

Abstract
Rationale
Hyperamylinemia is common in patients with obesity and insulin resistance, coincides with hyperinsulinemia, and results in amyloid deposition. Amylin amyloids are generally considered a pancreatic disorder in type-2 diabetes. However, elevated circulating levels of amylin may also lead to amylin accumulation and proteotoxicity in peripheral organs, including the heart.
Objective
To test whether amylin accumulates in the heart of obese and type-2 diabetic patients and to uncover the effects of amylin accumulation on cardiac morphology and function.
Methods and Results
We compared amylin deposition in failing and non-failing hearts from lean, obese, and type-2 diabetic humans using immunohistochemistry and western blots. We found significant accumulation of large amylin oligomers, fibrils and plaques in failing hearts from obese and diabetic patients, but not in normal hearts and failing hearts from lean, non-diabetic humans. Small amylin oligomers were even elevated in non-failing hearts from overweight/obese patients suggesting an early state of accumulation. Using a rat model of hyperamylinemia transgenic for human amylin, we observed that amylin oligomers attach to the sarcolemma, leading to myocyte Ca2+ dysregulation, pathological myocyte remodeling, and diastolic dysfunction, starting from pre-diabetes. In contrast, pre-diabetic rats expressing the same level of wild-type rat amylin, a non-amyloidogenic isoform, exhibited normal heart structure and function.
Conclusions
Hyperamylinemia promotes amylin deposition in the heart causing alterations of cardiac myocyte structure and function. We propose that detection and disruption of cardiac amylin buildup may be both a predictor of heart dysfunction and a novel therapeutic strategy in diabetic cardiomyopathy.
Keywords: hyperinsulinemia/hyperamylinemia, diabetic cardiomyopathy, calcium, HIP rat, UCD-T2DM rat
One third of adults and 17% of children in the US* are currently obese and at high risk of developing both type-2 diabetes and cardiovascular disease.1-3 Progression to overt type-2 diabetes may accelerate pathological changes in heart structure and function,4-7 independent of confounding factors such as coronary artery disease and hypertension.1-3 It is assumed1 that increases in body fat can affect the body's response to insulin, potentially leading to insulin resistance and subsequent impaired glucose and lipid homeostasis. As such, insulin resistance is unequivocally associated with heart disease.1-10 However, the myocardial insulin responsiveness in diabetic patients is surprisingly intact,11,12 suggesting that factors secondary to insulin resistance may critically contribute to cardiac dysfunction in type-2 diabetes.5,9,10 In addition to hyperglycemia and dyslipidemia, patients with obesity and insulin resistance present also hyperinsulinemia and hyperamylinemia.13-15 While the hyperinsulinemic response prevents a large fraction of insulin resistant patients from developing type-2 diabetes1, the coincident hyperamylinemia leads to proteotoxicity and amyloid deposition.13,14 More than 95% of patients with type-2 diabetes stain positive for amylin amyloids in pancreatic islets.14 Amylin deposition was also found in kidneys of obese and type-2 diabetic patients.16 Recently,17,18 we hypothesized that hyperamylinemia may favor cardiac amylin accumulation17 causing alterations of myocyte structure and function in ways that may contribute to progressive heart failure.18
Amylin is a 4 kDa hormone co-expressed and co-secreted with insulin by pancreatic β-cells.13,14 Human amylin, also known as islet amyloid polypeptide (IAPP), has aggregation properties similar to prions and amyloidogenic proteins that are associated with neurodegenerative diseases.19 At high secretion rates, amyloidogenic proteins readily form oligomers, fibrils and amyloid plaques. It is increasingly recognized that soluble oligomers rather than fibrils and plaques are the most toxic species of amyloids.20-28 They attach to cellular membranes causing Ca2+ dyshomeostasis, cell dysfunction, and apoptosis.20-28 Previous data27,28 indicated that cardiac myocyte-restricted expression and accumulation of amyloidogenic peptides, such as polyglutamine27 or presenilin28, can induce cytotoxicity and heart failure in mice. Presenilin oligomers coimmunoprecipitated with sarcoplasmic reticulum Ca-ATPase (SERCA) and altered Ca2+ handling in cardiac myocytes.28
To clarify whether amylin builds up in the heart and whether this could be associated with cardiac failure in obesity and type-2 diabetes, we assessed amylin deposition in hearts from lean, obese, and type-2 diabetic humans, with and without heart failure. Using a ‘humanized’ rat model of hyperamylinemia, we examined changes in cardiac structure and function in relation with cardiac amylin accumulation.
Methods
Detailed procedures, description of human tissue specimens and animal models are included in the Supplemental Material.
Human tissue specimens
Failing hearts from obese, type-2 diabetic, and non-diabetic patients were obtained at the time of orthotopic heart transplantation at the Hospital of University of Pennsylvania. Non-failing hearts from obese and lean individuals are from organ donation. Tissue specimens were obtained in accordance with the Institutional Review Board approval. Inclusion in tissue-based studies was not restricted on the basis of age, gender, race or ethnic status. Heart failure etiology, body mass index (BMI), age, gender and state of diabetes with respect to dependence on insulin and/or oral hypoglycemic agents of all cases studied here are summarized in the Online Table I.
Experimental animals
Studies were approved by the University of California, Davis Animal Research Committee. Because rodent amylin is not amyloidogenic and rodents do not accumulate amylin amyloids,29 most rodent models of type-2 diabetes are not adequate for this study. We used Sprague-Dawley (SD) rats transgenic for human amylin in the pancreatic β-cells (HIP rats).30 HIP rat breeding pairs were kindly provided by Pfizer. HIP rats show hyperamylinemia, leading to amylin deposits in pancreatic islets and gradual decline in β-cell mass.31 They develop insulin resistance at 5 months of age and diabetes by 10 months of age.31 As negative controls, we used obese, insulin resistant rats expressing only wild-type, non-amyloidogenic rat amylin, which does not form amyloids (UCD-T2DM rats).32 UCD-T2DM rats were obtained by breading obese SD rats with Zucker Diabetic Lean rats that lack the leptin receptor defect and have inherent ß-cell defects. 32 UCD-T2DM rats exhibit insulin resistance prior to the onset of diabetes,32 similar to HIP rats30 and humans.1 In the present study, we used age matched HIP (N=17) and UCD-T2DM (N=19) rats in the pre-diabetic state, i.e. non-fasting blood glucose level in the 150-200 mg/dl range.33 Wild-type littermates (N=16) served as non-diabetic controls for HIP rats. Age-matched SD rats (N=13, Charles Rivers Laboratory) were controls for UCD-T2DM rats.
Immunochemistry
Western blot analysis was performed on left ventricular homogenates, myocyte lysates, and blood serum. Immunohistochemistry was done on thin sections from paraffin blocks.
Insulin Signaling
Myocardial insulin responsiveness was determined by measuring the phosphorylation level of protein kinase B (Akt) and glycogen synthase kinase 3β (GSK3β) in hearts from rats injected (I.P.) with insulin (10 mU/g body weight) and sacrificed 10 min after injection.
Cardiac myocyte isolation and Ca2+ measurements
Rat ventricular myocytes were isolated by perfusion with 1 mg/ml collagenase on a Langendorff apparatus as previously reported.34 Intracellular Ca2+ level ([Ca2+]i) was measured with Fura2 or Fluo4.
Activation of Ca2+-dependent hypertrophic pathways
Activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII)-histone deacetylase (HDAC) and calcineurin-nuclear factor of activated T cells (NFAT) hypertrophic pathways was examined by determining the nuclear vs. cytosolic localization of HDAC4 and NFATc4 in cardiac myocytes using immunofluorescence.
In vivo echocardiography and hemodynamics
M-mode echo echocardiography and hemodynamic measurements were performed as described before.35
Electron microscopy
Aliquots of human/rat amylin aggregation reaction were imaged by a Philips CM 12 electron microscope, as previously described.36
Statistical Analysis
Data are expressed as mean ± SEM. Statistical discriminations were performed using two-tailed unpaired Student's t test, with P < 0.05 considered significant. One way analysis of variance (ANOVA) with the Dunnett post hoc test was used when comparing multiple groups.
