

Abstract
Sarcopenia, the age-related loss of skeletal muscle mass, is characterized by a deterioration of muscle quantity and quality leading to a gradual slowing of movement, a decline in strength and power, increased risk of fall-related injury, and, often, frailty. Since sarcopenia is largely attributed to various molecular mediators affecting fiber size, mitochondrial homeostasis, and apoptosis, the mechanisms responsible for these deleterious changes present numerous therapeutic targets for drug discovery. Resistance training combined with amino acid-containing supplements is often utilized to prevent age-related muscle wasting and weakness. In this review, we summarize more recent therapeutic strategies (myostatin or proteasome inhibition, supplementation with eicosapentaenoic acid (EPA) or ursolic acid, etc.) for counteracting sarcopenia. Myostatin inhibitor is the most advanced research with a Phase I/II trial in muscular dystrophy but does not try the possibility for attenuating sarcopenia. EPA and ursolic acid seem to be effective as therapeutic agents, because they attenuate the degenerative symptoms of muscular dystrophy and cachexic muscle. The activation of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) in skeletal muscle by exercise and/or unknown supplementation would be an intriguing approach to attenuating sarcopenia. In contrast, muscle loss with age may not be influenced positively by treatment with a proteasome inhibitor or antioxidant.
1. Introduction
Skeletal muscle contractions power human body movements and are essential for maintaining stability. Skeletal muscle tissue accounts for almost half of the human body mass and, in addition to its power-generating role, is a crucial factor in maintaining homeostasis. Given its central role in human mobility and metabolic function, any deterioration in the contractile, material, and metabolic properties of skeletal muscle has an extremely important effect on human health. Aging is associated with a progressive decline of muscle mass, quality, and strength, a condition known as sarcopenia [1]. The term sarcopenia, coined by I. H. Rosenberg, originates from the Greek words sarx (flesh) and penia (loss). Although this term is applied clinically to denote loss of muscle mass, it is often used to describe both a set of cellular processes (denervation, mitochondrial dysfunction, inflammatory and hormonal changes) and a set of outcomes such as decreased muscle strength, decreased mobility and function [2], increased fatigue, a greater risk of falls [3], and reduced energy needs [4]. In addition, reduced muscle mass in aged individuals has been associated with decreased survival rates following critical illness [5]. Estimates of the prevalence of sarcopenia range from 13% to 24% in adults over 60 years of age to more than 50% in persons aged 80 and older [2]. The estimated direct healthcare costs attributable to sarcopenia in the United States in 2000 were $18.5 billion ($10.8 billion in men and $7.7 billion in women), which represented about 1.5% of total healthcare expenditures for that year [6]. Therefore, age-related losses in skeletal muscle mass and function present an extremely important current and future public health issue.
Lean muscle mass generally contributes up to ~50% of total body weight in young adults but declines with aging to be 25% at 75–80 yr old [7, 8]. The loss of muscle mass is typically offset by gains in fat mass. The loss of muscle mass is most notable in the lower limb muscle groups, with the cross-sectional area of the vastus lateralis being reduced by as much as 40% between the age of 20 and 80 yr [9]. On a muscle fiber level, sarcopenia is characterized by specific type II muscle fiber atrophy, fiber necrosis, and fiber-type grouping [9–13]. In elderly men, Verdijk et al. [12] showed a reduction in type II muscle fiber satellite cell content with aging. Although various investigators support such an age-related decrease in the number of satellite cells [12–17], some reports [18–20] indicate no such change. In contrast, most studies point to an age-dependent reduction in muscle-regenerative capacity due to reduced satellite cell proliferation and differentiation.
Several possible mechanisms for age-related muscle atrophy have been described; however, the precise contribution of each is unknown. Age-related muscle loss is a result of reductions in the size and number of muscle fibers [21] possibly due to a multifactorial process that involves physical activity, nutritional intake, oxidative stress, and hormonal changes [3, 22]. The specific contribution of each of these factors is unknown, but there is emerging evidence that the disruption of several positive regulators (Akt and serum response factor) of muscle hypertrophy with age is an important feature in the progression of sarcopenia [23–25]. In contrast, many investigators have failed to demonstrate an age-related enhancement in levels of common negative regulators [atrophy gene-1 (Atrogin-1), myostatin, and calpain] in senescent mammalian muscles [24, 25].
Resistance training combined with amino acid-containing supplements is effective candidate to prevent age-related muscle wasting and weakness [24–26]. In particular, sarcopenia has been most attenuated by treatment with many essential amino acids plus high-amount leucine [24–26]. In addition, many researchers have focused on inhibiting myostatin for treating various muscle disorders such as muscular dystrophy, cachexia, and sarcopenia [27, 28]. Furthermore, more recent studies have indicated a possible application of new supplements to prevent muscle atrophy [29, 30]. This review aims to address several novel strategies for inhibiting the muscle wasting in particular sarcopenia.
2. Myostatin Inhibition
Growth and differentiation factor 8, otherwise known as myostatin, was first discovered during screening for members of a novel transforming growth factor-β (TGF-β ) superfamily and shown to act as a potent negative regulator of muscle growth [31, 32]. Studies indicate that myostatin inhibits cell-cycle progression and levels of myogenic regulatory factors, thereby controlling myoblastic proliferation and differentiation during developmental myogenesis [32–35]. Mutations in myostatin can lead to massive hypertrophy and/or hyperplasia in developing animals, as evidenced by knockout experiments in mice and by the phenotype seen in myostatin-null cattle [36] and humans [37]. Myostatin binds to and signals through a combination of Activin IIA/B receptors (ActRIIA/IIB) on the cell membrane; however, it has higher affinity for ActRIIB. On binding ActRIIB, myostatin forms a complex with a second surface type I receptor, either activin receptor-like kinase (ALK4 or ActRIB) or ALK5, to stimulate the phosphorylation of receptor Smad (Rsmad), and Smad2/3 transcription factors in the cytoplasm. Then Smad2/3 translocate and modulate nuclear gene transcription such as MyoD [33] via a TGF-β-like mechanism. In contrast, forkhead box O (FOXO) 1 and Smad2 appear to control the differentiation of C2C12 myoblasts by regulating myostatin mRNA and its promoters [38].
Studies measuring myostatin levels during aging have yielded conflicting results such as marked increases in humans at the mRNA and protein levels [39], no change in mice at the protein level [40], and a decrease in rats at the mRNA level [41]. The functional role of myostatin in aged mammalian muscle may be revealed by further descriptive analysis using other methods (ex. immunofluorescence) and examining the adaptive changes in downstream modulators (ex. ActRIIB, Smad3) of myostatin signaling.
