

Abstract
Epstein-Barr virus (EBV) is the causal agent in the etiology of Burkitt's lymphoma and nasopharyngeal carcinoma and is also associated with multiple human malignancies, including Hodgkin's and non-Hodgkin's lymphoma, and posttransplantation lymphoproliferative disease, as well as sporadic cancers of other tissues. A causal relationship of EBV to these latter malignancies remains controversial, although the episomic EBV genome in most of these cancers is clonal, suggesting infection very early in the development of the tumor and a possible role for EBV in the genesis of these diseases. Furthermore, the prognosis of these tumors is invariably poor when EBV is present, compared to their EBV-negative counterparts. The physical presence of EBV in these tumors represents a potential “tumor-specific” target for therapeutic approaches. While treatment options for other types of herpesvirus infections have evolved and improved over the last two decades, however, therapies directed at EBV have lagged. A major constraint to pharmacological intervention is the shift from lytic infection to a latent pattern of gene expression, which persists in those tumors associated with the virus. In this paper we provide a brief account of new virus-targeted therapeutic approaches against EBV-associated malignancies.
1. Introduction
Epstein-Barr virus (EBV) infection is ubiquitous in human populations worldwide. EBV infection in children and adolescents usually leads to a self-limiting lytic infection, designated as infectious mononucleosis (IM) [1, 2]. However, in immunocompromised individuals, such as those with X-linked lymphoproliferative disease (XLP) [3, 4], EBV infections often progress unchecked and are lethal. EBV is invariably associated with nasopharyngeal carcinoma (NPC) [5], African Burkitt's lymphoma (BL) [6], posttransplantation lymphoproliferative disease (PTLD) [7–10], and less often with a number of other human malignancies such as Hodgkin's lymphoma (HD) [11], and non-Hodgkin's lymphomas (NHL). In addition, EBV is found in a fraction of gastric carcinomas [12, 13] and carcinomas of the breast [14–16]. Although EBV has been identified in these latter tumors, it remains controversial whether EBV is causally-related to their development. Nonetheless, multiple studies have clearly demonstrated that the presence of EBV in these tumors confers a poorer prognosis [17–22].
In the mid-Eighties, the technique of random cleavage of the terminal repeat region of the EBV genome was employed as a method of identifying clonality of the virus episome population in infected cells or tissues [23, 24]. During EBV lytic replication (described later), long head-to-tail concatameric DNA is produced from the circular episomal DNA, which is then cleaved randomly within the terminal repeat region by viral-encoded terminase, leading to the production of virus particles with varying length of their terminal repeats [25]. Latently infected B-cells, however, do not produce virus particles and the circularized EBV episomal genome replicates in coordination with the division of cellular genome, producing exact copies of the viral genome in daughter cells. Multiple studies using this type of analysis clearly demonstrated that the EBV genome in many of the EBV-associated tumors, such as BL [26], NPC [23], and HD [11], is clonal in nature, strongly suggesting that these tumors developed from a single progenitor cell that was already infected with EBV, lending further support to the concept that EBV may be causally related to the genesis of many of these tumors.
2. EBV Infection and Replication
EBV transmission usually takes place through the mucosal secretions of the mouth of an infected individual. Primary infection of epithelial cells of the oropharynx leads to active production of virus particles with shedding of the virus in saliva. Although the EBV-epithelial cell attachment process is not fully understood, the neighboring B-cells subsequently become infected via interaction of the EBV surface protein gp350 with the lymphocyte receptor CD21, however, such infections are often nonproductive. Active or “lytic” replication of EBV induces lysis of infected cells concurrent with production of virus particles, whereas latent replication of EBV does not. EBV is a member of the gamma herpesvirus family, with a large 172 Kb double-stranded linear DNA genome encoding nearly 100 genes. Most of these genes are expressed during lytic-phase replication, whereas only a maximum of eleven viral genes are expressed during latent-phase replication. The up to eleven EBV gene products that are expressed in latently infected cells (the number depends upon the type of latency) include six nuclear antigens (EBNA1, EBNA2, EBNA3A-3B-3C, EBNA-LP), three latent membrane proteins (LMP1, LMP2A-2B), the BARF0 protein, generated from BART transcripts, and two small noncoding non-poly-A RNAs (EBER1 and EBER2). Primary EBV infection results in strong humoral and cellular immune responses. IgM antibodies against EBV surface protein (gp350) are easily detectable in the serum during primary infection, which is then eclipsed by a steady state level of IgG antibody over the ensuing months and beyond [27].
