothalamic synaptic plasticity. Gynecological Endocrinology. 1997;11(2):139–143. doi: 10.3109/09513599709152525. [DOI] [PubMed] [Google Scholar]
- 48.Horvath TL, Andrews ZB, Diano S. Fuel utilization by hypothalamic neurons: roles for ROS. Trends in Endocrinology and Metabolism. 2009;20(2):78–87. doi: 10.1016/j.tem.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Diano S. New aspects of melanocortin signaling: a role for PRCP in α-MSH degradation. Frontiers in Neuroendocrinology. 2011;32(1):70–83. doi: 10.1016/j.yfrne.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hinney A, Schmidt A, Nottebom K, et al. Several mutations in the melanocortin-4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans. Journal of Clinical Endocrinology and Metabolism. 1999;84(4):1483–1486. doi: 10.1210/jcem.84.4.5728. [DOI] [PubMed] [Google Scholar]
- 51.Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. Journal of Clinical Investigation. 2000;106(2):253–262. doi: 10.1172/JCI9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sahu A. Minireview: a hypothalamic role in energy balance with special emphasis on leptin. Endocrinology. 2004;145(6):2613–2620. doi: 10.1210/en.2004-0032. [DOI] [PubMed] [Google Scholar]
- 53.Pinto S, Roseberry AG, Liu H, et al. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304(5667):110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 54.Maejima Y, Sedbazar U, Suyama S, et al. Nesfatin-1-regulated oxytocinergic signaling in the paraventricular nucleus causes anorexia through a leptin-independent melanocortin pathway. Cell Metabolism. 2009;10(5):355–365. doi: 10.1016/j.cmet.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 55.Walsh D, Donnelly S, Rybicki L. The symptoms of advanced cancer: relationship to age, gender, and performance status in 1,000 patients. Supportive Care in Cancer. 2000;8(3):175–179. doi: 10.1007/s005200050281. [DOI] [PubMed] [Google Scholar]
- 56.Dewys WD, Begg C, Lavin PT, et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. American Journal of Medicine. 1980;69(4):491–497. doi: 10.1016/s0149-2918(05)80001-3. [DOI] [PubMed] [Google Scholar]
- 57.O’Gorman P, McMillan DC, McArdle CS. Impact of weight loss, appetite, and the inflammatory response on quality of life in gastrointestinal cancer patients. Nutrition and Cancer. 1998;32(2):76–80. doi: 10.1080/01635589809514722. [DOI] [PubMed] [Google Scholar]
- 58.Tisdale MJ. Cachexia in cancer patients. Nature Reviews Cancer. 2002;2(11):862–871. doi: 10.1038/nrc927. [DOI] [PubMed] [Google Scholar]
- 59.Plata-Salamán CR. Cytokines and feeding. International Journal of Obesity. 2001;25(supplement 5):S48–S52. doi: 10.1038/sj.ijo.0801911. [DOI] [PubMed] [Google Scholar]
- 60.Laviano A, Meguid MM, Rossi-Fanelli F. Cancer anorexia: clinical implications, pathogenesis, and therapeutic strategies. The Lancet Oncology. 2003;4(11):686–694. doi: 10.1016/s1470-2045(03)01247-6. [DOI] [PubMed] [Google Scholar]
- 61.Martignoni ME, Kunze P, Friess H. Cancer cachexia. Molecular Cancer. 2003;2, article 36 doi: 10.1186/1476-4598-2-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Broussard SR, Mccusker RH, Novakofski JE, et al. Cytokine-hormone interactions: tumor necrosis factor α impairs biologic activity and downstream activation signals of the insulin-like growth factor I receptor in myoblasts. Endocrinology. 2003;144(7):2988–2996. doi: 10.1210/en.2003-0087. [DOI] [PubMed] [Google Scholar]