Many researchers have focused on inhibiting myostatin for treating various muscle disorders. The use of neutralizing antibodies to myostatin improved muscle disorders in rodent models of Duchenne muscular dystrophy (DMD), limb girdle muscular dystrophy 2F (Sgcg−/−), and amyotrophic lateral sclerosis (SOD1G93A transgenic mouse) [27, 28, 42, 43]. Indeed, myostatin inhibition using MYO-029 was tested in a prospective, randomized, and US phase I/II trial in 116 adults with muscular dystrophy such as Becker muscular dystrophy, facioscapulohumeral muscular dystrophy, and limb-girdle muscular dystrophy [44]. On the other hand, inhibiting myostatin to counteract sarcopenia has also been investigated only in animals. A lack of myostatin caused by gene manipulation increased the number of satellite cells and enlarged the cross-sectional area of predominant type IIB/X fibers in tibialis anterior muscles of mice [45]. In addition, these myostatin-null mice showed prominent regenerative potential including accelerated fiber remodeling after an injection of notexin [45]. LeBrasseur et al. [46] reported several positive effects of 4 weeks of treatment with PF-354 (24 mg/Kg), a drug for myostatin inhibition, in aged mice. They showed that PF-354-treated mice exhibited significantly greater muscle mass (by 12%), and increased performance such as treadmill time, distance to exhaustion, and habitual activity. Furthermore, PF-354-treated mice exhibited decreased levels of phosphorylated Smad3 and muscle ring-finger protein 1 (MuRF1) in aged muscle. More recently, Murphy et al. [47] showed, by way of once weekly injections, that a lower dose of PF-354 (10 mg/Kg) significantly increased the fiber cross-sectional area (by 12%) and in situ force of tibialis anterior muscles (by 35%) of aged mice (21-mo-old). In addition, this form of treatment reduced markers of apoptosis by 56% and reduced caspase3 mRNA levels by 65%. Blocking myostatin enhances muscle protein synthesis [48] by potentially relieving the inhibition normally imposed on the Akt/mammalian target of rapamycin- (mTOR) signaling pathway by myostatin [49]. The blockade may also attenuate muscle protein degradation by inhibiting the ubiquitin-proteasome system, which is controlled, in part, by Akt [50, 51] although the mechanism involved has not been demonstrated. In contrast, a microarray analysis of the skeletal muscle of myostatin knockout mice showed an increased expression of antiapoptotic genes compared with that in control mice [51]. These lines of evidence clearly highlight the therapeutic potential of antibody-directed inhibition of myostatin for treating sarcopenia by inhibiting protein degradation and/or apoptosis.
3. Ursolic Acid
A water-insoluble pentacyclic triterpenoid, ursolic acid is the major waxy component in apple peels [52]. It is also found in many other edible plants. Interestingly, because it exerts beneficial effects in animal models of diabetes and hyperlipidemia [53, 54], ursolic acid is thought to be the active component in a variety of folkloric antidiabetic herbal medicines [53, 55]. As predicted by connectivity mapping, Kunkel et al. [29] found that ursolic acid reduced skeletal muscle atrophy in the setting of two-distinct atrophy-inducing stresses (fasting and muscle denervation). A major strength of the connectivity map is that it takes into account positive and negative changes in mRNA expression that together constitute an authentic mRNA expression signature. Thus, by querying the connective map with signatures of muscle atrophy, Kunkel et al. [29] were, in effect, querying with the reciprocal signature of muscle atrophy but also induced muscle hypertrophy.
Ursolic acid might increase muscle mass by inhibiting atrophy-associated skeletal muscle gene expression. Indeed, Kunkel et al. [29] found that acute ursolic acid treatment of fasted mice reduced Atrogin-1 and MuRF1 mRNA levels in association with reduced muscle atrophy. Similarly, chronic ursolic acid treatment of unstressed mice reduced Atrogin-1 and MuRF1 expression and induced muscle hypertrophy. Although ursolic acid increased skeletal muscle Akt phosphorylation in vivo, the experiments could not determine if it acted directly on skeletal muscle, how quickly it acted, and if the effect required insulin-like growth factor-I (IGF-I) or insulin, which are always present in healthy animals, even during fasting. To address these issues, Kunkel et al. [29] studied serum-starved skeletal myotubes and found that ursolic acid rapidly stimulated IGF-I receptor and insulin receptor activity, but only if IGF-I or insulin was also present. Taken together, their data suggest that ursolic acid first enhances the capacity of preexisting IGF-I and insulin to activate skeletal muscle IGF-I receptors and insulin receptors, respectively. Importantly, ursolic acid alone was not sufficient to increase phosphorylation of the IGF-I receptor or insulin receptor. Rather, its effects also required IGF-I and insulin, respectively. This suggests that ursolic acid either facilitates hormone-mediated receptor autophosphorylation or inhibits receptor dephosphorylation. The latter possibility is supported by previous in vitro data showing that ursolic acid directly inhibits PTP1B [56], a tyrosine phosphatase that dephosphorylates (inactivates) the IGF-I and insulin receptors [57]. Further research is needed to elucidate the effect of supplementation with ursolic acid in skeletal muscle and to attenuate muscle wasting (ex. sarcopenia).
4. Eicosapentaenoic Acid
Eicosapentaenoic acid (EPA) is a 20-carbon omega (n)-3 polyunsaturated fatty acid with anti-inflammatory properties, which is synthesized from ingested alpha-linolenic acid or is consumed in fish and fish oil such as cod liver, sardine, and salmon oil [58]. There is no established Dietary Reference Intake for n-3 fatty acids; yet, adequate intake (AI) is set at 1.6 and 1.1 g/d for men and women, respectively. While intake in the United States occurs at levels much lower than the proposed AI and no signs of deficiency are observed, the AI is proposed to provide optimal health benefits associated with consuming omega-3 polyunsaturated fatty acids [59]. Several clinical trials have reported potential health benefits of omega-3 polyunsaturated fatty acids in many diseases, including cardiovascular diseases [60], epilepsy, inflammatory bowel disease, exercise-trained subjects [61], and cancer-associated cachexia [62]. In particular, the administration of omega-3 fatty acids and EPA capsules or supplements with EPA has been shown to be associated with weight stabilization, gains in lean body mass, and improvements in quality of life markers in weight-losing patients with advanced pancreatic cancer. In addition, EPA has also been shown to inhibit the proinflammatory transcription factor nuclear factor kappaB (NF-κB) [62, 63], to reduce tumor necrosis factor-α (TNF-α) production by macrophages [64] and to prevent the damaging effects of TNF-α during skeletal muscle differentiation in vitro [65]. Furthermore, short-term treatment with EPA (16 day, 100 mg/Kg) attenuates the muscle degeneration of mdx mice, a model of DMD [66]. EPA treatment decreased creatine kinase levels and attenuated myonecrosis (decrease in Evans-blue dye-positive fibers and a concomitant increase in peripheral nucleated fibers), and reduced the levels of TNF-α.
Some evidence suggests omega-3 polyunsaturated fatty acids to be also a potentially useful therapeutic agent for the treatment and prevention of sarcopenia. In a more recent study [30], sixteen healthy, older adults have been randomly assigned to receive either omega-3 fatty acids or corn oil for 8 week. In their study, the rate of muscle protein synthesis and the phosphorylation of key elements of the anabolic-signaling pathway were evaluated in three different conditions. Smith et al. [30] found that omega-3 fatty acid supplementation had no effect on the basal rate of muscle protein synthesis but augmented the hyperaminoacidemia- hyperinsulinemia-induced increase in the rate of muscle protein synthesis probably due to a greater increase in muscle p70S6K Thr389 phosphorylation.
5. Angiotensin-Converting Enzyme Inhibitors
Angiotensin-converting enzyme (ACE) inhibitors have long been used as a treatment in primary and secondary prevention in cardiovascular disease as well as secondary stroke prevention. It has now been suggested that ACE inhibitors may have a beneficial effect on skeletal muscle. ACE inhibitors may exert their beneficial effects on skeletal muscles through a number of different mechanisms. ACE inhibitors may improve muscle function through improvements in endothelial function, metabolic function, anti-inflammatory effects, and angiogenesis thereby improving skeletal muscle blood flow. ACE inhibitors can increase mitochondrial numbers and IGF-I levels thereby helping to counter sarcopenia [67–69].