The symptoms of acute infection, such as IM, subside within few weeks as EBV enters a latent replication mode. EBV subsequently generates a life-long persistent infection in all infected individuals. Seroepidemiological surveys demonstrate that more than 90% of humans are positive for EBV antibody [28, 29]. The majority of infected individuals bear cytotoxic T cells directed against the virus, and at any given time only a small proportion of resting B cells are latently infected with the virus (one or two in a million) [30, 31]. EBV infection of primary human B lymphocytes in vitro leads to their immortalization and the development of continuously growing lymphoblastoid cell lines (LCL) [32]. In a healthy individual, however, a strict equilibrium between proliferation of EBV-infected B cells and immune surveillance is maintained [33]. In the setting of immunodeficiencies (whether hereditary or induced), however, this equilibrium is lost and the resulting unchecked proliferation of virus-immortalized B cells can then lead to the development of lymphoma, such as XLP or PTLD.
3. Therapies against EBV Diseases
Existing therapeutic approaches for EBV-associated diseases are broadly categorized into three groups, as shown in Table 1.
Table 1.
Available therapies for EBV diseases.
| Modalities | Classification | Comments |
|---|---|---|
| Drug treatment | Acyclovir/ganciclovir | Inhibits virus replication and induces cell killing. Effective only against lytic EBV infection. |
|
| ||
| Immunotherapy | Vaccination | Recombinant gp350-based vaccine, CTL epitope peptide-based vaccine. |
| Treatment of disease | Monoclonal antibody against CD21, anti CD-20 antibody (Rituximab); EBV-specific cytotoxic T cells from donor or ex vivo amplification of CTLs from patients and infusion back to the patient. | |
|
| ||
| Virus-directed approaches | Utilization of presence of virus in the tumor | Various cell-killing strategies that are dependent on EBV gene expression. |
3.1. Pharmacological Therapy
Nucleoside-analog anti-herpesvirus drugs, such as ganciclovir, acyclovir, or famcyclovir, are moderately efficient in suppressing virus replication and virus shedding during diseases characterized by acute or lytic replication of EBV. They are not active in EBV-associated malignancies, however, because in EBV-associated malignant diseases the EBV maintains a latent state of replication. These commonly-used antiviral agents are prodrugs, and require conversion to their active form by virus-encoded kinases before they can be effective (see Figure 1). For the Epstein-Barr virus, however, these viral kinases are expressed only during lytic replication.
Figure 1.
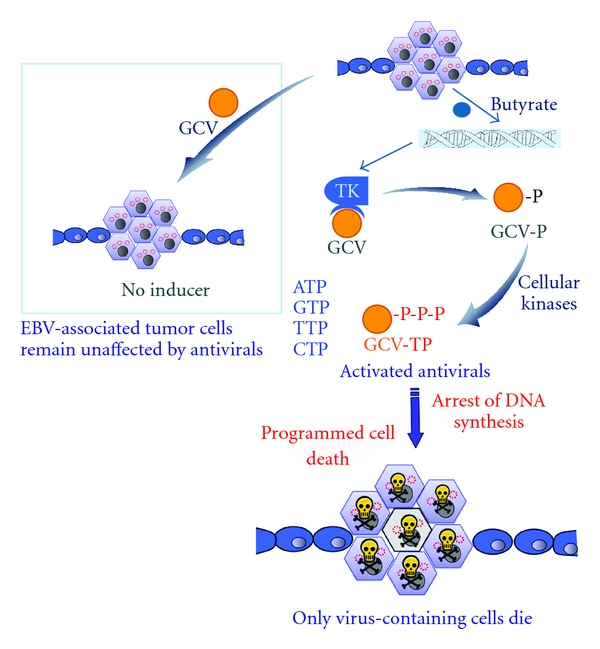
Schematic diagram of combination therapy approaches in EBV malignancies. EBV maintains latent replication in tumor cells and these tumor cells are not susceptible to anti-herpesvirus prodrugs, such as GCV. In the presence of lytic-phase gene expression-inducing agents such as butyrate, the latent EBV expresses thymidine kinase (TK) which converts the prodrug GCV to GCV-P, which is then converted to the (cytotoxic) triphosphate form by cellular kinases. During DNA replication, the triphosphate form of GCV is then incorporated into genomic and viral DNA, causing chain termination, cell-cycle arrest, and apoptosis of the EBV-infected cells.