Results
Patients with obesity and type-2 diabetes present cardiac amylin accumulation
We examined left ventricular tissue from 53 human hearts divided in five pathologically distinct groups (Online Table I). These included failing hearts from patients with type-2 diabetes (N=25) and obese patients that developed overt type-2 diabetes within one year post-transplantation (N=8). These hearts were expected to show significant amylin accumulation. To uncover the early stage of amylin buildup in the heart, a third group included non-failing hearts from overweight/obese humans (N=8). Lastly, non-failing hearts from lean, healthy patients (N=5) and failing hearts from lean patients without diabetes (N=7), which should not accumulate amylin, served as negative controls.
To assess the level and size distribution of cardiac amylin aggregates, we performed western blots with an anti-amylin antibody on left ventricular protein homogenates. We found molecular weight bands corresponding to amylin trimers (12 kDa), tetramers (16 kDa) and two additional larger molecular weight structures at ∼32 kDa (octamers) and ∼64 kDa (16-mers) (Fig. 1A-C). Negative controls showed that these bands are specific (Online Fig. I). Intensity signal analysis (Fig. 1D-F) indicated that amylin oligomer accumulation is markedly larger in failing hearts from patients with type-2 diabetes and overweight/obesity than in normal hearts and failing hearts from patients without diabetes (controls). Intriguingly, large amylin oligomers, i.e. >32kDa, are abundant in failing hearts from diabetic and obese patients (Fig. 1A,B,F), but not in non-failing hearts from overweight/obese individuals (Fig. 1C,F). Smaller amylin oligomers were already elevated in non-failing hearts from overweight/obese patients (Fig. 1C-E) indicating an early stage of cardiac amylin accumulation. The results are consistent with the idea that accumulation of large amylin oligomers can induce deleterious cardiac effects. Amylin tetramers are also present in failing hearts from non-diabetic patients (Fig. 1E), which might indicate undiagnosed insulin resistance in those patients (as commonly seen in aging).
Figure 1.
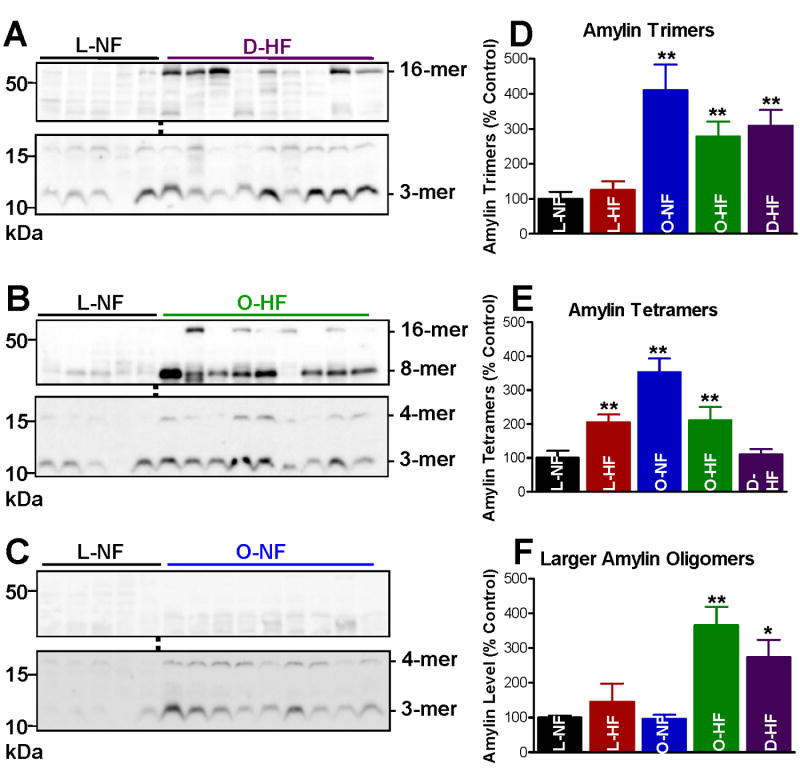
(A-C) Amylin oligomer size distribution in failing (HF) and non-failing (NF) hearts from diabetic (D) and overweight/obese (O) patients vs. lean (L) controls. Representative (of ≥4 experiments) western blots with anti-amylin antibody of left ventricle protein homogenates from hearts in the D-HF (A), O-HF (B) and O-NF (C) groups. In each case, comparisons are made with hearts from the L-NF group. Specific molecular weight bands correspond to amylin trimers (12 kDa), tetramers (16 kDa) and two larger molecular weight structures at ∼32 kDa (octamers) and ∼64 kDa (16-mers). (D-F) Intensity signal analysis of the 12, 16 and the integrated 32-64 kDa bands. Heart samples in the D-HF (N=25), O-HF (N=8) and O-NF (N=8) groups contain markedly higher amylin oligomer levels than the controls L-NF (N=5) and L-HF (N=7). Large size amylin octamers and 16-mers are abundantly present only in the failing heart O-HF and D-HF groups. *P < 0.05; **P < 0.01.
Immunohistochemistry with an anti-amylin antibody shows large amylin deposits in failing hearts from type-2 diabetic patients (Fig. 2A-D), similar to those found in the pancreas of type-2 diabetic patients (Fig. 2F). Amylin plaques (Fig. 2A,C, D) and fibrillar tangles (Fig. 2B) are scattered through the entire heart. Amylin deposits are often seen at sites with myocyte multinucleation, variation in nuclear size and infiltrating cells, which usually occur with fibrotic and infiltrative diseases. In contrast, sections from normal hearts (Fig. 2E) do not show amylin deposition and structural abnormalities. To quantify the extent of amylin deposition in large plaques and fibrils, pellets from heart protein homogenates were treated with formic acid and guanidine hydrochloride to partially break down the amylin oligomers. Dot blots showed significantly increased amylin levels (Online Fig. II) indicating that fragmenting of large amylin aggregates enhanced detection by the anti-amylin antibody. This also implies that blots exhibiting higher order oligomers (Fig. 1) likely underestimate the amount of amylin in these aggregates.
Figure 2.
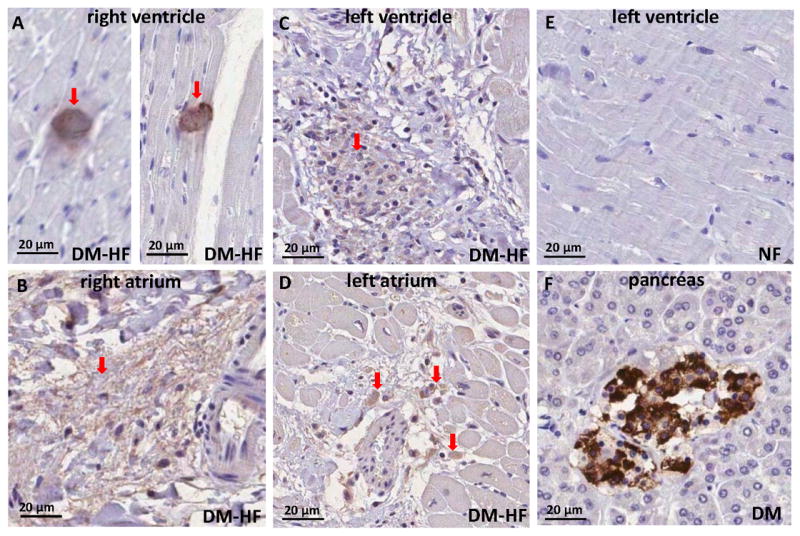
Amylin deposition in failing diabetic hearts demonstrated by immunohistochemistry with an anti-amylin antibody on thin heart sections. Amylin plaques (A,C, and D, arrows) and tangles (B, arrow) are scattered through the entire heart. (E) Left ventricle section from a non-failing heart; no amylin deposits are revealed. (F) Positive control for amylin deposition in a pancreas from a diabetic patient.