Observational studies have shown that the long-term use of ACE inhibitors was associated with a lower decline in muscle strength and walking speed in older hypertensive people and a greater lower limb lean muscle mass when compared with users of other antihypertensive agents [70]. Several studies have shown that ACE inhibitors improved exercise capacity in both younger and older people with heart failure [70, 71] but caused no improvement in grip strength [72]. Although this could be largely attributed to improvements in cardiac function, skeletal muscle atrophy is also associated with chronic heart failure so the evidence of muscle gains should not be discounted. Few interventional studies using ACE inhibitors for physical function have been undertaken. One study looking at functionally impaired older people without heart failure has shown that ACE inhibitors increase 6-minute walking distance to a degree comparable to that achieved after 6 months of exercise training [73]. Another found that ACE inhibitors increased exercise time in older hypertensive men [74]. However, a study comparing the effects of nifedipine with ACE inhibitors in older people found no difference between treatments in muscle strength, walking distance, or functional performance [75]. It is possible that frailer subjects with slower walking speeds, who have a tendency to more cardiovascular problems, benefit more. Further evidence is required before recommending ACE inhibitors to counter the effects of sarcopenia. However, ACE inhibitors are associated with cardiovascular benefits, and as older people frequently have underlying cardiovascular problems, these agents are already commonly prescribed.
6. Proteasome Inhibitors
In a variety of conditions such as cancer, diabetes, denervation, uremia, sepsis, disuse, and fasting, skeletal muscles undergo atrophy through degradation of myofibrillar proteins via the ubiquitin-proteasome pathway [76]. Recent advances have asserted that muscle atrophy in these conditions shares a common mechanism in the induction of the muscle-specific E3 ubiquitin ligases Atrogin-1 and MuRF1 [77–79]. Only very indirect measurements (small increases in mRNA levels encoding some components of the ubiquitin-proteasome pathway [80–82] or ubiquitin-conjugate accumulation [83]) in old muscles of rodents or humans suggested a modest activation of this pathway. Atrogin-1 and/or MuRF1 mRNA levels in aged muscle are reportedly unchanged in humans [83, 84], increased in rats [80, 85], or decreased in rats [82, 86, 87]. When even the mRNA expression of these atrogenes increased in sarcopenic muscles, the induction was very limited (1.5–2.5-fold) as compared with other catabolic situations (10-fold). In addition, the major peptidase activities of the proteasome (i.e., the chymotrypsin-like, trypsin-like, and caspase-like activities) were always reduced (as reported in other tissues [88]) or unchanged with aging [78, 81, 88, 89]. Altogether, these observations clearly suggest that activation of the ubiquitin-proteasome system contributed little to the establishment of sarcopenia in accordance with the very slow muscle mass erosion.
There are several chemical classes of compounds that inhibit proteasomal activity, including peptide analogs of substrates with different C-terminal groups, such as aldehydes, epoxyketones, boronic acids, and vinyl sulfones [90]. A selective boronic acid proteasome inhibitor, Velcade (also known as PS-341 and bortezomib), directly inhibits the proteasome complex without direct effects on ubiquitination. Velcade is well distributed in the body and does not cross the blood-brain barrier. In addition to being useful research tools for dissecting the roles of the proteasome, proteasome inhibitors have potential applications in biotechnology and medicine. For example, through their ability to block the activation of NF-κB, proteasome inhibitors can dramatically reduce in vitro and in vivo the production of inflammatory mediators as well as of various leukocyte adhesion molecules, which play a crucial role in many diseases. Indeed, Velcade is orally active and is presently approved by the Food and Drug Administration and the European Medicines Agency and well tolerated for treating multiple myeloma [91, 92]. Bonuccelli et al. [93] had indicated that Velcade, once injected locally into the gastrocnemius muscles of mdx mice, could upregulate the expression and membrane localization of dystrophin and members of the dystrophin-glycoprotein complex. Gazzerro et al. [94] suggested that treatment with Velcade (0.8 mg/Kg) over a 2-week period reduced muscle degeneration and necrotic features in mdx muscle fibers, as evaluated with Evans blue dye. In addition, they observed many myotubes and/or immature myofibers expressing embryonic myosin heavy chain in mdx muscle after Velcade administration probably due to the upregulation of several myogenic differentiating modulators (MyoD and Myf-5). These effects of Velcade on muscle degeneration would differ dependent on muscle-fiber type. Beehler et al. [95] demonstrated selective attenuation on treatment with Velcade (3 mg/Kg, 7 days) for atrophy of denervated slow-type muscle (soleus), but not fast-type muscle (EDL) of rats. In contrast, MG-132 exerts an inhibitory effect on both the proteasome and the calpain system. More recently, Gazzerro et al. [94] have clearly demonstrated that MG-132 increased dystrophin, alpha-sarcoglycan, and beta-dystroglycan protein levels in explants from Becker muscular dystrophy patients, whereas it increased the proteins of the dystrophin glycoprotein complex in DMD cases. Strangely, there is no rodent study examining the effect of these proteasome inhibitors to prevent muscle atrophy with aging. As indicated previously [80, 82, 88, 89], almost no studies demonstrated an enhancement of proteasome-linked modulators for protein degradation in sarcopenic mammalian muscles. Proteasome inhibitors may not act to attenuate muscle wasting in cases of sarcopenia.
7. Cyclophilin Inhibitor (Debio-025)
Ca2+ overload is known to cause cellular necrosis by directly inducing the opening of the mitochondrial permeability transition (MPT) pore [96, 97]. The MPT pore spans the inner and outer membranes of the mitochondria and, when opened for prolonged periods of time, leads to loss of ATP generation, swelling, rupture, and induction of cell death [96, 97]. Cyclophilin D is a mitochondrial matrix prolyl cis-trans isomerase that directly regulates calcium—and reactive oxygen species-dependent MPT and cellular necrosis. Indeed, mice lacking Ppif (the gene-encoding cyclophilin D) show protection from necrotic cell death in the brain and heart after ischemic injury, and mitochondria isolated from these mice are resistant to calcium-induced swelling [98, 99]. Additionally, genetic deletion of Ppif attenuated various dystrophic symptoms (fiber atrophy, fiber loss, invasion by inflammatory cells, and swollen mitochondria) of mice lacking δ-sarcoglycan and the α2-chain of laminin-2 [100]. Millay et al. [100] demonstrated that the subcutaneous injection of Debio-025, a potent inhibitor of the cyclophilin family, improved calcium overload-induced swelling of mitochondria and reduced manifestations of necrotic disease such as fibrosis and central nuclei, in mdx mice, a model of DMD. In addition, treatment with Debio-025 prevented mitochondrial dysfunction and normalized the apoptotic rates and ultrastructural lesions of myopathic Col6a1−/− mice, a model of human Ullrich congenital muscular dystrophy and Bethlem myopathy [101]. More recently, orally administered Debio-025 reduced creatine kinase blood levels and improved grip strength in mdx mice after 6 weeks of treatment [102]. This effect on muscular dystrophy was greater than that of prednisone, currently the standard for treatment of DMD [103, 104]. However, it had not been examined until now whether Debio-025 also has a therapeutic effect on the loss and/or atrophy of muscle fiber with aging in rodents as well as humans. Since there are many symptoms in common between muscular dystrophy and sarcopenia, treatment with Debio-025 may counteract sarcopenic symptoms.