3.2. Immunotherapy
Immunotherapeutic approaches have been studied in clinical trials for a number of years, with success in some cases. Normally, the host's CD4+ and CD8+ cytotoxic T cells and natural killer (NK) cells play an important role in killing EBV-infected cells during primary infection. Although EBV can often be cultured from throat washings of previously infected individual, continuing CTL immunosurveillance in normal individuals is quite efficient in controlling subsequent reactivation of EBV infection. Vaccination with recombinant gp350 viral glycoprotein or CTL epitope-based peptide has been successful in generating viral immunity in animal models [34–36] and may in the future prove useful in areas that are endemic for EBV malignancies (such as China and southeast Asia). Clinical vaccine trials in healthy individuals demonstrated the appearance of neutralizing anti-EBV antibodies in vaccinated individuals [37, 38]. However, the ubiquitous nature of EBV infection but low incidence of malignancies arising from the infected individuals makes prevention of EBV-associated malignancies of lesser importance than the control of the malignancy once it has occurred. Adoptive transfer of EBV-specific CTLs from an EBV-positive donor to the transplant recipient has been utilized in a limited fashion in the treatment of PTLDs and other solid tumors [39–42]. CTLs may also be isolated from a recipient's own lymphocytes, expanded in vitro, and infused back into the patient [43, 44]. These approaches have provided some clinical benefit in certain highly selected patients, particularly in the treatment of PTLD [40, 45, 46]. However, adoptive transfer of EBV-specific CTLs has not been as effective in patients with NPC or HD [47]. This approach is constrained by the availability of donor lymphocytes, and the long time required for the in vitro processing and expansion of the CTL. Furthermore, the requirement for prior lymphodepletion for in vivo CTL expansion is also a major obstacle. Radiation and chemotherapy-induced lymphodepletion often lead to multiple unwanted side effects. Recently, CD45 monoclonal antibodies are being used to induce a short-term lymphodepleted environment without unwanted side effects, allowing subsequent expansion of infused EBV-specific CTLs [48]. A recent comprehensive review of EBV-specific T-cell therapies currently under investigation is available [49].
4. Virus-Targeted Therapies
In most EBV-associated malignancies, all or nearly all of the tumor cells contain the viral genome. Furthermore, at any given time, the number of EBV-infected nontumor cells present in other physiological compartments of the host is usually very low, and for B cells is on the order of one in a million. This provides a unique opportunity to develop therapeutic strategies utilizing the presence of the viral genome of EBV in the tumors as an essentially “tumor-specific” target. One of the virus-targeted therapeutic strategies is based on the concept that EBV-containing cells will die if lytic replication can be induced. Other strategies employ selective expression of toxins in EBV-infected cells or preventing the function of EBV latent gene products that are linked to oncogenesis (Table 2). Elimination of episomal EBV genomes by low dose hydroxyurea treatment has been shown to decrease the tumorigenic potential of Akata cells of BL origin, both in vitro and in SCID mice [50]. When two patients with AIDS-related (EBV-positive) primary lymphoma of the central nervous system were treated with low dose hydroxyurea, their median survival compared to historical controls increased by almost 18 months [51]. The effectiveness of this approach in a controlled clinical trial, however, has yet to be evaluated. Expression of antisense RNA against the EBV LMP-1 protein has been shown to reduce LMP-1 expression in LCLs and concomitantly inhibit cell proliferation and stimulate apoptosis [52]. As EBNA1 is a viral transactivator expressed in all latently EBV-infected tumor cells and utilizes the OriP promotor for its activity, several studies have utilized an OriP-based vector to direct the expression of cellular toxins, such as driving cytosine deaminase expression (which converts the prodrug 5-flurocytosine to cytotoxic 5-flurouracil), or the herpes simplex virus TK, to make the cells susceptible to nucleoside analog antiviral drugs [53, 54]. Targeted delivery of these EBV-dependent vectors specifically to the tumors cells, however, remains a serious and unresolved challenge.
Table 2.