- 63.Frost RA, Lang CH. Alteration of somatotropic function by proinflammatory cytokines. Journal of Animal Science. 2004;82:E100–E109. doi: 10.2527/2004.8213_supplE100x. [DOI] [PubMed] [Google Scholar]
- 64.Lee GH, Proenca R, Montez JM, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379(6566):632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 65.Schwartz MW, Morton GJ. Keeping hunger at bay. Nature. 2002;418(6898):595–597. doi: 10.1038/418595a. [DOI] [PubMed] [Google Scholar]
- 66.Grunfeld C, Zhao C, Fuller J, et al. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. Journal of Clinical Investigation. 1996;97(9):2152–2157. doi: 10.1172/JCI118653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wisse BE, Ogimoto K, Morton GJ, et al. Physiological regulation of hypothalamic IL-1β gene expression by leptin and glucocorticoids: implications for energy homeostasis. American Journal of Physiology. 2004;287(6):E1107–E1113. doi: 10.1152/ajpendo.00038.2004. [DOI] [PubMed] [Google Scholar]
- 68.Luheshi GN, Gardner JD, Rushforth DA, Loudon AS, Rothwell NJ. Leptin actions on food intake and body temperature are mediated by IL-1. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(12):7047–7052. doi: 10.1073/pnas.96.12.7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sergeyev V, Broberger C, Hökfelt T. Effect of LPS administration on the expression of POMC, NPY, galanin, CART and MCH mRNAs in the rat hypothalamus. Molecular Brain Research. 2001;90(2):93–100. doi: 10.1016/s0169-328x(01)00088-2. [DOI] [PubMed] [Google Scholar]
- 70.Marks DL, Ling N, Cone RD. Role of the central melanocortin system in cachexia. Cancer Research. 2001;61(4):1432–1438. [PubMed] [Google Scholar]
- 71.Mantovani G, Macciò A, Mura L, et al. Serum levels of leptin and proinflammatory cytokines in patients with advanced-stage cancer at different sites. Journal of Molecular Medicine. 2000;78(10):554–561. doi: 10.1007/s001090000137. [DOI] [PubMed] [Google Scholar]
- 72.Baumann H, Gauldie J. The acute phase response. Immunology Today. 1994;15(2):74–80. doi: 10.1016/0167-5699(94)90137-6. [DOI] [PubMed] [Google Scholar]
- 73.Laviano A, Meguid MM, Yang ZJ, Gleason JR, Cangiano C, Fanelli FR. Cracking the riddle of cancer anorexia. Nutrition. 1996;12(10):706–710. doi: 10.1016/s0899-9007(96)00164-5. [DOI] [PubMed] [Google Scholar]
- 74.Jang PG, Namkoong C, Kang GM, et al. NF-κB activation in hypothalamic pro-opiomelanocortin neurons is essential in illness- and leptin-induced anorexia. Journal of Biological Chemistry. 2010;285(13):9706–9715. doi: 10.1074/jbc.M109.070706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thaler JP, Choi SJ, Schwartz MW, Wisse BE. Hypothalamic inflammation and energy homeostasis: resolving the paradox. Frontiers in Neuroendocrinology. 2010;31(1):79–84. doi: 10.1016/j.yfrne.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 76.Mohamed-Ali V, Goodrick S, Rawesh A, et al. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-α, in vivo. Journal of Clinical Endocrinology and Metabolism. 1997;82(12):4196–4200. doi: 10.1210/jcem.82.12.4450. [DOI] [PubMed] [Google Scholar]