8. PGC-1α
Although the mechanisms by which calorie restriction (CR, 30–40%) delays the aging process remain to be fully elucidated, CR is intricately involved in regulating cellular and systemic redox status and in modulating the expression of genes related to macromolecule and organelle turnover, energy metabolism, and cell death and survival [109–111]. Several studies indicate the protection of age-related functional decline and loss of muscle fibers by CR [109, 110]. These protective effects are likely attributable to the ability of CR to reduce the incidence of mitochondrial abnormalities (mitochondrial proton leak), attenuate oxidative stress [reactive oxidative species (ROS) generation], and counteract the age-related increases in proapoptotic signaling in skeletal muscle [109, 112]. Therefore, several lines of evidence suggest mitochondrial involvement in sarcopenia. Therapeutic strategies for sarcopenia like endurance exercise [113] and CR [114] result in increased mitochondrial capacity in the muscle. A key player controlling mitochondrial function is the peroxisome proliferator-activated receptor γ coactivator α (PGC-1α), a master regulator of mitochondrial biogenesis. In skeletal muscle, PGC-1α can also prevent muscle wasting by regulating autophagy [115] and stabilization of the neuromuscular junction program [116] in the context of muscle atrophy during disease. Thereby, PGC-1α links mitochondrial function to muscle integrity [115]. PGC-1α levels in skeletal muscle decrease during aging [116]. The health-promoting effects of increased PGC-1α expression in skeletal muscle have been shown in different mouse models with affected muscle such as DMD [117], denervation-induced atrophy [115], and mitochondrial myopathy [118]. Indeed, Wenz et al. [118] showed that elevated PGC-1α expression in skeletal muscle enhanced oxidative phosphorylation function in a mouse model of mitochondrial myopathy, delaying the onset of the myopathy and markedly prolonging lifespan.
Adipose tissue infiltration of skeletal muscle also increases with age [119, 120]. Sarcopenia may be linked with increased obesity in the elderly [121]. Indeed, persons who are obese and sarcopenic are reported, independent of age, ethnicity, smoking, and comorbidity, to have worse outcomes, including functional impairment, disabilities, and falls, than do those who are nonobese and sarcopenic [122]. Recent work has demonstrated that mitochondrial damage occurs in obese individuals due to enhanced ROS and chronic inflammation caused by increased fatty acid load [123–125]. Specifically, in skeletal muscle, the expression of PGC-1α drives not only mitochondrial biogenesis and the establishment of oxidative myofibers, but also vascularization [126, 127]. It was found that a high-fat diet or fatty acid treatment caused a reduction in the expression of PGC-1α and other mitochondrial genes in skeletal muscle [128], which may be a mechanism through which excess caloric intake impairs skeletal muscle function. A recent study has also demonstrated that transgenic overexpression of PGC-1α in skeletal muscle improved sarcopenia and obesity associated with aging in mice [129]. Endurance training has been shown to upregulate the amount of PGC-1α to elicit mitochondrial biogenesis [130]. The well-known sarcopenia-attenuating effects by endurance training may be attributable to the protection for mitochondrial disorders (apoptosis, oxidative damage, etc.) by the increase of PGC-1α amount. Figure 1 provides an overview of the molecular pathways of muscle hypertrophy and novel strategies for counteracting sarcopenia.
Figure 1.
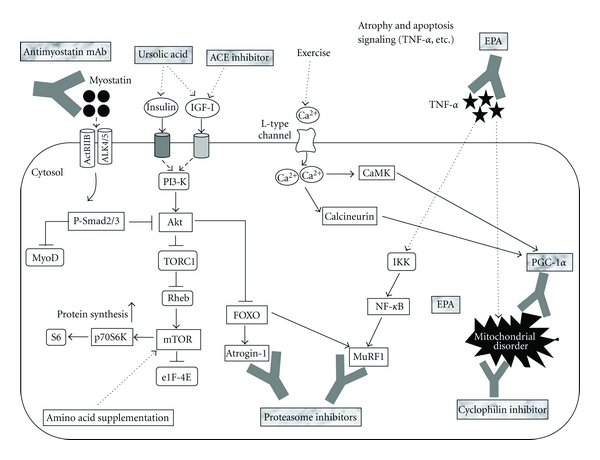
Myostatin signals through the ActRIIB-ALK4/5 heterodimer activate Smad2/3 with blocking of MyoD transactivation in an autoregulatory feedback loop. In addition, Smad3 sequesters MyoD in the cytoplasm to prevent it from entering the nucleus and activating the stem cell population. Recent findings [105, 106] have suggested that myostatin-Smad pathway inhibits protein synthesis probably due to blocking the functional role of Akt. Supplementation with ursolic acid upregulates the amount of IGF-I and insulin and then stimulates protein synthesis by activating Akt/mTOR/p70S6K pathway [29]. Treatment with ACE inhibitor also enhances IGF-I level in muscle. Amino acid supplementation enhances protein synthesis by stimulating mTOR [107]. Akt blocks the nuclear translocation of FOXO to inhibit the expression of Atrogin-1 and MuRF1 and the consequent protein degradation. Proteasome inhibitors combat the ubiquitin-proteasome signaling activated by these atrogenes. In cachexic muscle, supplementation with EPA downregulates the amount of TNF-α and NF-κB [63, 64]. Endurance exercise increases the amount of PGC-1α through calcineurin- or CaMK-dependent signaling [108]. Both activated PGC-1α, and cyclophilin inhibitor protects several mitochondrial disorders (apoptosis, oxidative damage, etc.) elicited by the increase in NF-κB and Bax and/or the decrease in Bcl-2 in senescent muscle. ACE; angiotensin-converting enzyme, ActRIIB; activin receptor IIB, ALK4/5; activin-like kinase 4/5, CaMK; calmodulin kinase, eIF4E; eukaryotic initiation factor 4E, EPA; eicosapentaenoic acid, FOXO; Forkhead box O, IGF-I; insulin-like growth factor-I, IKK; inhibitor of κB kinase, mTOR; mammalian target of rapamycin, MuRF1; muscle ring-finger protein 1, NF-κB; nuclear factor of kappa B, PGC-1α; peroxisome proliferator-activated receptor γ coactivator α, PI3-K; phosphatidylinositol 3-kinase, Rheb; Ras homolog enriched in brain, TNF-α; tumor nectosis factor-α, TORC1; a component of TOR-signaling complex 1.
9. Conclusions and Perspectives
The advances in our understanding of muscle biology that have occurred over the past decade have led to new hopes for pharmacological treatment of muscle wasting. These treatments will be tested in humans in the coming years and offer the possibility of treating sarcopenia/frailty. These treatments should be developed in the setting of appropriate dietary and exercise strategies. Currently, resistance training combined with amino acid-containing supplements would be the best way to prevent age-related muscle wasting and weakness. Supplementation with ursolic acid and EPA seems to be more intriguing candidates combating sarcopenia although systematic and fundamental research in these treatments has not been conducted even in rodent. The well-known sarcopenia-attenuating effects by endurance training may be attributable to the protection for mitochondrial disorders by the increase of PGC-1α amount.
Acknowledgment
This work was supported by a research Grant-in-Aid for Scientific Research C (no. 23500778) from the Ministry of Education, Science, Culture, Sports, Science, and Technology of Japan.
Abbreviations
- ACE:
Angiotensin-converting enzyme
- ActRIIB:
Activin receptor IIB
- AI:
Adequate intake
- ALK:
Activin receptor-like kinase
- Atrogin-1:
Atrophy gene-1
- CR:
Calorie restriction
- DMD:
Duchenne muscular dystrophy
- EPA:
Eicosapentaenoic acid
- FOXO:
Forkhead box O
- IGF-I:
Insulin-like growth factor-I
- MPT:
Mitochondrial permeability transition
- mTOR:
Mammalian target of rapamycin
- NF-κB:
Nuclear factor-kappaB
- PGC-1α:
Peroxisome proliferator-activated receptor γ coactivator 1α
- ROS:
Reactive oxidative species
- TGF-β:
Transforming growth factor-β
- TNF-α:
Tumor necrosis factor-α.