Virus-directed novel approaches.
| Classes | Comments | Reference |
|---|---|---|
| Targeting EBV episome | Low dose hydroxyurea treatment | Chodosh et al. [50], Slobod et al. [51] |
|
| ||
| Inhibition of EBV transforming protein | Antisense RNA against LMP-1 oncoprotein | Kenney et al. [52] |
| EBV-dependent expression of cellular toxins | Expression of detrimental cellular proteins through OriP dependent expression vector | Hirai et al. [53], Kenney et al. [54] |
|
| ||
| Combination therapy | Induction of EBV lytic replication + cytotoxic drugs | Numerous, Listed in Table 3 |
The most significant advancement in virus-targeted therapies for EBV malignancies is undoubtedly the combination therapy that is based on artificial induction of EBV lytic-phase gene expression, followed by exposure of the tumor cells to anti-herpesvirus drugs. It was established thirty years ago that induced expression of the herpes simplex virus TK enzyme renders cells susceptible to prodrugs such as ganciclovir or acyclovir [55]. Gutiérrez et al. first utilized this approach in EBV lymphoma cells by transfecting a Zta expression plasmid under the control of an OriP promoter. This plasmid also expressed HSV-TK. Transfection of this plasmid in the EBV-positive BL cell line P3HR1 induced lytic-phase gene expression, and in the presence of ACV, significantly reduced the growth of P3HR1 cells [56]. In two other studies, adenoviral vectors containing a BZLF1 or BRLF1 expression cassette under the control of the CMV promoter were used to infect the EBV-positive BL cell line, an NPC cell line, and an EBV-transformed gastric carcinoma cell lines [57, 58]. The BZLF1 and BRLF1 proteins both initiate the lytic replication cycle of EBV. In both of these studies, adenoviral vector-mediated BZLF1 and BRLF1 expression induced EBV-lytic phase proteins, such as BMRF1, in all the cell lines tested, whereas the empty adenovirus vector (expressing only GFP) had no such effect. When these adenovirus vectors were injected directly into xenografted tumors in SCID mice induced by the BL or NPC lines, BZLF1 and BLRF1 expression in the tumors was readily detected. There was a substantial coincident reduction of xenograft tumor size in the BZLF1- or BRLF1-adenovirus vector-injected tumors compared to controls [58]. The addition of GCV, however, did not appear to have any additional effects on tumor burden. The authors speculated that high level expression of EBV immediate-early genes, such as BZLF1 or BRLF1, in normal cells could be toxic and acknowledged that there is no reliable gene delivery system yet available to target only EBV-containing cells [59].
The mechanisms underlying induction of EBV lytic replication out of latency using chemical inducers such as phorbol esters (PMA), 5-Azacytidine (5-Aza-C), sodium butyrate, and other agents have been studied for decades [60–64]. These specific agents are known to induce or support the expression of EBV immediate-early genes. In the case of PMA, this effect is the result of protein kinase C-mediated activation of the Jun-Fos proteins and their interaction with AP-1 binding sites on the regulatory elements of the EBV IE genes [65]. Exposure to 5-Aza-C removes the transcriptional block on expression of the EBV genome during latency, caused by extensive CpG methylation [66]. Butyrate facilitates reactivation of latent EBV by allowing remodeling of the chromatin-like structure of the EBV genome, through its histone deacetylase (HDAC) inhibitory activity [67, 68]. The recent discovery that butyrate and other HDAC inhibitors (HDACi) can also induce demethylation and reactivation of methylated, silenced genes through repression of DNA methyltransferase 1 DNMT1 [69] may also contribute to their activity in inducing EBV lytic-phase gene expression. Combination therapy using a chemical inducer of EBV lytic-phase gene expression (arginine butyrate) and the anti-herpesvirus prodrug GCV was first tested on lymphoma cells derived from the EBV-positive lymphoma from a lung transplantation patient [70]. Arginine butyrate exposure induced the lytic-phase gene and protein TK in these lymphoma cells, and, in combination with GCV, inhibited the proliferation of the tumor cells in a dose-dependent manner. This in vitro activity led to a protocol for treatment of the lung transplant patient (who was already receiving GCV with no effect on the lymphoma) with 750 mg/kg/day arginine butyrate therapy for 15 days. The combination therapy was well tolerated by the patient, as no additional toxicity was observed. Although the patient eventually succumbed to an unrelated Aspergillus infection, pathological examination revealed substantial necrosis of the tumor following the treatment [70]. This study demonstrated for the first time the feasibility of combination therapy in the treatment of EBV malignancies in human subjects. Shortly thereafter, other inducers of EBV lytic replication were investigated in vitro and also in xenografted EBV tumor models in mice. Westphal et al. employed γ-radiation or sodium butyrate treatment to induce EBV lytic-phase gene expression in LCLs, BL cells Akata and Jijoye, and in an EBV-transformed gastric carcinoma cell line AGS [71]. Although the extent of EBV reactivation was variable within different cell lines, on average, 400 cG irradiation induced the immediate-early BMRF1 protein in all of the lines. Induction of BMRF1 by sodium butyrate (up to 2.5 mM) was inconsistent in this study, although a combination of butyrate and irradiation markedly induced BMRF1, irrespective of the cell lines tested. When xenografted tumors in SCID mice induced by these EBV-positive cell lines were exposed to a single dose of γ-radiation (400 cG), or were injected intraperitoneally with 500 μL of 50 mM sodium butyrate, the BMRF1 protein was induced in the tumors, although less efficiently by butyrate than by γ-irradiation. A subset of the xenografted animals that were exposed to γ-radiation were also treated with GCV and followed over a month. Tumors in three out of four mice in this group did not progress in size. This study therefore supported the earlier findings in the clinical trial that the combination of induction of EBV lytic-phase proteins and treatment with anti-herpesvirus drugs may be a useful therapeutic approach.