- 77.Kim F, Pham M, Maloney E, et al. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(11):1982–1988. doi: 10.1161/ATVBAHA.108.169722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Souza CT, Araujo EP, Bordin S, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146(10):4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- 79.Posey KA, Clegg DJ, Printz RL, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. American Journal of Physiology. 2009;296(5):E1003–E1012. doi: 10.1152/ajpendo.90377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ozcan L, Ergin AS, Lu A, et al. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metabolism. 2009;9(1):35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 82.Milanski M, Degasperi G, Coope A, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. Journal of Neuroscience. 2009;29(2):359–370. doi: 10.1523/JNEUROSCI.2760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Burstow M, Kelly T, Panchani S, et al. Outcome of palliative esophageal stenting for malignant dysphagia: a retrospective analysis. Diseases of the Esophagus. 2009;22(6):519–525. doi: 10.1111/j.1442-2050.2009.00948.x. [DOI] [PubMed] [Google Scholar]
- 84.Suzuki H, Hashimoto H, Kawasaki M, et al. Similar changes of hypothalamic feeding-regulating peptides mRNAs and plasma leptin levels in PTHrP-, LIF-secreting tumors-induced cachectic rats and adjuvant arthritic rats. International Journal of Cancer. 2011;128(9):2215–2223. doi: 10.1002/ijc.25535. [DOI] [PubMed] [Google Scholar]
- 85.Ding Q, Mracek T, Gonzalez-Muniesa P, et al. Identification of macrophage inhibitory cytokine-1 in adipose tissue and its secretion as an adipokine by human adipocytes. Endocrinology. 2009;150(4):1688–1696. doi: 10.1210/en.2008-0952. [DOI] [PubMed] [Google Scholar]
- 86.Weryńska B, Kosacka M, Gołecki M, Jankowska R. Leptin serum levels in cachectic and non-cachectic lung cancer patients. Pneumonologia i Alergologia Polska. 2009;77(6):500–506. [PubMed] [Google Scholar]
- 87.Smiechowska J, Utech A, Taffet G, Hayes T, Marcelli M, Garcia JM. Adipokines in patients with cancer anorexia and cachexia. Journal of Investigative Medicine. 2010;58(3):554–559. doi: 10.231/JIM.0b013e3181cf91ca. [DOI] [PubMed] [Google Scholar]
- 88.Bing C, Taylor S, Tisdale MJ, Williams G. Cachexia in MAC16 adenocarcinoma: suppression of hunger despite normal regulation of leptin, insulin and hypothalamic neuropeptide Y. Journal of Neurochemistry. 2001;79(5):1004–1012. doi: 10.1046/j.1471-4159.2001.00639.x. [DOI] [PubMed] [Google Scholar]
- 89.Macciò A, Madeddu C, Massa D, et al. Interleukin-6 and leptin as markers of energy metabolicchanges in advanced ovarian cancer patients. Journal of Cellular and Molecular Medicine. 2009;13(9):3951–3959. doi: 10.1111/j.1582-4934.2008.00408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Costelli P, Carbo N, Tessitore L, et al. Tumor necrosis factor-α mediates changes in tissue protein turnover in a rat cancer cachexia model. Journal of Clinical Investigation. 1993;92(6):2783–2789. doi: 10.1172/JCI116897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goldberg RM, Loprinzi CL, Mailliard JA, et al. Pentoxifylline for treatment of cancer anorexia and cachexia? A randomized, double-blind, placebo-controlled trial. Journal of Clinical Oncology. 1995;13(11):2856–2859. doi: 10.1200/JCO.1995.13.11.2856. [DOI] [PubMed] [Google Scholar]
- 92.Bruera E, Neumann CM, Pituskin E, Calder K, Ball G, Hanson J. Thalidomide in patients with cachexia due to terminal cancer: preliminary report. Annals of Oncology. 1999;10(7):857–859. doi: 10.1023/a:1008329821941. [DOI] [PubMed] [Google Scholar]