References
- 1.Candow DG, Chilibeck PD. Differences in size, strength, and power of upper and lower body muscle groups in young and older men. Journals of Gerontology Series A: Biological Science Medical Science. 2005;60(2):148–156. doi: 10.1093/gerona/60.2.148. [DOI] [PubMed] [Google Scholar]
- 2.Melton LJ, 3rd, Khosla S, Crowson CS, O'Fallon WM, Riggs BL. Epidemiology of sarcopenia. Journal of the American Geriatrics Society. 2000;48(6):625–630. [PubMed] [Google Scholar]
- 3.Baumgartner RN, Waters DL, Gallagher D, Morley JE, Garry PJ. Predictors of skeletal muscle mass in elderly men and women. Mechanisms of Ageing and Development. 1999;107(2):123–136. doi: 10.1016/s0047-6374(98)00130-4. [DOI] [PubMed] [Google Scholar]
- 4.Poehlnan ET, Toth MJ, Fonong T. Exercise, substrate utilization and energy requirements in the elderly. International Journal of Obesity. 1995;19(4):S93–S96. [PubMed] [Google Scholar]
- 5.Griffiths RD. Muscle mass, survival, and the elderly ICU patient. Nutrition. 1996;12(6):456–458. doi: 10.1016/s0899-9007(96)00141-4. [DOI] [PubMed] [Google Scholar]
- 6.Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R. The healthcare costs of sarcopenia in the United States. Journal of the American Geriatrics Society. 2004;52(1):80–85. doi: 10.1111/j.1532-5415.2004.52014.x. [DOI] [PubMed] [Google Scholar]
- 7.Short KR, Nair KS. The effect of age on protein metabolism. Current Opinion in Clinical Nutrition and Metabolic Care. 2000;3(1):39–44. doi: 10.1097/00075197-200001000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Short KR, Vittone JL, Bigelow JL, Proctor DN, Nair KS. Age and aerobic exercise training effects on whole body and muscle protein metabolism. American Journal of Physiology: Endocrinology and Metabolism. 2004;286(1):E92–E101. doi: 10.1152/ajpendo.00366.2003. [DOI] [PubMed] [Google Scholar]
- 9.Lexell J. Human aging, muscle mass, and fiber type composition. Journals of Gerontology Series A: Biological Science and Medical Science. 1995;50:11–16. doi: 10.1093/gerona/50a.special_issue.11. [DOI] [PubMed] [Google Scholar]
- 10.Larsson L. Morphological and functional characteristics of the ageing skeletal muscle in man. A cross-sectional study. Acta Physiologica Scandinavica, Supplement. 1978;457:1–36. [PubMed] [Google Scholar]
- 11.Larsson L, Sjödin B, Karlsson J. Histochemical and biochemical changes in human skeletal muscle with age in sedentary males age 22-65 years. Acta Physiologica Scandinavica. 1978;103(1):31–39. doi: 10.1111/j.1748-1716.1978.tb06187.x. [DOI] [PubMed] [Google Scholar]
- 12.Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. American Journal of Physiology: Endocrinology and Metabolism. 2007;292(1):E151–E157. doi: 10.1152/ajpendo.00278.2006. [DOI] [PubMed] [Google Scholar]
- 13.Verdijk LB, Gleeson BG, Jonkers RAM, et al. Skeletal muscle hypertrophy following resistance training is accompanied by a fiber type-specific increase in satellite cell content in elderly men. Journals of Gerontology Series A: Biological Science and Medical Sciences. 2009;64(3):332–339. doi: 10.1093/gerona/gln050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brack AS, Bildsoe H, Hughes SM. Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. Journal of Cell Science. 2005;118(20):4813–4821. doi: 10.1242/jcs.02602. [DOI] [PubMed] [Google Scholar]
- 15.Collins CA, Zammit PS, Ruiz AP, Morgan JE, Partridge TA. A population of myogenic stem cells that survives skeletal muscle aging. Stem Cells. 2007;25(4):885–894. doi: 10.1634/stemcells.2006-0372. [DOI] [PubMed] [Google Scholar]
- 16.Day K, Shefer G, Shearer A, Yablonka-Reuveni Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Developmental Biology. 2010;340(2):330–343. doi: 10.1016/j.ydbio.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shefer G, van de Mark DP, Richardson JB, Yablonka-Reuveni Z. Satellite-cell pool size does matter: defining the myogenic potency of aging skeletal muscle. Developmental Biology. 2006;294(1):50–66. doi: 10.1016/j.ydbio.2006.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302(5650):1575–1577. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- 19.Roth SM, Martel GF, Ivey FM, et al. Skeletal muscle satellite cell populations in healthy young and older men and women. Anatomical Record. 2000;260(4):351–358. doi: 10.1002/1097-0185(200012)260:4<350::AID-AR30>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 20.Wagers AJ, Conboy IM. Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell. 2005;122(5):659–667. doi: 10.1016/j.cell.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 21.Lexell J. Ageing and human muscle: observations from Sweden. Canadian Journal of Applied Physiology. 1993;18(1):2–18. doi: 10.1139/h93-002. [DOI] [PubMed] [Google Scholar]
- 22.Roubenoff R, Hughes VA. Sarcopenia: current concepts. Journals of Gerontology Series A: Biologial Science and Medical Science. 2000;55(12):M716–M724. doi: 10.1093/gerona/55.12.m716. [DOI] [PubMed] [Google Scholar]
- 23.Sakuma K, Akiho M, Nakashima H, Akima H, Yasuhara M. Age-related reductions in expression of serum response factor and myocardin-related transcription factor A in mouse skeletal muscles. Biochimica et Biophysica Acta Molecular Basis of Disease. 2008;1782(7-8):453–461. doi: 10.1016/j.bbadis.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Sakuma K, Yamaguchi A. Molecular mechanisms in aging and current strategies to counteract sarcopenia. Current Aging Science. 2010;3(2):90–101. doi: 10.2174/1874609811003020090. [DOI] [PubMed] [Google Scholar]
- 25.Sakuma K, Yamaguchi A. Cell Aging. Huntington, NY, USA: Nova Science Publishers; 2011. Sarcopenia: molecular mechanisms and current therapeutic strategy. [Google Scholar]
- 26.Paddon-Jones D, Rasmussen BB. Dietary protein recommendations and the prevention of sarcopenia. Current Opinion in Clinical Nutrition and Metabolic Care. 2009;12(1):86–90. doi: 10.1097/MCO.0b013e32831cef8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradley L, Yaworsky PJ, Walsh FS. Myostatin as a therapeutic target for musculoskeletal disease. Cellular and Molecular Life Sciences. 2008;65(14):2119–2124. doi: 10.1007/s00018-008-8077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakuma K, Yamaguchi A. Inhibitors of myostatin- and proteasome-dependent signaling for attenuating muscle wasting. Recent Patents on Regenerative Medicine. 2011;1(3):284–298. [Google Scholar]
- 29.Kunkel SD, Suneja M, Ebert SM, et al. mRNA expression signatures of human skeletal muscle atrophy identify a natural compound that increases muscle mass. Cell Metabolism. 2011;13(6):627–638. doi: 10.1016/j.cmet.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith GI, Atherton P, Reeds DN, et al. Dietary omega-3 fatty acid supplementation increases the rate of muscle protein synthesis in older adults: a randomized controlled trial. American Journal of Clinical Nutrition. 2011;93(2):402–412. doi: 10.3945/ajcn.110.005611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature. 1997;387(6628):83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 32.Lee SJ. Regulation of muscle mass by myostatin. Annual Review of Cell and Developmental Biology. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]
- 33.Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. Journal of Biological Chemistry. 2002;277(51):49831–49840. doi: 10.1074/jbc.M204291200. [DOI] [PubMed] [Google Scholar]
- 34.Thomas M, Langley B, Berry C, et al. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast differentiation. Journal of Biological Chemistry. 2000;275(51):40235–40243. doi: 10.1074/jbc.M004356200. [DOI] [PubMed] [Google Scholar]