Human but not rat amylin alters cardiac myocyte structure and function ex vivo
Amylin oligomers elevate [Ca2+]i in pancreatic β-cells leading to cellular dysfunction and apoptosis.37 Similarly, β-amyloid oligomers induce neuron dysfunction and death in Alzheimer's disease through a mechanism involving increased [Ca2+]i.20,24,25 Thus, we examined Ca2+ cycling in cardiac myocytes incubated with exogenous amylin oligomers. Rat cardiac myocytes were incubated for ∼2 hours with 5 or 50 μmol/L exogenous human (amyloidogenic) or rat (non-amyloidogenic) amylin. Electron microscopy showed that at 50 μmol/L and 2 hours incubation time, human but not rat amylin forms oligomers (Online Fig. III). 50 μmol/L human amylin significantly increased Ca2+ transient amplitude (Fig. 3A,C). In contrast, same concentration of non-amyloidogenic rat amylin had only a modest, not significant effect (Fig. 3B). Incubation with either human or rat amylin (50 μmol/L) did not alter Ca2+ transient decay and diastolic [Ca2+]i (Online Fig. IV). This suggests that amylin does not directly affect SERCA function.
Figure 3.
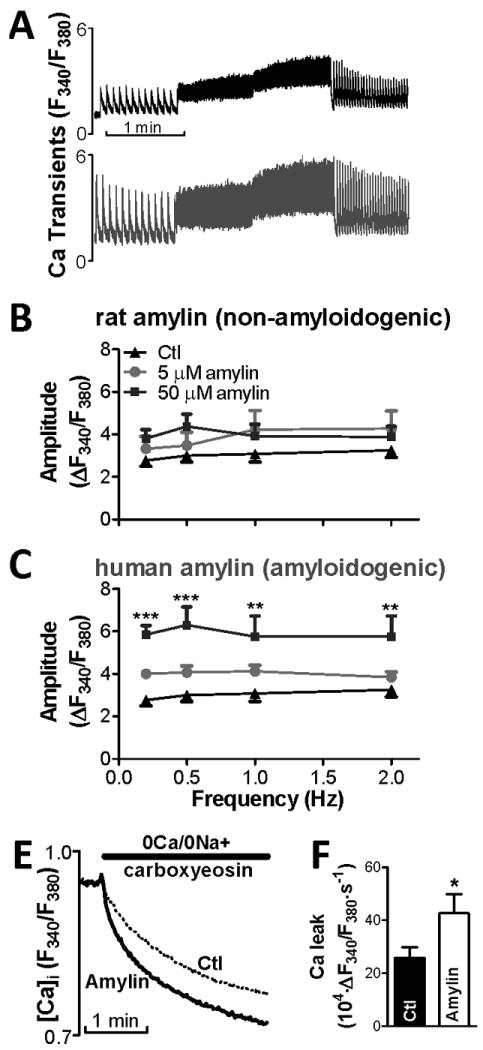
Exogenous human amylin oligomers increase Ca2+ transient amplitude in isolated rat cardiac myocytes. (A) Representative Ca2+ transient measurements in a control (top panel) cell and a myocyte pre-incubated with 50 μmol/L human amylin (bottom panel). (B-C) Effect of 5 and 50 μmol/L of rat (B) and human (C) amylin on Ca2+ transient amplitude. Oligomerization of human amylin resulted in a marked Ca2+ transient increase. (D) Passive sarcolemmal Ca2+ leak measurements as initial slope of [Ca2+]i decline upon reducing external [Ca2+] from 1 mmol/L to 0, with the SR, Na/Ca exchanger and sarcolemmal Ca2+-ATPase blocked by pre-treatment with thapsigargin, 0Na+/0Ca+ solution and 20 μmol/L carboxyeosin, respectively. (E) Passive trans-sarcolemmal Ca2+ leak is significantly larger in myocytes pre-treated with 50 μmol/L human amylin vs. control. For each group, measurements were done on ≥6 myocytes from 3 different rats.
To determine if amylin oligomers elevate [Ca2+]i by increasing sarcolemmal Ca2+ permeability, we measured the effect of human amylin oligomers on the passive sarcolemmal Ca2+ leak. We measured the initial rate of [Ca2+]i decline upon reducing [Ca2+]0 from 1 to 0 mmol/L, with the SR, Na+/Ca+ exchanger and sarcolemmal Ca2+-ATPase blocked (Fig. 3D). Trans-sarcolemmal Ca2+ leak was significantly larger in myocytes pre-incubated with 50 μmol/L human amylin vs. control (Fig. 3E), suggesting alteration of sarcolemmal processes. Incubation of isolated myocytes with fluorescent human amylin showed that amylin attaches to the sarcolemma (Online Fig. V-B). Human amylin monomers, dimers, and trimers were present in lysates of myocytes pre-incubated with 50 μmol/L human amylin (Online Fig. V-A), in agreement with human amylin attachment to sarcolemma. Thus, amylin oligomers attach to sarcolemma and raise cellular Ca2+ load in cardiac myocytes, an effect generated also in neurons25 and pancreatic β-cells.26,37
Cardiac amylin accumulation alters Ca2+ cycling in HIP rats
To test whether in vivo cardiac accumulation of human amylin affects Ca2+ cycling, we used pre-diabetic HIP rats. Age-matched, pre-diabetic UCD-T2DM rats expressing only the native, non-amyloidogenic rat amylin were used as negative control. Using pre-diabetic rats has the advantage that one can dissociate the effect of cardiac amylin accumulation from other confounding factors that affect cardiac Ca2+ cycling during late diabetes.7,38,39 Immunohistochemistry (Fig. 4A) and dot blots (Fig. 4B) with an anti-amylin antibody that recognizes both human and rat amylin (the latter with higher avidity) show that amylin significantly accumulates only in HIP rat hearts. Western blots on left ventricular homogenates and cardiac myocyte lysates from HIP rats (Fig. 4C) show amylin multimers similar to those detected in humans (Fig. 1A-C) in all groups. These data indicate that amylin oligomer accumulation in HIP rat hearts starts from pre-diabetes. The presence of amylin oligomers in cardiac myocyte lysates suggests that they attach to sarcolemma and/or enter the myocytes.
Figure 4.
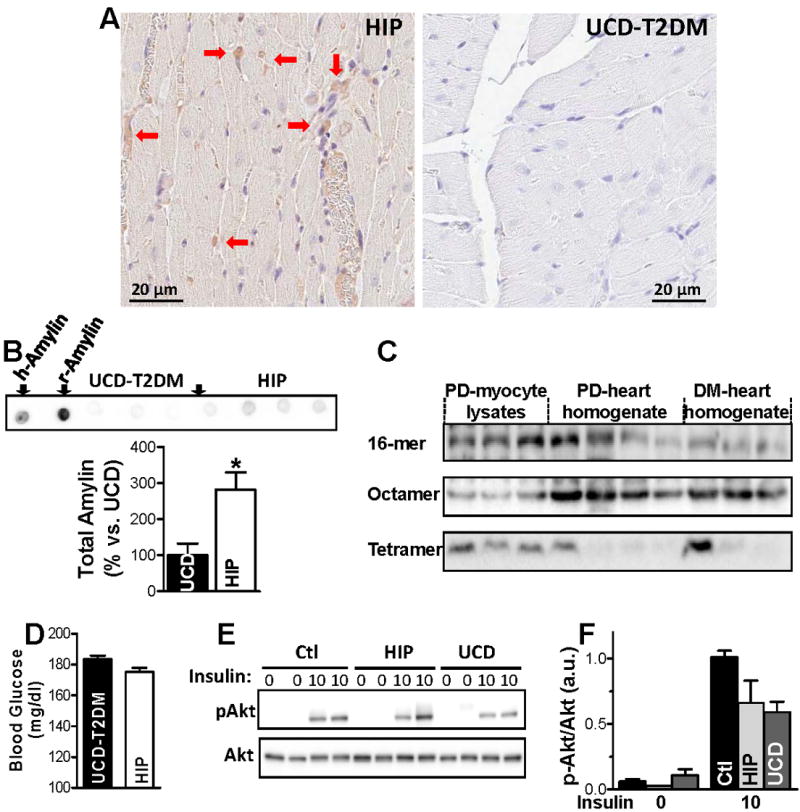
(A) Immunohistochemistry with an anti-amylin antibody on thin heart sections demonstrating amylin deposition in cardiac tissues from pre-diabetic HIP but not UCD-T2DM rats (20X). (B) Dot blots with the anti-amylin antibody comparing total amylin level in HIP vs. UCD-T2DM rats. The first two dots on the left show positive controls using 5 ng of recombinant human (h-Amylin) and rat (r-Amylin) amylin. The antibody binds r-Amylin with significantly higher affinity than h-amylin; The bottom panel shows the average signal intensity in hearts from pre-diabetic HIP vs. UCD-T2DM rats. The experiment was performed in triplicate. (C) Representative western blot with anti-amylin primary antibody on ventricular myocyte lysates from pre-diabetic HIP rats, and left ventricle protein homogenates from pre-diabetic (PD) and diabetic (DM) HIP rats. (D) Blood glucose levels in age-matched pre-diabetic HIP rats (N=14) and UCD-T2DM rats (N=16). (E-F) Akt phosphorylation in hearts from pre-diabetic HIP and UCD-T2DM rats and littermate controls under basal conditions (0 insulin) and following stimulation with insulin (10 mU/g body weight). Representative example (E) and mean values for the ratio between phosphorylated and total Akt (F). N=3 rats for each group.