- 93.Mori K, Fujimoto-Ouchi K, Ishikawa T, Sekiguchi F, Ishitsuka H, Tanaka Y. Murine interleukin-12 prevents the development of cancer cachexia in a murine model. International Journal of Cancer. 1996;67(6):849–855. doi: 10.1002/(SICI)1097-0215(19960917)67:6<849::AID-IJC15>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 94.Carbó N, López-Soriano J, Costelli P, et al. Interleukin-15 antagonizes muscle protein waste in tumour-bearing rats. British Journal of Cancer. 2000;83(4):526–531. doi: 10.1054/bjoc.2000.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mantovani G, MacCiò A, Madeddu C, et al. Phase II nonrandomized study of the efficacy and safety of COX-2 inhibitor celecoxib on patients with cancer cachexia. Journal of Molecular Medicine. 2010;88(1):85–92. doi: 10.1007/s00109-009-0547-z. [DOI] [PubMed] [Google Scholar]
- 96.Anker SD, Negassa A, Coats AJS, et al. Prognostic importance of weight loss in chronic heart failure and the effect of treatment with angiotensin-converting-enzyme inhibitors: an observational study. The Lancet. 2003;361(9363):1077–1083. doi: 10.1016/S0140-6736(03)12892-9. [DOI] [PubMed] [Google Scholar]
- 97.Anker SD, Ponikowski P, Varney S, et al. Wasting as independent risk factor for mortality in chronic heart failure. The Lancet. 1997;349(9058):1050–1053. doi: 10.1016/S0140-6736(96)07015-8. [DOI] [PubMed] [Google Scholar]
- 98.Gibbs CR, Jackson G, Lip GYH. ABC of heart failure: non-drug management. British Medical Journal. 2000;320(7231):366–369. doi: 10.1136/bmj.320.7231.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. New England Journal of Medicine. 1990;323(4):236–241. doi: 10.1056/NEJM199007263230405. [DOI] [PubMed] [Google Scholar]
- 100.Filippatos GS, Tsilias K, Venetsanou K, et al. Leptin serum levels in cachectic heart failure patient. Relationship with tumor necrosis factor-α system. International Journal of Cardiology. 2000;76(2-3):117–122. doi: 10.1016/s0167-5273(00)00397-1. [DOI] [PubMed] [Google Scholar]
- 101.Murdoch DR, Rooney E, Dargie HJ, Shapiro D, Morton JJ, McMurray JJV. Inappropriately low plasma leptin concentration in the cachexia associated with chronic heart failure. Heart. 1999;82(3):352–356. doi: 10.1136/hrt.82.3.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Toth MJ, Gottlieb SS, Fisher ML, Ryan AS, Nicklas BJ, Poehlman ET. Plasma leptin concentrations and energy expenditure in heart failure patients. Metabolism. 1997;46(4):450–453. doi: 10.1016/s0026-0495(97)90065-2. [DOI] [PubMed] [Google Scholar]
- 103.Paolisso G, Rizzo MR, Mazziotti G, et al. Lack of association between changes in plasma leptin concentration and in food intake during the menstrual cycle. European Journal of Clinical Investigation. 1999;29(6):490–495. doi: 10.1046/j.1365-2362.1999.00488.x. [DOI] [PubMed] [Google Scholar]
- 104.Nickola MW, Wold LE, Colligan PB, Wang GJ, Samson WK, Ren J. Leptin attenuates cardiac contraction in rat ventricular myocytes role of NO. Hypertension. 2000;36(4):501–505. doi: 10.1161/01.hyp.36.4.501. [DOI] [PubMed] [Google Scholar]
- 105.McGaffin KR, Moravec CS, McTiernan CF. Leptin signaling in the failing and mechanically unloaded human heart. Circulation. 2009;2(6):676–683. doi: 10.1161/CIRCHEARTFAILURE.109.869909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schulze PC, Kratzsch J, Linke A, et al. Elevated serum levels of leptin and soluble leptin receptor in patients with advanced chronic heart failure. European Journal of Heart Failure. 2003;5(1):33–40. doi: 10.1016/s1388-9842(02)00177-0. [DOI] [PubMed] [Google Scholar]