- 35.Yang W, Zhang Y, Li Y, Wu Z, Zhu D. Myostatin induces cyclin D1 degradation to cause cell cycle arrest through a phosphatidylinositol 3-kinase/AKT/GSK-3β pathway and is antagonized by insulin-like growth factor 1. Journal of Biological Chemistry. 2007;282(6):3799–3808. doi: 10.1074/jbc.M610185200. [DOI] [PubMed] [Google Scholar]
- 36.McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(23):12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuelke M, Wagner KR, Stolz LE, et al. Myostatin mutation associated with gross muscle hypertrophy in a child. New England Journal of Medicine. 2004;350(26):2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 38.Allen DL, Unterman TG. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. American Journal of Physiology: Cell Physiology. 2007;292(1):C188–C199. doi: 10.1152/ajpcell.00542.2005. [DOI] [PubMed] [Google Scholar]
- 39.Léger B, Derave W, de Bock K, Hespel P, Russell AP. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Research. 2008;11(1):163–175. doi: 10.1089/rej.2007.0588. [DOI] [PubMed] [Google Scholar]
- 40.Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454(7203):528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haddad F, Adams GR. Aging-sensitive cellular and molecular mechanisms associated with skeletal muscle hypertrophy. Journal of Applied Physiology. 2006;100(4):1188–1203. doi: 10.1152/japplphysiol.01227.2005. [DOI] [PubMed] [Google Scholar]
- 42.Bogdanovich S, Krag TO, Barton ER, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420(6914):418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 43.Holzbaur EL, Howland DS, Weber N, et al. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiology of Disease. 2006;23(3):697–707. doi: 10.1016/j.nbd.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 44.Wagner KR, Fleckenstein JL, Amato AA, et al. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Annals of Neurology. 2008;63(5):561–571. doi: 10.1002/ana.21338. [DOI] [PubMed] [Google Scholar]
- 45.Siriett V, Platt L, Salerno MS, Ling N, Kambadur R, Sharma M. Prolonged absence of myostatin reduces sarcopenia. Journal of Cellular Physiology. 2006;209(3):866–873. doi: 10.1002/jcp.20778. [DOI] [PubMed] [Google Scholar]
- 46.LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA. Myostatin inhibition enhances the effects of exercise on performance and metabolic outcomes in aged mice. Journals of Gerontology Series A: Biological Science and Medical Sciences. 2009;64(9):940–948. doi: 10.1093/gerona/glp068. [DOI] [PubMed] [Google Scholar]
- 47.Murphy KT, Koopman R, Naim T, et al. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB Journal. 2010;24(11):4433–4442. doi: 10.1096/fj.10-159608. [DOI] [PubMed] [Google Scholar]
- 48.Welle S, Burgess K, Mehta S. Stimulation of skeletal muscle myofibrillar protein synthesis, p70 S6 kinase phosphorylation, and ribosomal protein S6 phosphorylation by inhibition of myostatin in mature mice. American Journal of Physiology: Endocrinology and Metabolism. 2009;296(3):E567–E572. doi: 10.1152/ajpendo.90862.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Amirouche A, Durieux AC, Banzet S, et al. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology. 2009;150(1):286–294. doi: 10.1210/en.2008-0959. [DOI] [PubMed] [Google Scholar]
- 50.McFarlane C, Plummer E, Thomas M, et al. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-κB-independent, FoxO1-dependent mechanism. Journal of Cellular Physiology. 2006;209(2):501–514. doi: 10.1002/jcp.20757. [DOI] [PubMed] [Google Scholar]
- 51.Chelh I, Meunier B, Picard B, et al. Molecular profiles of Quadriceps muscle in myostatin-null mice reveal PI3K and apoptotic pathways as myostatin targets. BMC Genomics. 2009;10, article 196:13 pages. doi: 10.1186/1471-2164-10-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frighetto RTS, Welendorf RM, Nigro EN, Frighetto N, Siani AC. Isolation of ursolic acid from apple peels by high speed counter-current chromatography. Food Chemistry. 2008;106(2):767–771. [Google Scholar]
- 53.Liu J. Pharmacology of oleanolic acid and ursolic acid. Journal of Ethnopharmacology. 1995;49(2):57–68. doi: 10.1016/0378-8741(95)90032-2. [DOI] [PubMed] [Google Scholar]
- 54.Wang ZH, Hsu CC, Huang CN, Yin MC. Anti-glycative effects of oleanolic acid and ursolic acid in kidney of diabetic mice. European Journal of Pharmacology. 2010;628(1–3):255–260. doi: 10.1016/j.ejphar.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 55.Liu J. Oleanolic acid and ursolic acid: research perspectives. Journal of Ethnopharmacology. 2005;100(1-2):92–94. doi: 10.1016/j.jep.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 56.Zhang W, Hong D, Zhou Y, et al. Ursolic acid and its derivative inhibit protein tyrosine phosphatase 1B, enhancing insulin receptor phosphorylation and stimulating glucose uptake. Biochimica et Biophysica Acta. 2006;1760(10):1505–1512. doi: 10.1016/j.bbagen.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 57.Kenner KA, Anyanwu E, Olefsky JM, Kusari J. Protein-tyrosine phosphatase 1B is a negative regulator of insulin- and insulin-like growth factor-I-stimulated signaling. Journal of Biological Chemistry. 1996;271(33):19810–19816. doi: 10.1074/jbc.271.33.19810. [DOI] [PubMed] [Google Scholar]
- 58.Arterburn LM, Hall EB, Oken H. Distribution, interconversion, and dose response of n-3 fatty acids in humans. American Journal of Clinical Nutrition. 2006;83(6):1467S–1476S. doi: 10.1093/ajcn/83.6.1467S. [DOI] [PubMed] [Google Scholar]
- 59.Fearon KC, von Meyenfeldt MF, Moses AG, et al. Effect of a protein and energy dense n-3 fatty acid enriched oral supplement on loss of weight and lean tissue in cancer cachexia: a randomised double blind trial. Gut. 2003;52(10):1479–1486. doi: 10.1136/gut.52.10.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harper CR, Jacobson TA. Usefulness of omega-3 fatty acids and the prevention of coronary heart disease. American Journal of Cardiology. 2005;96(11):1521–1529. doi: 10.1016/j.amjcard.2005.07.071. [DOI] [PubMed] [Google Scholar]
- 61.Bloomer RJ, Larson DE, Fisher-Wellman KH, Galpin AJ, Schilling BK. Effect of eicosapentaenoic and docosahexaenoic acid on resting and exercise-induced inflammatory and oxidative stress biomarkers: a randomized, placebo controlled, cross-over study. Lipids in Health and Disease. 2009;8, article 36 doi: 10.1186/1476-511X-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Babcock TA, Helton WS, Espat NJ. Eicosapentaenoic acid (EPA): an antiinflammatory ω-3 fat with potential clinical applications. Nutrition. 2000;16(11-12):1116–1118. doi: 10.1016/s0899-9007(00)00392-0. [DOI] [PubMed] [Google Scholar]
- 63.Singer P, Shapiro H, Theilla M, Anbar R, Singer J, Cohen J. Anti-inflammatory properties of omega-3 fatty acids in critical illness: novel mechanisms and an integrative perspective. Intensive Care Medicine. 2008;34(9):1580–1592. doi: 10.1007/s00134-008-1142-4. [DOI] [PubMed] [Google Scholar]
- 64.Babcock TA, Helton WS, Hong D, Espat NJ. Omega-3 fatty acid lipid emulsion reduces LPS-stimulated macrophage TNF-α production. Surgical Infections. 2002;3(2):145–149. doi: 10.1089/109629602760105817. [DOI] [PubMed] [Google Scholar]