To test whether cardiac amylin accumulation affects myocardial insulin responsiveness, we compared the activation status of Akt and GSK-3β, key components of the cardiac insulin signaling pathway, in HIP, littermate control, and UCD-T2DM rats. For this test, HIP and UCD-T2DM rats were matched for age and non-fasting blood glucose level (Fig. 4D). The ratio between basal levels of phosphorylated Akt and total Akt is not statistically different among the three groups (Fig. 4E,F). Whereas insulin stimulation significantly increased the phosphorylation of Akt in all rats, no statistical difference among HIP, UCD-T2DM, and control rat groups was observed (Fig. 4E). GSK-3β displays a similar response to stimulation by insulin (Online Fig. VI).
Cardiac amylin accumulation in pre-diabetic HIP rats alters myocyte Ca2+ cycling (Fig. 5). At low stimulation frequencies, Ca2+ transient amplitude is significantly larger (4.7±0.5 vs. 3.5±0.3 at 0.5 Hz) in myocytes from pre-diabetic HIP rats versus control rats (Figs. 5A,D). In contrast, cardiac myocytes from age-matched, pre-diabetic UCD-T2DM rats show no change in Ca2+ transient amplitude (Fig. 5G and Online Fig. VII). Thus, cardiac amylin accumulation may be the cause for the larger Ca2+ transient amplitude in pre-diabetic HIP rats, in agreement with our results using exogenous human amylin oligomers (Fig. 3). Different from littermate controls, the amplitude of Ca2+ transients in myocytes from pre-diabetic HIP rats decreases with increasing the stimulation frequency (negative staircase), so that at 2 Hz the amplitude is similar to that recorded in control rats (Fig. 5B,D). This is probably due to deficiencies in Ca2+ re-uptake into the SR. Indeed, Ca2+ transient decline, which is mostly due to SR Ca2+ re-uptake via the SR Ca-ATPase (SERCA), is significantly slower in pre-diabetic HIP rats vs. control (τ=0.71±0.07 vs. 0.55±0.04 s at a stimulation rate of 0.5 Hz; Fig. 5C,E). In contrast, Ca2+ transient decay remains unchanged in myocytes from age-matched, pre-diabetic UCD-T2DM rats (Fig. 5H). Despite slower Ca2+ transient relaxation, the SR Ca2+ load, assessed as the amplitude of Ca2+ transient produced by 10 mmol/L caffeine, is similar in myocytes from control and pre-diabetic HIP rats paced at 2 Hz (ΔF/F0=8.5±0.4 vs. 8.6±0.5). However, the slower Ca2+ transient relaxation in pre-diabetic HIP rats results in elevated diastolic [Ca2+]i at higher pacing rates (Fig. 5B,F). Diastolic [Ca2+]i is unaltered in pre-diabetic UCD-T2DM rats (Fig. 5I). Similar to myocytes incubated with human amylin, the sarcolemmal Ca2+ leak was significantly larger in pre-diabetic HIP rats vs. control (41.6±4.5 vs. 29.2±2.4 104·ΔF340/F380·s-1, P<0.05). We infer that amylin oligomers can acutely increase Ca2+ leak into myocytes, causing elevated diastolic [Ca2+]i and Ca2+ transients, but that reduced SERCA function may be a longer term effect, as in heart failure.
Figure 5.
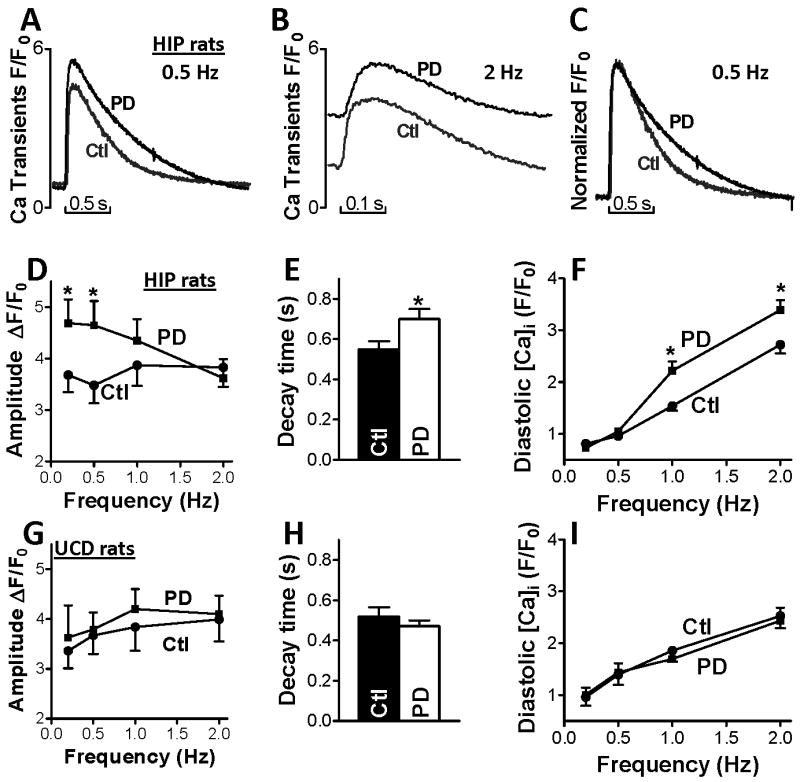
Altered Ca2+ cycling in cardiac myocytes from pre-diabetic HIP but not pre-diabetic UCD-T2DM rats. Representative Ca2+ transients in myocytes from control (Ctl) and pre-diabetic (PD) HIP rats paced at 0.5 Hz (A) and 2 Hz (B). (C) Normalized Ca2+ transients in myocytes from control and pre-diabetic HIP rats (0.5 Hz) indicate slower Ca2+ transient relaxation in pre-diabetic HIP rats vs. control. (D) Mean amplitude of Ca2+ transients recorded in cardiac myocytes from control rats (20 myocytes, 4 rats) and pre-diabetic HIP rats (18 cells, 4 rats) paced at 0.2, 0.5, 1 and 2 Hz. At 0.2 and 0.5 Hz, Ca2+ transient amplitude is significantly larger in myocytes from pre-diabetic HIP rats vs. control. This difference disappears at higher stimulation frequencies. (E) Ca2+ transient decay time in cardiac myocytes from control and pre-diabetic HIP rats paced at 0.5 Hz. (F) Diastolic [Ca2+]i in cardiac myocytes from control rats and pre-diabetic HIP rats paced at 0.2, 0.5, 1 and 2 Hz. At higher frequencies, diastolic [Ca2+]i is significantly higher in myocytes from pre-diabetic HIP vs. control rats. (G) Mean amplitude of Ca2+ transients in myocytes from control rats (22 myocytes, 6 rats) and pre-diabetic UCD-T2DM rats (21 cells, 4 rats) paced at 0.2, 0.5, 1 and 2 Hz. (H) Ca transient decay time in myocytes from control and pre-diabetic UCD-T2DM rats paced at 0.5 Hz. (I) Diastolic [Ca2+]i in myocytes from control and pre-diabetic UCD-T2DM rats paced at 0.2, 0.5, 1 and 2 Hz. *P<0.05
HDAC and NFAT translocation in pre-diabetic HIP rats and myocytes incubated with human amylin
Larger Ca2+ transients and elevated diastolic [Ca2+]i may activate Ca2+-dependent hypertrophic signaling, such as CaMKII-HDAC and calciuneurin-NFAT pathways.40,41 High [Ca2+]i activates CaMKII, which phosphorylates HDAC. Normally, HDAC represses transcriptional activation. However, HDAC phosphorylation causes its export from the nucleus, which activates hypertrophic gene expression. In the calcineurin-NFAT pathway, upon activation by Ca2+/calmodulin, calcineurin dephosphorylates NFAT, causing NFAT import into the nucleus, which activates hypertrophic gene transcription. Using immunofluorescence, we found lower nuclear-to-cytosolic ratio of HDAC4 in myocytes from pre-diabetic HIP vs. littermate controls (Fig. 6A,C), indicating nuclear HDAC export. In contrast, the nuclear-to-cytosolic ratio of NFATc4 is elevated in pre-diabetic HIP rats (Fig. 6B,D), suggesting the nuclear import of NFAT. Thus, both CaMKII-HDAC and calciuneurin-NFAT hypertrophic pathways may be activated in pre-diabetic HIP rats.