- 107.Leyva F, Anker SD, Egerer K, Stevenson JC, Kox WJ, Coats AJS. Hyperleptinaemia in chronic heart failure. Relationships with insulin. European Heart Journal. 1998;19(10):1547–1551. doi: 10.1053/euhj.1998.1045. [DOI] [PubMed] [Google Scholar]
- 108.Tsutamoto T, Hisanaga T, Wada A, et al. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. Journal of the American College of Cardiology. 1998;31(2):391–398. doi: 10.1016/s0735-1097(97)00494-4. [DOI] [PubMed] [Google Scholar]
- 109.Sharma V, McNeill JH. The emerging roles of leptin and ghrelin in cardiovascular physiology and pathophysiology. Current Vascular Pharmacology. 2005;3(2):169–180. doi: 10.2174/1570161053586868. [DOI] [PubMed] [Google Scholar]
- 110.Haynes WG, Sivitz WI, Morgan DA, Walsh SA, Mark AL. Sympathetic and cardiorenal actions of leptin. Hypertension. 1997;30(3):619–623. doi: 10.1161/01.hyp.30.3.619. [DOI] [PubMed] [Google Scholar]
- 111.Haynes WG, Morgan DA, Walsh SA, Sivitz WI, Mark AL. Cardiovascular consequences of obesity: role of leptin. Clinical and Experimental Pharmacology and Physiology. 1998;25(1):65–69. doi: 10.1111/j.1440-1681.1998.tb02147.x. [DOI] [PubMed] [Google Scholar]
- 112.Lembo G, Vecchione C, Fratta L, et al. Leptin induces direct vasodilation through distinct endothelial mechanisms. Diabetes. 2000;49(2):293–297. doi: 10.2337/diabetes.49.2.293. [DOI] [PubMed] [Google Scholar]
- 113.Anker SD, Von Haehling S. Inflammatory mediators in chronic heart failure: an overview. Heart. 2004;90(4):464–470. doi: 10.1136/hrt.2002.007005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ferrari R, Bachetti T, Confortini R, et al. Tumor necrosis factor soluble receptors in patients with various degrees of congestive heart failure. Circulation. 1995;92(6):1479–1486. doi: 10.1161/01.cir.92.6.1479. [DOI] [PubMed] [Google Scholar]
- 115.Rauchhaus M, Doehner W, Francis DP, et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000;102(25):3060–3067. doi: 10.1161/01.cir.102.25.3060. [DOI] [PubMed] [Google Scholar]
- 116.Kirchgessner TG, Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Tumor necrosis factor-α contributes to obesity-related hyperleptinemia by regulating leptin release from adipocytes. Journal of Clinical Investigation. 1997;100(11):2777–2782. doi: 10.1172/JCI119824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kovačić D, Marinšek M, Gobec L, Lainščak M, Podbregar M. Effect of selective and non-selective β-blockers on body weight, insulin resistance and leptin concentration in chronic heart failure. Clinical Research in Cardiology. 2008;97(1):24–31. doi: 10.1007/s00392-007-0571-3. [DOI] [PubMed] [Google Scholar]
- 118.Schutz Y, Woringer V. Obesity in switzerland: a critical assessment of prevalence in children and adults. International Journal of Obesity. 2002;26(supplement 2):S3–S11. doi: 10.1038/sj.ijo.0802122. [DOI] [PubMed] [Google Scholar]
- 119.Sridhar MK, Carter R, Lean MEJ, Banham SW. Resting energy expenditure and nutritional state of patients with increased oxygen cost of breathing due to emphysema, scoliosis and thoracoplasty. Thorax. 1994;49(8):781–785. doi: 10.1136/thx.49.8.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hugli O, Schutz Y, Fitting JW. The cost of breathing in stable chronic obstructive pulmonary disease. Clinical Science. 1995;89(6):625–632. doi: 10.1042/cs0890625. [DOI] [PubMed] [Google Scholar]