- 65.Magee P, Pearson S, Allen J. The omega-3 fatty acid, eicosapentaenoic acid (EPA), prevents the damaging effects of tumour necrosis factor (TNF)-alpha during murine skeletal muscle cell differentiation. Lipids in Health and Disease. 2008;7, article 24 doi: 10.1186/1476-511X-7-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Machado RV, Mauricio AF, Taniguti APT, Ferretti R, Neto HS, Marques MJ. Eicosapentaenoic acid decreases TNF-α and protects dystrophic muscles of mdx mice from degeneration. Journal of Neuroimmunology. 2011;232(1-2):145–150. doi: 10.1016/j.jneuroim.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 67.Fabre JE, Rivard A, Magner M, Silver M, Isner JM. Tissue inhibition of angiotensin-converting enzyme activity stimulates angiogenesis in vivo. Circulation. 1999;99(23):3043–3049. doi: 10.1161/01.cir.99.23.3043. [DOI] [PubMed] [Google Scholar]
- 68.de Cavanagh EM, Piotrkowski B, Basso N, et al. Enalapril and losartan attenuate mitochondrial dysfunction in aged rats. The FASEB Journal. 2003;17(9):1096–1098. doi: 10.1096/fj.02-0063fje. [DOI] [PubMed] [Google Scholar]
- 69.Maggio M, Ceda GP, Lauretani F, et al. Relation of angiotensin converting enzyme inhibitor treatment to insulin-like growth factor-1 serum levels in subjects. 65 years of age (the InCHIANTI study) . American Journal of Cardiology. 2006;97(10):1525–1529. doi: 10.1016/j.amjcard.2005.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Onder G, Penninx BWJH, Balkrishnan R, et al. Relation between use of angiotensin-converting enzyme inhibitors and muscle strength and physical function in older women: an observational study. Lancet. 2002;359(9310):926–930. doi: 10.1016/s0140-6736(02)08024-8. [DOI] [PubMed] [Google Scholar]
- 71.Dossegger L, Aldor E, Baird MG, et al. Influence of angiotensin converting enzyme inhibition on exercise performance and clinical symptoms in chronic heart failure—a multicentre, double-blind, placebo-controlled trial. European Heart Journal. 1993;14:18–23. doi: 10.1093/eurheartj/14.suppl_c.18. [DOI] [PubMed] [Google Scholar]
- 72.Schellenbaum GD, Smith NL, Heckbert SR, et al. Weight loss, muscle strength, and angiotensin-converting enzyme inhibitors in older adults with congestive heart failure or hypertension. Journal of the American Geriatrics Society. 2005;53(11):1996–2000. doi: 10.1111/j.1532-5415.2005.53568.x. [DOI] [PubMed] [Google Scholar]
- 73.Sumukadas D, Witham MD, Struthers AD, McMurdo MET. Effect of perindopril on physical function in elderly people with functional impairment: a randomized controlled trial. Canadian Medical Association Journal. 2007;177(8):867–874. doi: 10.1503/cmaj.061339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leonetti G, Mazzola C, Pasotti C, et al. Treatment of hypertension in the elderly—effects on blood pressure, heart rate, and physical fitness. American Journal of Medicine. 1991;90:S12–S13. doi: 10.1016/0002-9343(91)90429-2. [DOI] [PubMed] [Google Scholar]
- 75.Bunout D, Barrera G, de la Maza PM, Leiva L, Backhouse C, Hirsch S. Effects of enalapril or nifedipine on muscle strength or functional capacity in elderly subjects. A double blind trial. Journal of the Renin-Angiotensin-Aldosterone System. 2009;10(2):77–84. doi: 10.1177/1470320309105338. [DOI] [PubMed] [Google Scholar]
- 76.Cai D, Frantz JD, Tawa NE, Jr., et al. IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell. 2004;119(2):285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 77.Mitch WE, Goldberg AL. Mechanisms of disease: mechanisms of muscle wasting: the role of the ubiquitin-proteasome pathway. New England Journal of Medicine. 1996;335(25):1897–1905. doi: 10.1056/NEJM199612193352507. [DOI] [PubMed] [Google Scholar]
- 78.Sandri M, Sandri C, Gilbert A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117(3):399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stitt TN, Drujan D, Clarke BA, et al. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Molecular Cell. 2004;14(3):395–403. doi: 10.1016/s1097-2765(04)00211-4. [DOI] [PubMed] [Google Scholar]
- 80.Bossola M, Pacelli F, Costelli P, Tortorelli A, Rosa F, Doglietto GB. Proteasome activities in the rectus abdominis muscle of young and older individuals. Biogerontology. 2008;9(4):261–268. doi: 10.1007/s10522-008-9135-9. [DOI] [PubMed] [Google Scholar]
- 81.Pattison JS, Folk LC, Madsen RW, Childs TE, Booth FW. Transcriptional profiling identifies extensive downregulation of extracellular matrix gene expression in sarcopenic rat soleus muscle. Physiological Genomics. 2004;15:34–43. doi: 10.1152/physiolgenomics.00040.2003. [DOI] [PubMed] [Google Scholar]
- 82.Combaret L, Dardevet D, Béchet D, Taillandier D, Mosoni L, Attaix D. Skeletal muscle proteolysis in aging. Current Opinion in Clinical Nutrition and Metabolic Care. 2009;12(1):37–41. doi: 10.1097/MCO.0b013e32831b9c31. [DOI] [PubMed] [Google Scholar]
- 83.DeRuisseau KC, Kavazis AN, Powers SK. Selective downregulation of ubiquitin conjugation cascade mRNA occurs in the senescent rat soleus muscle. Experimental Gerontology. 2005;40(6):526–531. doi: 10.1016/j.exger.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 84.Welle S, Brooks AL, Delehanty JM, Needler N, Thornton CA. Gene expression profile of aging in human muscle. Physiological Genomics. 2003;14:149–159. doi: 10.1152/physiolgenomics.00049.2003. [DOI] [PubMed] [Google Scholar]
- 85.Whitman SA, Wacker MJ, Richmond SR, Godard MP. Contributions of the ubiquitin-proteasome pathway and apoptosis to human skeletal muscle wasting with age. Pflügers Archiv European Journal of Physiology. 2005;450(6):437–446. doi: 10.1007/s00424-005-1473-8. [DOI] [PubMed] [Google Scholar]
- 86.Clavel S, Coldefy AS, Kurkdjian E, Salles J, Margaritis I, Derijard B. Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat Tibialis Anterior muscle. Mechanisms of Ageing and Development. 2006;127(10):794–801. doi: 10.1016/j.mad.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 87.Edström E, Altun M, Hägglund M, Ulfhake B. Atrogin-1/MAFbx and MuRF1 are downregulated in aging-related loss of skeletal muscle. Journals of Gerontology: Series A Biological Sciences and Medical Sciences. 2006;61(7):663–674. doi: 10.1093/gerona/61.7.663. [DOI] [PubMed] [Google Scholar]
- 88.Attaix D, Mosoni L, Dardevet D, Combaret L, Mirand PP, Grizard J. Altered responses in skeletal muscle protein turnover during aging in anabolic and catabolic periods. International Journal of Biochemistry and Cell Biology. 2005;37(10):1962–1973. doi: 10.1016/j.biocel.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 89.Husom AD, Peters EA, Kolling EA, Fugere NA, Thompson LV, Ferrington DA. Altered proteasome function and subunit composition in aged muscle. Archives of Biochemistry and Biophysics. 2004;421(1):67–76. doi: 10.1016/j.abb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 90.Lee DH. Proteasome inhibitors: valuable new tools for cell biologists. Trends in Cell Biology. 1998;8(10):397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 91.Adams J, Palombella VJ, Sausville EA, et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Research. 1999;59(11):2615–2622. [PubMed] [Google Scholar]
- 92.Orlowski RZ. Proteasome inhibitors in cancer therapy. Methods in Molecular Biology. 1997;100:197–203. doi: 10.1385/1-59259-895-1:339. [DOI] [PubMed] [Google Scholar]
- 93.Bonuccelli G, Sotgia F, Capozza E, Gazzerro E, Minetti C, Lisanti MP. Localized treatment with a novel FDA-approved proteasome inhibitor blocks the degradation of dystrophin and dystrophin-associated proteins in mdx mice. Cell Cycle. 2007;6(10):1242–1248. doi: 10.4161/cc.6.10.4182. [DOI] [PubMed] [Google Scholar]
- 94.Gazzerro E, Assereto S, Bonetto A, et al. Therapeutic potential of proteasome inhibition in Duchenne and Becker muscular dystrophies. American Journal of Pathology. 2010;176(4):1863–1877. doi: 10.2353/ajpath.2010.090468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Beehler BC, Sleph PG, Benmassaoud L, Grover GJ. Reduction of skeletal muscle atrophy by a proteasome inhibitor in a rat model of denervation. Experimental Biology and Medicine. 2006;231(3):335–341. doi: 10.1177/153537020623100315. [DOI] [PubMed] [Google Scholar]