Figure 6.

HDAC4 nuclear export, NFATc4 nuclear import, reduced SERCA and increased BNP in hearts from pre-diabetic HIP rats. (A-B) Representative immunofluorescence images showing the distribution of HDAC4 and, respectively, NFATc4 in myocytes from pre-diabetic HIP rats and age-matched WT rats. (C-D) The nuclear-to-cytosolic ratio of HDAC4 (C) and NFATc4 (D) demonstrates the nuclear export of HDAC4 and nuclear import of NFATc4 in pre-diabetic HIP rats. Experiments were done on more than 15 cells from 3 rats for both groups. (E) Increased expression of the hyperthrophic marker BNP in hearts from pre-diabetic (PD) and diabetic (DM) HIP rats. (F) Alterations in the protein expression of SERCA, phospholamban and Na/Ca exchanger in hearts from pre-diabetic and diabetic HIP rats vs. control, non-diabetic rats (Ctl). Ctl – 5 hearts; PD – 5 hearts, DM – 5 hearts.
NFATc4 was also translocated to the nucleus in control isolated rats myocytes incubated with 50 μmol/L human amylin for 2 h (Online Fig. VIII). At this concentration, human amylin forms oligomers and fibrils and elevates Ca2+ transients, as discussed in above. The distribution of HDAC4 was not altered by this acute amylin exposure (Online Fig. VIII). We conclude that Ca2+-dependent nuclear signaling initiated by amylin oligomers is capable of inducing hypertrophic transcriptional effects.
Cardiac amylin accumulation accelerates cardiac hypertrophy and remodeling
Elevated natriuretic peptide levels are thought to reflect cardiac dysfunction and have been used as a “biomarker” of cardiac hypertrophy.42,43 We found that the level of brain natriuretic peptide (BNP) is elevated (by 100±30%) in hearts from pre-diabetic HIP rats vs. littermate controls and further increases with diabetes development (Fig. 6E). This suggests hormonal alterations specific to the onset of cardiac hypertrophy in HIP rats. In contrast, the BNP level is not altered in pre-diabetic UCD-T2DM rats and only increases after the full development of diabetes (Online Fig. IX-A). Of note, a previous study found that external human amylin induces hypertrophy in isolated cardiac myocytes.44 However, the heart weight/body weight ratio in pre-diabetic HIP rats (2.72±0.22 g) versus control rats (2.71±0.2 g) did not change, showing the lack of overt cardiac hypertrophy in this early disease state.
Activation of Ca2+-dependent transcriptional pathways may also alter the transcription of key Ca2+ transport and regulatory proteins, which cause further alterations in Ca2+ cycling. Thus, we measured the protein expression of SERCA, phospholamban (the endogenous SERCA inhibitor), and Na+/Ca2+ exchanger, the main Ca2+ extrusion pathway in HIP rats (Fig. 6F). We found that SERCA expression is reduced by 20% and 30% in pre-diabetic and diabetic HIP rats, respectively (Fig. 6F). In contrast, SERCA expression was unchanged in pre-diabetic UCD-T2DM rats (Online Fig. IX-B). Protein expressions of phospholamban and Na+/Ca2+ exchanger are unaltered in pre-diabetic HIP rats (Fig. 6F).
Pre-diabetic HIP rats show diastolic dysfunction
To determine how amylin accumulation affects cardiac function, we performed in vivo echocardiography and hemodynamic measurements on pre-diabetic HIP rats (Table 1). We found significantly slower relaxation (reduced –dP/dtmin values) in pre-diabetic HIP rats vs. control. This suggests that cardiac amylin oligomer accumulation may accelerate the occurrence of heart dysfunction, particularly diastolic dysfunction, a typical sign of diabetic cardiomyopathy.2-10,38,39 Furthermore, the left ventricular end diastolic volume is increased in pre-diabetic HIP rats, which, combined with the unchanged fractional shortening, suggests dilation of the heart (Table 1).
Table 1. Echo and hemodynamic parameters in pre-diabetic HIP rats (n=6) vs. control (n=10). LVEDD-Left-ventricular end diastolic diameter.
| Ctl. | HIP | |
|---|---|---|
| Heart rate (BPM) | 208±14 | 231±17 |
| Fractional shortening (%) | 66.7±3.4 | 57.8±2.8 |
| LVEDD (mm) | 6.4±0.3 | 7.2±0.2* |
| dP/dtmax (mmHg/s) | 7277±247 | 6597±672 |
| -dP/dtmin (mmHg/s) | 6237±411 | 4822±344* |
| LVmax systolic pressure (mmHg) | 107±2 | 99±3* |
| LV end-diastolic pressure (mmHg) | 6.8±0.8 | 5.8±1.4 |
Discussion
We found significant accumulation of large amylin oligomers (>octamers, Figs. 1A,B,F), fibrillar tangles (Fig. 2B), and plaques (Fig. 2A,C,D) in failing hearts from patients with obesity and type-2 diabetes, but not in normal hearts and failing hearts from lean humans without diabetes (Figs. 1C,F). Small amylin aggregates are even elevated in non-failing hearts from overweight/obese patients (Fig. 1C-E), suggesting that cardiac amylin buildup starts in the early state of insulin resistance/pre-diabetes. HIP rats, which express human amylin in the pancreas, accumulate amylin oligomers in the heart (Fig. 4A-C) starting also in pre-diabetes. In pre-diabetic HIP rats, the interaction of amylin oligomers with cardiac myocytes results in larger sarcolemmal Ca2+ leak and Ca2+ transients (Figs. 5A,D) leading to activation of Ca2+-mediated hypertrophic pathways (Fig. 6A-D), pathological heart remodeling (Fig. 6E,F), and diastolic dysfunction (Fig. 5 6E,F, Table 1). In contrast, UCD-T2DM rats, which are matched for age, blood glucose level (Fig. 4D), and myocardial insulin responsiveness (Fig. 4E,F), but lack amylin deposition (Fig. 4A,B), have normal cardiac structure (Fig. 4A,B) and function (Fig. 5G-I). These results suggest that cardiac dysfunction in HIP rats is most likely an amylin-mediated effect. Hence, hyperamylinemia and consequent amylin deposition, a toxic effect generally assumed to contribute to pancreatic β-cell dysfunction and development of type-2 diabetes,13,14,31,45,46 may also be causally implicated in cardiac dysfunction.