- 121.Di Francia M, Barbier D, Mege JL, Orehek J. Tumor necrosis factor-alpha levels and weight loss in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 1994;150(5):1453–1455. doi: 10.1164/ajrccm.150.5.7952575. [DOI] [PubMed] [Google Scholar]
- 122.De Godoy I, Donahoe M, Calhoun WJ, Mancino J, Rogers RM. Elevated TNF-α production by peripheral blood monocytes of weight-losing COPD patients. American Journal of Respiratory and Critical Care Medicine. 1996;153(2):633–637. doi: 10.1164/ajrccm.153.2.8564110. [DOI] [PubMed] [Google Scholar]
- 123.Sarraf P, Frederich RC, Turner EM, et al. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. Journal of Experimental Medicine. 1997;185(1):171–175. doi: 10.1084/jem.185.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zumbach MS, Boehme MWJ, Wahl P, Stremmel W, Ziegler R, Nawroth PP. Tumor necrosis factor increases serum leptin levels in humans. Journal of Clinical Endocrinology and Metabolism. 1997;82(12):4080–4082. doi: 10.1210/jcem.82.12.4408. [DOI] [PubMed] [Google Scholar]
- 125.Takabatake N, Nakamura H, Abe S, et al. Circulating leptin in patients with chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine. 1999;159(4):1215–1219. doi: 10.1164/ajrccm.159.4.9806134. [DOI] [PubMed] [Google Scholar]
- 126.Takabatake N, Nakamura H, Minamihaba O, et al. A novel pathophysiologic phenomenon in cachexic patients with chronic obstructive pulmonary disease: the relationship between the circadian rhythm of circulating leptin and the very low-frequency component of heart rate variability. American Journal of Respiratory and Critical Care Medicine. 2001;163(6):1314–1319. doi: 10.1164/ajrccm.163.6.2004175. [DOI] [PubMed] [Google Scholar]
- 127.O’Donnell CP, Schaub CD, Haines AS, et al. Leptin prevents respiratory depression in obesity. American Journal of Respiratory and Critical Care Medicine. 1999;159(5):1477–1484. doi: 10.1164/ajrccm.159.5.9809025. [DOI] [PubMed] [Google Scholar]
- 128.Tankersley CG, O’Donnell C, Daood MJ, et al. Leptin attenuates respiratory complications associated with the obese phenotype. Journal of Applied Physiology. 1998;85(6):2261–2269. doi: 10.1152/jappl.1998.85.6.2261. [DOI] [PubMed] [Google Scholar]
- 129.Groeben H, Meier S, Brown RH, O’Donnell CP, Mitzner W, Tankersley CG. The effect of leptin on the ventilatory response to hyperoxia. Experimental Lung Research. 2004;30(7):559–570. doi: 10.1080/01902140490489144. [DOI] [PubMed] [Google Scholar]
- 130.Tankersley C, Kleeberger S, Russ B, Schwartz A, Smith P. Modified control of breathing in genetically obese (ob/ob) mice. Journal of Applied Physiology. 1996;81(2):716–723. doi: 10.1152/jappl.1996.81.2.716. [DOI] [PubMed] [Google Scholar]
- 131.Grosfeld A, André J, Mouzon SH-D, Berra E, Pouysségur J, Guerre-Millo M. Hypoxia-inducible factor 1 transactivates the human leptin gene promoter. Journal of Biological Chemistry. 2002;277(45):42953–42957. doi: 10.1074/jbc.M206775200. [DOI] [PubMed] [Google Scholar]
- 132.Budweiser S, Heinemann F, Meyer K, Wild PJ, Pfeifer M. Weight gain in cachectic COPD patients receiving noninvasive positive-pressure ventilation. Respiratory Care. 2006;51(2):126–132. [PubMed] [Google Scholar]
- 133.Tracey KJ. The inflammatory reflex. Nature. 2002;420(6917):853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- 134.Mak RH, Cheung W, Cone RD, Marks DL. Leptin and inflammation-associated cachexia in chronic kidney disease. Kidney International. 2006;69(5):794–797. doi: 10.1038/sj.ki.5000182. [DOI] [PubMed] [Google Scholar]