- 96.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiological Reviews. 1999;79(4):1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 97.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora's box opens. Nature Reviews Molecular Cell Biology. 2001;2(1):67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 98.Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434(7033):652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 99.Schinzel AC, Takeuchi O, Huang Z, et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(34):12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Millay DP, Sargent MA, Osinska H, et al. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nature Medicine. 2008;14(4):442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wissing ER, Millay DP, Vuagniaux G, Molkentin JD. Debio-025 is more effective than prednisone in reducing muscular pathology in mdx mice. Neuromuscular Disorders. 2010;20(11):753–760. doi: 10.1016/j.nmd.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tiepolo T, Angelin A, Palma E, et al. The cyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1 −/−myopathic mice. British Journal of Pharmacology. 2009;157(6):1045–1052. doi: 10.1111/j.1476-5381.2009.00316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Angelini C. The role of corticosteroids in muscular dystrophy: a critical appraisal. Muscle and Nerve. 2007;36(4):424–435. doi: 10.1002/mus.20812. [DOI] [PubMed] [Google Scholar]
- 104.Balaban B, Matthews DJ, Clayton GH, Carry T. Corticosteroid treatment and functional improvement in Duchenne muscular dystrophy long-term effect. American Journal of Physical Medicine and Rehabilitation. 2005;84(11):843–850. doi: 10.1097/01.phm.0000184156.98671.d0. [DOI] [PubMed] [Google Scholar]
- 105.Morissette MR, Cook SA, Buranasombati C, Rosenberg MA, Rosenzweig A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. American Journal of Physiology: Cell Physiology. 2009;297(5):C1124–C1132. doi: 10.1152/ajpcell.00043.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. American Journal of Physiology: Cell Physiology. 2009;296(6):C1258–C1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- 107.Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Molecular Cell. 2010;40(2):323–332. doi: 10.1016/j.molcel.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Holloszy JO. Regulation by exercise of skeletal muscle. Journal of Physiology and Pharmacology. 2008;59(7):5–18. [PubMed] [Google Scholar]
- 109.Dirks AJ, Leeuwenburgh C. Aging and lifelong calorie restriction result in adaptations of skeletal muscle apoptosis repressor, apoptosis-inducing factor, X-linked inhibitor of apoptosis, caspase-3, and caspase-12. Free Radical Biology and Medicine. 2004;36(1):27–39. doi: 10.1016/j.freeradbiomed.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 110.López-Lluch G, Hunt N, Jones B, et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(6):1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Payne AM, Dodd SL, Leeuwenburgh C. Life-long calorie restriction in Fischer 344 rats attenuates age-related loss in skeletal muscle-specific force and reduces extracellular space. Journal of Applied Physiology. 2003;95(6):2554–2562. doi: 10.1152/japplphysiol.00758.2003. [DOI] [PubMed] [Google Scholar]
- 112.Philips T, Leeuwenburgh C. Muscle fiber specific apoptosis and TNF-α signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB Journal. 2005;19(6):668–670. doi: 10.1096/fj.04-2870fje. [DOI] [PubMed] [Google Scholar]
- 113.Taaffe DR. Sarcopenia—exercise as a treatment strategy. Australian Family Physician. 2006;35(3):130–134. [PubMed] [Google Scholar]
- 114.Marzetti E, Lees HA, Wohlgemuth SE, Leeuwenburgh C. Sarcopenia of aging: underlying cellular mechanisms and protection by calorie restriction. BioFactors. 2009;35(1):28–35. doi: 10.1002/biof.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sandri M, Lin J, Handschin C, et al. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(44):16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Anderson R, Prolla T. PGC-1α in aging and anti-aging interventions. Biochimica et Biophysica Acta. 2009;1790(10):1059–1066. doi: 10.1016/j.bbagen.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Handschin C, Kobayashi YM, Chin S, Seale P, Campbell KP, Spiegelman BM. PGC-1α regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes and Development. 2007;21(7):770–783. doi: 10.1101/gad.1525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wenz T, Diaz F, Hernandez D, et al. Activation of the PPAR/PGC-1α pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy. Journal of Applied Physiology. 2008;106:1712–1719. [Google Scholar]
- 119.Goodpaster BH, Carlson CL, Visser M, et al. Attenuation of skeletal muscle and strength in the elderly: the health ABC study. Journal of Applied Physiology. 2001;90(6):2157–2165. doi: 10.1152/jappl.2001.90.6.2157. [DOI] [PubMed] [Google Scholar]
- 120.Song M-Y, Ruts E, Kim J, Janumala I, Heymsfield S, Gallagher D. Sarcopenia and increased adipose tissue infiltration of muscle in elderly African American women. American Journal of Clinical Nutrition. 2004;79(5):874–880. doi: 10.1093/ajcn/79.5.874. [DOI] [PubMed] [Google Scholar]
- 121.Zamboni M, Mazzali G, Fantin F, Rossi A, di Francesco V. Sarcopenic obesity: a new category of obesity in the elderly. Nutrition, Metabolism and Cardiovascular Diseases. 2008;18(5):388–395. doi: 10.1016/j.numecd.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 122.Morley JE, Baumgartner RN, Roubenoff R, Mayer J, Nair KS. Sarcopenia. Journal of Laboratory and Clinical Medicine. 2001;137(4):231–243. doi: 10.1067/mlc.2001.113504. [DOI] [PubMed] [Google Scholar]
- 123.Roth RJ, Le AM, Zhang L, et al. MAPK phosphatase-1 facilitates the loss of oxidative myofibers associated with obesity in mice. Journal of Clinical Investigation. 2009;119(12):3817–3829. doi: 10.1172/JCI39054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. Journal of Clinical Investigation. 2009;119(3):573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bonnard C, Durand A, Peyrol S, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. Journal of Clinical Investigation. 2008;118(2):789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Arany Z, Foo SY, Ma Y, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α . Nature. 2008;451(7181):1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 127.Handschin C, Chin S, Li P, et al. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1α muscle-specific knock-out animals. Journal of Biological Chemistry. 2007;282(41):30014–30021. doi: 10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- 128.Crunkhorn S, Dearie F, Mantzoros C, et al. Peroxisome proliferator activator receptor γ coactivator-1 expression is reduced in obesity: potential pathogenic role of saturated fatty acids and p38 mitogen-activated protein kinase activation. Journal of Biological Chemistry. 2007;282(21):15439–15450. doi: 10.1074/jbc.M611214200. [DOI] [PubMed] [Google Scholar]
- 129.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(48):20405–20410. doi: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 130.Arany Z. PGC-1 coactivators and skeletal muscle adaptations in health and disease. Current Opinion in Genetics and Development. 2008;18(5):426–434. doi: 10.1016/j.gde.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]