Pathologically important form of amylin
Conditions underlying amylin oligomerization13-15,45,46 and proteotoxicity21,22 are complex and only poorly understood. Amyloidogenicity of human amylin promotes the attachment to the sarcolemma (Online Fig. V-B) and oligomer formation, two apparently independent processes. Small oligomers develop rapidly at the sarcolemma, i.e. in only 1-2 hours (Online Fig. V), which correlates with an increase in sarcolemmal permeability to Ca2+ (Fig. 3D,E) and elevated Ca2+ transients (Fig. 3A,C), with consequent activation of Ca2+-mediated hypertrophic pathways (Online Fig. VIII). In contrast, rat amylin, a non-amyloidogenic isoform of amylin29 (Online Fig. III), does not affect Ca2+ cycling when incubated with cardiac myocytes (Fig. 3B). These results suggest that the oligomers may be the pathologically important forms of amylin. Amylin oligomerization at the sarcolemma may act as seeds for further amyloid growth. Fibril growth at the membrane amplifies structural alteration of the membrane22 and Ca 2+ dysregulation, aggravating the deleterious effects in the heart. This might be the case for the large amylin oligomers in the 32-64 kDa size range that are abundant in failing hearts from diabetic and obese patients (Figs. 1A,B,F) and in HIP rat hearts (Fig.4C).
There is increasing support for the toxic oligomer hypothesis in amyloid-related diseases,20-28 including cardiomyopathies caused by other amyloidogenic proteins that infiltrate the heart, i.e. transthyretin, immunoglobulin light chain, and serum amyloid.47 Data48,49 suggest that the infiltration of amyloidogenic proteins in the heart may induce cardiotoxicity even before amyloid fibril formation. Moreover, intracellular accumulation of amyloid oligomers, such as those formed by polyglutamine27 or presenilin28, induced cytotoxicity and heart failure in mice. Presenilin oligomers coimmunoprecipitated with SERCA and altered Ca2+ handling.28 Our data from human and HIP rat hearts suggest that amylin oligomer accumulation in the heart, which is an outside-inside cardiac event, is cardiotoxic and may represent an early pathogenic mechanism linking type-2 diabetes with cardiac dysfunction.
Cardiac amylin accumulation and altered myocyte Ca2+ cycling
Our data show that the primary effect of cardiac amylin oligomer accumulation is an increase in myocyte Ca2+ and Ca2+ transients (schematic in Fig. 7). This effect was observed both in cardiac myocytes exposed acutely to human amylin and in pre-diabetic HIP rats, which accumulate amylin oligomers in the heart, but not in pre-diabetic UCD-T2DM rats that express only non-amyloidogenic rat amylin and thus lack cardiac oligomeric amylin accumulation. Such an effect agrees well with previous data showing that the toxicity associated with amyloidogenic proteins is mediated by an initial increase in [Ca2+]i.20,25,37 While the mechanisms underlying the increase of [Ca2+]i are not fully elucidated, our data suggest that an augmented passive trans-sarcolemmal Ca2+ flux is partly responsible. Amylin oligomers may also modulate the function of Ca2+ channels, as proposed in the pathology of Alzheimer's disease.20,50 Elevated [Ca2+]i is involved in transcriptional regulation and hypertrophic signaling in the heart.40,41 Both CaMKII-HDAC and calciuneurin-NFAT hypertrophic pathways are activated in cardiac myocytes from pre-diabetic HIP rats (Fig. 6A-D). Moreover, the calciuneurin-NFAT pathway can be activated even by acute exposure of isolated myocytes to human amylin oligomers (Online Fig. VIII). These data implicate the amylin oligomers as a trigger of hypertrophic and remodeling maladaptive changes in the heart (Fig. 7). Pre-diabetic HIP rats show SERCA downregulation (Fig. 6F), a common occurrence in diabetic cardiomyopathy,51 and increased level of the pro-hypertrophic hormone BNP (Fig. 6E). SERCA downregulation causes further alterations in cardiac myocyte Ca2+ cycling by impairing Ca2+ transient relaxation, which leads to negative force-frequency relationship (Fig. 5D) and elevated diastolic [Ca2+]i (Fig. 5F). Slower Ca2+ transient relaxation and elevated diastolic [Ca2+]i may further activate the CaMKII-HDAC and calcineurin-NFAT transcriptional regulation/hypertrophic pathways and thus aggravate the cardiac hypertrophy and remodeling (Fig. 7). Furthermore, impaired Ca2+ transient relaxation and elevated diastolic [Ca2+]i cause diastolic dysfunction in pre-diabetic HIP rats (Table 1). None of these alterations were present in pre-diabetic UCD-T2DM rats. Alterations in function and/or expression of proteins involved in cardiac Ca2+ cycling, including SERCA, and diastolic dysfunction have been reported in other rodent models of type-2 diabetes, but only after the onset of full-blown type-2 diabetes.7,38,39 Thus, our data indicate that cardiac amylin oligomer accumulation accelerates the occurrence of cardiac dysfunction and remodeling in diabetes.
Figure 7.

Proposed mechanism for amylin oligomer-induced cardiac dysfunction. Amylin oligomers elevate Ca2+ transients, which results in activation of Ca2+-dependent CaMKII-HDAC and calcineurin-NFAT transcriptional regulation/hypertrophic pathways. This may reduce SERCA expression, which further alters myocyte Ca2+ cycling by impairing Ca2+ transient relaxation leading to higher diastolic [Ca2+]i. Impaired Ca2+ transient relaxation and elevated diastolic [Ca2+]i may further activate the Ca2+-dependent transcriptional regulation/hypertrophic pathways (positive feedback) and cause diastolic dysfunction in HIP rats. With the advancement of the disease, reduced SERCA function may cause SR unloading, consequent reduction in Ca2+ transient amplitude, and systolic dysfunction.
Amylin oligomer-induced cardiac phenotype
At the whole heart level, HIP rats show pathological signs of an infiltrative disease52 (Table 1). This is characterized by diastolic dysfunction (significantly reduced -dP/dtmin) and unchanged fractional shortening, which is one type of diastolic heart failure.52 Diastolic heart failure often progresses to systolic failure. Hemodynamics and echocardiographic measurements in HIP rats show indeed significantly reduced maximum rate of pressure fall (-dP/dtmin), an index of diastolic dysfunction, along with unchanged fractional shortening (Table 1). The left-ventricular maximum systolic pressure is also significantly reduced in HIP rats (Table 1). Hence, pre-diabetic HIP rats display cardiac changes resembling the cardiac infiltrative disease in humans.52 Diastolic dysfunction is also important in the pathogenesis of diabetic cardiomyopathy.2-10,38,39 The molecular mechanisms underlying diastolic dysfunction in diabetic patients are poorly understood.2-10,38,39 Present data suggest that cardiac amylin oligomer accumulation is linked to Ca2+ dysregulation and pathological cardiac hypertrophy, which may accelerate the onset of diastolic dysfunction in diabetes.
The pathogenesis of diabetic cardiomyopathy is multifactorial and includes metabolic components 2-10,38,39 which have not been studied here. Additional work to address any potential influences of the amylin oligomers on key metabolic processes in the heart is needed.
In conclusion, our data show that patients with obesity and type-2 diabetes accumulate amylin oligomers in the heart and suggest that this accelerates the development of cardiac dysfunction. We propose that detection and disruption of cardiac amylin buildup may be a predictor of myocardial dysfunction and a novel therapeutic target in diabetic cardiomyopathy.
Supplementary Material
Novelty and Significance.
What is known?
Patients with obesity and insulin resistance have elevated circulating levels of amylin, an amyloidogenic hormone co-expressed and co-secreted with insulin by pancreatic β-cells.