- 135.Cheung WW, Paik KH, Mak RH. Inflammation and cachexia in chronic kidney disease. Pediatric Nephrology. 2010;25(4):711–724. doi: 10.1007/s00467-009-1427-z. [DOI] [PubMed] [Google Scholar]
- 136.Scholze A, Rattensperger D, Zidek W, Tepel M. Low serum leptin predicts mortality in patients with chronic kidney disease stage 5. Obesity. 2007;15(6):1617–1622. doi: 10.1038/oby.2007.191. [DOI] [PubMed] [Google Scholar]
- 137.Beberashvili I, Sinuani I, Azar A, et al. Longitudinal study of leptin levels in chronic hemodialysis patients. Nutrition Journal. 2011;10, article 68(1) doi: 10.1186/1475-2891-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Stenvinkel P. Leptin—a new hormone of definite interest for the nephrologist. Nephrology Dialysis Transplantation. 1998;13(5):1099–1101. doi: 10.1093/ndt/13.5.1099. [DOI] [PubMed] [Google Scholar]
- 139.Aguilera A, Sánchez-Tomero JA, Selgas R. Brain activation in uremic anorexia. Journal of Renal Nutrition. 2007;17(1):57–61. doi: 10.1053/j.jrn.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 140.Suffredini AF, Fantuzzi G, Badolato R, Oppenheim JJ, O’Grady NP. New insights into the biology of the acute phase response. Journal of Clinical Immunology. 1999;19(4):203–214. doi: 10.1023/a:1020563913045. [DOI] [PubMed] [Google Scholar]
- 141.Pecoits-Filho R, Sylvestre LC, Stenvinkel P. Chronic kidney disease and inflammation in pediatric patients: from bench to playground. Pediatric Nephrology. 2005;20(6):714–720. doi: 10.1007/s00467-005-1891-z. [DOI] [PubMed] [Google Scholar]
- 142.Kalantar-Zadeh K, Stenvinkel P, Pillon L, Kopple JD. Inflammation and nutrition in renal insufficiency. Advances in Renal Replacement Therapy. 2003;10(3):155–169. doi: 10.1053/j.arrt.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 143.Mak RH, Cheung W. Energy homeostasis and cachexia in chronic kidney disease. Pediatric Nephrology. 2006;21(12):1807–1814. doi: 10.1007/s00467-006-0194-3. [DOI] [PubMed] [Google Scholar]
- 144.Mak RH, Cheung W. Adipokines and gut hormones in end-stage renal disease. Peritoneal Dialysis International. 2007;27(supplement 2):S298–S302. [PubMed] [Google Scholar]
- 145.Cheung W, Yu PX, Little BM, Cone RD, Marks DL, Mak RH. Role of leptin and melanocortin signaling in uremia-associated cachexia. Journal of Clinical Investigation. 2005;115(6):1659–1665. doi: 10.1172/JCI22521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Scarpace PJ, Matheny M, Moore RL, Tümer N. Impaired leptin responsiveness in aged rats. Diabetes. 2000;49(3):431–435. doi: 10.2337/diabetes.49.3.431. [DOI] [PubMed] [Google Scholar]
- 147.Wolden-Hanson T. Mechanisms of the anorexia of aging in the Brown Norway rat. Physiology and Behavior. 2006;88(3):267–276. doi: 10.1016/j.physbeh.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 148.Shek EW, Scarpace PJ. Resistance to the anorexic and thermogenic effects of centrally administrated leptin in obese aged rats. Regulatory Peptides. 2000;92(1–3):65–71. doi: 10.1016/s0167-0115(00)00151-8. [DOI] [PubMed] [Google Scholar]
- 149.Gruenewald DA, Naai MA, Marck BT, Matsumoto AM. Age-related decrease in neuropeptide-Y gene expression in the arcuate nucleus of the male rat brain is independent of testicular feedback. Endocrinology. 1994;134(6):2383–2389. doi: 10.1210/endo.134.6.8194464. [DOI] [PubMed] [Google Scholar]
- 150.Horrillo D, Sierra J, Arribas C, et al. Age-associated development of inflammation in Wistar rats: effects of caloric restriction. Archives of Physiology and Biochemistry. 2011;117(3):140–150. doi: 10.3109/13813455.2011.577435. [DOI] [PubMed] [Google Scholar]