At increased concentrations, amylin readily form amyloids, which are cytotoxic and contribute to the development of type-2 diabetes.
The most toxic species of amylin amyloids are the soluble oligomers, which attach to cellular membranes causing Ca2+ dyshomeostasis, cell dysfunction, and apoptosis.
What new information does this article contribute?
Amylin oligomers accumulate in the heart and are associated with cardiac failure in patients with obesity and type-2 diabetes.
Amylin oligomer buildup in the heart of rats transgenic for human amylin is linked to myocyte Ca2+ dysregulation, pathological cardiac hypertrophy and remodeling, and diastolic dysfunction.
Cardiac amylin accumulation accelerates the onset of diabetic cardiomyopathy.
Summary
Obesity and insulin resistance increase the risk for both type-2 diabetes and cardiac disease, but the underlying mechanisms remain poorly understood. In addition to hyperglycemia and dyslipidemia, patients with obesity and insulin resistance present also hyperinsulinemia and hyperamylinemia. While the hyperinsulinemic response prevents a large fraction of insulin resistant patients from developing type-2 diabetes, the coincident hyperamylinemia leads to proteotoxicity and amyloid deposition in pancreatic islets. Here we show that amylin oligomers, fibrils and plaques also accumulate in failing hearts from obese and diabetic patients, but not in non-failing hearts and failing hearts from lean, non-diabetic humans. Using rats transgenic for human amylin, we show that cardiac amylin oligomer accumulation causes myocyte Ca2+ dysregulation, activation of Ca2+-dependent pathological cardiac hypertrophy and remodeling, and diastolic dysfunction. Our data suggest that cardiac amylin accumulation accelerates the onset of diabetic cardiomyopathy. The present results show for the first time that amylin oligomers are a direct pathogenic link between pancreatic and cardiac disorders and an independent contributor to the multifactorial pathogenesis of diabetic cardiomyopathy. We propose that detection and disruption of cardiac amylin buildup may be a predictor of myocardial dysfunction and a novel therapeutic target in diabetic cardiomyopathy.
Acknowledgments
We thank Ms. Christine Malloy, RN and Mr. James Graham, MSc for technical help.
Sources of Funding: Work was supported in part by AHA (BGIA2220165 to FD), NSF (CBET 1133339 to FD), NIH (RO1-HL109501 to SD; RO1-HL089847, RO1-AG017022 to KBM; RO1-HL077281, RO1-HL079071 to AAK; HL075675, HL091333, AT003645, DK087307, HL107256 to PJH; RO1-HL073162, RO1-HL061483 to HT; P01-HL080101 to DMB), a Multicampus Award from the University of California, Office of the President (PJH), and a Vision Grant from University of California-Davis Health System (FD).
Non-standard Abbreviations and Acronyms
- BMI
body mass index
- BNP
brain natriuretic peptide
- HDAC
histone deacetylase
- HF
heart failure
- HIP rats
type-2 diabetic rat model transgenic for human amylin
- IAPP
islet amyloid polypeptide
- NF
non-failing
- NFAT
nuclear factor of activated T cells
- OB
obese
- OW
overweight
- PD
pre-diabetic
- UCD-T2DM rats
type-2 diabetic rat model expressing only wild-type rat amylin
Footnotes
National Center for Health Statistics (2009)
Disclosures: None
References
- 1.Biddinger SB, Kahn CR. From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol. 2006;68:123–158. doi: 10.1146/annurev.physiol.68.040104.124723. [DOI] [PubMed] [Google Scholar]
- 2.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 3.Reaven GM. Relationships among insulin resistance, type 2 diabetes, essential hypertension, and cardiovascular disease: similarities and differences. J Clin Hypertens. 2011;13:238–243. doi: 10.1111/j.1751-7176.2011.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Battiprolu PK, Gillette TG, Wang ZV, Lavandero S, Hill JA. Diabetic Cardiomyopathy: Mechanisms and Therapeutic Targets. Drug Discov Today Dis Mech. 2010;7:e135–e143. doi: 10.1016/j.ddmec.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guha A, Harmancey R, Taegtmeyer H. Nonischemic heart failure in diabetes mellitus. Curr Opin Cardiol. 2008;23:241–248. doi: 10.1097/HCO.0b013e3282fcc2fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–39. doi: 10.1007/s11154-010-9131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lebeche D, Davidoff AJ, Hajjar RJ. Interplay between impaired calcium regulation and insulin signaling abnormalities in diabetic cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5:715–724. doi: 10.1038/ncpcardio1347. [DOI] [PubMed] [Google Scholar]
- 8.Young LH. Diet-induced obesity obstructs insulin signaling in the heart. Am J Physiol Heart Circ Physiol. 2010;298:H306–307. doi: 10.1152/ajpheart.01088.2009. [DOI] [PubMed] [Google Scholar]
- 9.Swan JW, Anker SD, Walton C, Godsland IF, Clark AL, Leyva F, Stevenson JC, Coats AJ. Insulin resistance in chronic heart failure: relation to severity and etiology of heart failure. J Am Coll Cardiol. 1997;30:527–532. doi: 10.1016/s0735-1097(97)00185-x. [DOI] [PubMed] [Google Scholar]
- 10.Ashrith G, Algahim MF, Taegtmeyer H. Insulin resistance: marker or mediator? Am J Med. 2009;122:e13–e15. doi: 10.1016/j.amjmed.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Utriainen T, Takala T, Luotolahti M, Ronnemaa T, Laine H, Ruotsalainen U, Haaparanta M, Nuutila P, Yki-Jarvien H. Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia. 1998;41:555–559. doi: 10.1007/s001250050946. [DOI] [PubMed] [Google Scholar]
- 12.Jagasia D, Whiting JM, Concato J, Pfau S, McNulty PH. Effect of non-insulin-dependent diabetes mellitus on myocardial insulin responsiveness in patients with ischemic heart disease. Circulation. 2001;103:1734–1739. doi: 10.1161/01.cir.103.13.1734. [DOI] [PubMed] [Google Scholar]
- 13.Johnson KH, O'Brien TD, Jordan K, Westermark P. Impaired glucose tolerance is associated with increased islet amyloid polypeptide (IAPP) immunoreactivity in pancreatic beta cells. Am J Pathol. 1989;135:245–250. [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson KH, O'Brien TD, Betsholtz C, Westermark P. Islet amyloid, islet amyloid polypeptide and diabetes mellitus. N Engl J Med. 1989;321:513–518. doi: 10.1056/NEJM198908243210806. [DOI] [PubMed] [Google Scholar]
- 15.Enoki S, Mitsukawa T, Takemura J, Nakazato M, Aburaya J, Toshimori H, Matsukara S. Plasma islet amyloid polypeptide levels in obesity, impaired glucose tolerance and non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract. 1992;15:97–102. doi: 10.1016/0168-8227(92)90074-2. [DOI] [PubMed] [Google Scholar]
- 16.Gong W, Liu ZH, Zeng CH, Peng A, Chen HP, Zhou H, Li LS. Amylin deposition in the kidney of patients with diabetic nephropathy. Kidney Int. 2007;72:213–218. doi: 10.1038/sj.ki.5002305. [DOI] [PubMed] [Google Scholar]
- 17.Despa S, Chen L, Cummings B, Havel PJ, Margulies KB, Knowlton AA, Bers DM, Despa F. Cardiac consequences of increased amylin secretion in diabetics. Circulation. 2009;120:S457. [Google Scholar]
- 18.Despa S, Bers DM, Despa F. Accumulation of islet amyloid polypetide (IAPP) oligomers in the heart in type-2 diabetes alters Ca cycling in myocytes. Circulation. 2010;122:A18345. [Google Scholar]
- 19.Colby DW, Prusiner B. Prions. Cold Spring Harb Perspect Biol. 2011;3(1) doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 21.Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48:491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- 22.Engel MF, Khemtémourian L, Kleijer CC, Meeldijk HJ, Jacobs J, Verkleij AJ, de Kruijff B, Killian JA, Höppener JW. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc Natl Acad Sci USA. 2008;105:6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anguiano M, Nowak RJ, Lansbury PT., Jr Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry. 2002;41:11338–11343. doi: 10.1021/bi020314u. [DOI] [PubMed] [Google Scholar]
- 24.Mattson MP, Goodman Y. Different amyloidogenic peptides share a similar mechanism of neurotoxicity involving reactive oxygen species and calcium. Brain Res. 1995;676:219–224. doi: 10.1016/0006-8993(95)00148-j. [DOI] [PubMed] [Google Scholar]
- 25.Kawahara M, Kuroda Y, Arispe N, Rojas E. Alzheimer's beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem. 2000;275:14077–14083. doi: 10.1074/jbc.275.19.14077. [DOI] [PubMed] [Google Scholar]
- 26.Casas S, Novials A, Reimann F, Gomis R, Gribble FM. Calcium elevation in mouse pancreatic beta cells evoked by extracellular human islet amyloid polypeptide involves activation of the mechanosensitive ion channel TRPV4. Diabetologia. 2008;51:2252–2262. doi: 10.1007/s00125-008-1111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pattison JS, Sanbe A, Maloyan A, Osinska H, Klevitsky R, Robbins J. Cardiomyocyte Expression of a Polyglutamine Preamyloid Oligomer Causes Heart Failure. Circulation. 2008;117:2743–2751. doi: 10.1161/CIRCULATIONAHA.107.750232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gianni D, Li A, Tesco G, McKay KM, Moore J, Raygor K, Rota M, Gwathmey JK, Dec GW, Aretz T, Leri A, Semigran MJ, Anversa P, Macgillivray TE, Tanzi RE, del Monte F. Protein aggregates and novel presenilin gene variants in idiopathic dilated cardiomyopathy. Circulation. 2010;121:1216–1226. doi: 10.1161/CIRCULATIONAHA.109.879510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westermark P, Engström U, Johnson KH, Westermark GT, Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc Natl Acad Sci USA. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matveyenko AV, Butler PC. Islet amyloid polypeptide (IAPP) transgenic rodents as models for Type 2 Diabetes. ILAR Journal. 2006;47:225–233. doi: 10.1093/ilar.47.3.225. [DOI] [PubMed] [Google Scholar]
- 31.Matveyenko AV, Butler PC. β-cell deficit due to increased apoptosis in the human islet amyloid polypeptide transgenic (HIP) rat recapitulates the metabolic defects present in type-2 diabetes. Diabetes. 2006;55:2106–2114. doi: 10.2337/db05-1672. [DOI] [PubMed] [Google Scholar]
- 32.Cummings BP, Digitale EK, Stanhope KL, Graham JL, Baskin DG, Reed BJ, Sweet IR, Griffen SC, Havel PJ. Development and characterization of a novel rat model of type 2 diabetes mellitus: the UC Davis type 2 diabetes mellitus UCD-T2DM rat. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1782–1793. doi: 10.1152/ajpregu.90635.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.http://www.nlm.nih.gov/medlineplus/ency/article/000313.htm
- 34.Despa S, Bers DM. Functional analysis of Na/K-ATPase isoform distribution in rat ventricular myocytes. Am J Physiol - Cell Physiol. 2007;293:C321–C327. doi: 10.1152/ajpcell.00597.2006. [DOI] [PubMed] [Google Scholar]
- 35.Lin L, Kim SC, Wang Y, Gupta S, Davis B, Simon SI, Torre-Amione G, Knowlton AA. HSP60 in heart failure: abnormal distribution and role in cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. 2007;293:H2238–H2247. doi: 10.1152/ajpheart.00740.2007. [DOI] [PubMed] [Google Scholar]
- 36.Walton JH, Berry RS, Despa F. Amyloid oligomer formation probed by water proton magnetic resonance spectroscopy. Biophys J. 2010;100:2302–2308. doi: 10.1016/j.bpj.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang CJ, Gurlo T, Haataja L, Costes S, Daval M, Ryazantsev S, Wu X, Butler AE, Butler PC. Calcium-activated calpain-2 is a mediator of beta cell dysfunction and apoptosis in type 2 diabetes. J Biol Chem. 2010;285:339–348. doi: 10.1074/jbc.M109.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Netticadan T, Temsah RM, Kent A, Elimban V, Dhalla NS. Depressed levels of Ca2+-cycling proteins may underlie sarcoplasmic reticulum dysfunction in the diabetic heart. Diabetes. 2001;50:2133–2138. doi: 10.2337/diabetes.50.9.2133. [DOI] [PubMed] [Google Scholar]
- 39.Pereira L, Matthes J, Schuster I, Valdivia HH, Herzig S, Richard S, Gómez AM. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes. 2006;55:608–615. doi: 10.2337/diabetes.55.03.06.db05-1284. [DOI] [PubMed] [Google Scholar]
- 40.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 41.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–80. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 42.Luchner A, Stevens TL, Borgeson DD, Redfield M, Wei CM, Porter JG, Burnett JC., Jr Differential atrial and ventricular expression of myocardial BNP during evolution of heart failure. Am J Physiol Heart Circ Physiol. 1998;274:H1684–H1689. doi: 10.1152/ajpheart.1998.274.5.H1684. [DOI] [PubMed] [Google Scholar]
- 43.Ellmers LJ, Knowles JW, Kim HS, Smithies O, Maeda N, Cameron VA. Ventricular expression of natriuretic peptides in Npr1(-/-) mice with cardiac hypertrophy and fibrosis. Am J Physiol Heart Circ Physiol. 2002;283:H707–H714. doi: 10.1152/ajpheart.00677.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bell D, Schluter KD, Zhou XJ, McDermott BJ, Piper HM. Hypertrophic effects of calcitonin gene related peptide (CGRP) and amylin on adult mammalian ventricular cardiomyocytes. J Mol Cell Cardiol. 1995;27:2433–2443. doi: 10.1006/jmcc.1995.0231. [DOI] [PubMed] [Google Scholar]
- 45.Ionescu-Tirgoviste C, Despa F. Biophysical Alteration of the Secretory Track in β-Cells Due to Molecular Overcrowding: The Relevance for Diabetes. Integr Biol (Camb) 2011;3:173–179. doi: 10.1039/c0ib00029a. [DOI] [PubMed] [Google Scholar]
- 46.Despa F. Endoplasmic reticulum overcrowding as a mechanism of beta-cell dysfunction in diabetes. Biophys J. 2010;98:1641–1648. doi: 10.1016/j.bpj.2009.12.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52:347–361. doi: 10.1016/j.pcad.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 48.Palladini G, Lavatelli F, Russo P, Perlini S, Perfetti V, Bosoni T, Obici L, Bradwell AR, D'Eril GM, Fogari R, Moratti R, Merlini G. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood. 2006;107:3854–3858. doi: 10.1182/blood-2005-11-4385. [DOI] [PubMed] [Google Scholar]
- 49.Liao R, Jain M, Teller P, Connors LH, Ngoy S, Skinner M, Falk RH, Apstein CS. Infusion of light chains from patients with cardiac amyloidosis causes diastolic dysfunction in isolated mouse hearts. Circulation. 2001;104:1594–1597. [PubMed] [Google Scholar]
- 50.Green KN, LaFerla FM. Linking calcium to abeta and Alzheimer's disease. Neuron. 2008;59:190–194. doi: 10.1016/j.neuron.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 51.Razeghi P, Young ME, Cockrill TC, Frazier OH, Taegtmeyer H. Downregulation of myocardial myocyte enhancer factor 2C and myocyte enhancer factor 2C-regulated gene expression in diabetic patients with nonischemic heart failure. Circulation. 2002;106:407–11. doi: 10.1161/01.cir.0000026392.80723.dc. [DOI] [PubMed] [Google Scholar]
- 52.Seward JB, Casaclang-Verzosa G. Infiltrative cardiovascular diseases: cardiomyopathies that look alike. J Am Coll Cardiol. 2010;55:1769–79. doi: 10.1016/j.jacc.2009.12.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.