- 151.Carrero JJ, Nakashima A, Qureshi AR, et al. Protein-energy wasting modifies the association of ghrelin with inflammation, leptin, and mortality in hemodialysis patients. Kidney International. 2011;79(7):749–756. doi: 10.1038/ki.2010.487. [DOI] [PubMed] [Google Scholar]
- 152.van Tellingen A, Grooteman MPC, Schoorl M, et al. Enhanced long-term reduction of plasma leptin concentrations by super-flux polysulfone dialysers. Nephrology Dialysis Transplantation. 2004;19(5):1198–1203. doi: 10.1093/ndt/gfh122. [DOI] [PubMed] [Google Scholar]
- 153.Javor ED, Cochran EK, Musso C, Young JR, DePaoli AM, Gorden P. Long-term efficacy of leptin replacement in patients with generalized lipodystrophy. Diabetes. 2005;54(7):1994–2002. doi: 10.2337/diabetes.54.7.1994. [DOI] [PubMed] [Google Scholar]
- 154.Newman AB, Yanez D, Harris T, Duxbury A, Enright PL, Fried LP. Weight change in old age and its association with mortality. Journal of the American Geriatrics Society. 2001;49(10):1309–1318. doi: 10.1046/j.1532-5415.2001.49258.x. [DOI] [PubMed] [Google Scholar]
- 155.Hauser G, Neumann M. Aging with quality of life—a challenge for society. Journal of Physiology and Pharmacology. 2005;56(supplement 2):35–48. [PubMed] [Google Scholar]
- 156.Kmiec Z. Central regulation of food intake in ageing. Journal of Physiology and Pharmacology. 2006;57(supplement 6):7–16. [PubMed] [Google Scholar]
- 157.Fernández-Galaz C, Fernández-Agulló T, Campoy F, et al. Decreased leptin uptake in hypothalamic nuclei with ageing in Wistar rats. Journal of Endocrinology. 2001;171(1):23–32. doi: 10.1677/joe.0.1710023. [DOI] [PubMed] [Google Scholar]
- 158.Gruenewald DA, Marck BT, Matsumoto AM. Fasting-induced increases in food intake and neuropeptide Y gene expression are attenuated in aging male brown Norway rats. Endocrinology. 1996;137(10):4460–4467. doi: 10.1210/endo.137.10.8828508. [DOI] [PubMed] [Google Scholar]
- 159.Wolden-Hanson T, Marck BT, Matsumoto AM. Blunted hypothalamic neuropeptide gene expression in response to fasting, but preservation of feeding responses to AgRP in aging male Brown Norway rats. American Journal of Physiology. 2004;287(1):R138–R146. doi: 10.1152/ajpregu.00465.2003. [DOI] [PubMed] [Google Scholar]
- 160.Bouillanne O, Dupont-Belmont C, Hay P, Hamon-Vilcot B, Cynober L, Aussel C. Fat mass protects hospitalized elderly persons against morbidity and mortality. American Journal of Clinical Nutrition. 2009;90(3):505–510. doi: 10.3945/ajcn.2009.27819. [DOI] [PubMed] [Google Scholar]
- 161.Arai Y, Takayama M, Abe Y, Hirose N. Adipokines and aging. Journal of Atherosclerosis and Thrombosis. 2011;18(7):545–550. doi: 10.5551/jat.7039. [DOI] [PubMed] [Google Scholar]
- 162.Zamboni M, Zoico E, Fantin F, et al. Relation between leptin and the metabolic syndrome in elderly women. Journals of Gerontology. 2004;59(4):396–400. doi: 10.1093/gerona/59.4.m396. [DOI] [PubMed] [Google Scholar]
- 163.Enriori PJ, Evans AE, Sinnayah P, et al. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metabolism. 2007;5(3):181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 164.Hubbard RE, O MS, Calver BL, Woodhouse KW. Nutrition, inflammation, and leptin levels in aging and frailty. Journal of the American Geriatrics Society. 2008;56(2):279–284. doi: 10.1111/j.1532-5415.2007.01548.x. [DOI] [PubMed] [Google Scholar]