Based on the results of our previous combination treatment approach for the EBV lymphoma in a lung transplant recipient [70], we initiated a phase I/II clinical trial with ten patients, all of whom had EBV-positive lymphoid tumors which were refractory to conventional chemotherapeutics or radiation therapy. The types of tumors in the patients enrolled in the trial included PTLD, B-cell NHL, cutaneous T-cell lymphomas, T/NK lymphomas, and HD. The trial design utilized an intrapatient dose escalation of arginine butyrate combined with a standard dose of GCV. Preliminary results of this study, published in 2001 [72], demonstrated that five out of 10 patients had complete clinical responses and two additional patients had partial responses. Complete necrosis of the EBV lymphoma was noted in two out of the three patients in which pathological analyses were carried out. Analysis of the patient-derived tumor cells in culture again confirmed induction of EBV TK expression and resulting susceptibility to GCV [73]. A complete report of this multicenter multinational trial, with five additional patients included, was reported more recently [74]. Altogether, 10 out of 15 patients, all of whom had tumors refractory to all conventional therapies, showed significant anti-tumor responses, with 4 complete responses and 6 partial responses. Although complications from rapid lysis of the tumors did occur in 3 patients, this study demonstrated that the combination of arginine butyrate and GCV was well tolerated in a broad spectrum of patients. The maximum tolerated dose of arginine butyrate was found to be 1250 mg/kg/day. This controlled phase I/II trial paved the way for studies of this combination therapy approach in larger cohorts. It is also noteworthy that response to this regimen occurred in patients with multiple diverse types of lymphomas, including both lymphomas in which EBV was clearly a causative agent (PTLD), and in tumors where the etiological role of the virus was less clear (B-cell NHL, cutaneous T-cell lymphomas, and T/NK lymphomas). It appears that the presence of the virus in latent form alone is sufficient to render the tumor cells sensitive to this therapeutic approach. In vitro studies, in which the virus is artificially introduced into the tumor cells, have confirmed this (Ghosh and Faller, unpublished).
Several other studies have investigated the potential of other lytic-phase gene expression-inducing agents in preclinical studies, using EBV+ lymphoma cell lines. Some of these studies also utilized mouse models to test the reproducibility of their cell culture findings. DNA methyl transferase inhibitors, HDACi, radiation therapy, proteasomal inhibitors, B-cell receptor-blocking antibodies, and chemotherapeutic drugs were evaluated in these reports. A list of these studies is presented in Table 3. The studies all demonstrated varying degrees of activity in the various model cell lines. The study of Fu et al. [84] included an interesting variation which utilized an 125I-labeled nucleoside analogue, 2′-fluoro-2′-deoxy-ß-D-5-iodouracil-arabinofuranoside (FIAU), to visualize antiviral agent uptake in EBV+ tumors, by specialized CT-imaging. In this study, EBV lytic-phase gene expression in xenografted EBV+ BL tumors in SCID mice was induced by the chemotherapeutic proteasomal inhibitor bortezomib. Biodistribution analyses indicated preferential accumulation of FIAU in the tumors of the bortezomib-treated animals by 48–72 hr, but not in the vehicle-treated animals. The experimental radiopharmaceutical 131I-FIAU was used in a later study by this group which demonstrated xenografted EBV-tumor regression in SCID mice in response to combination treatment with 131I-FIAU and bortezomib by CT imaging [81].
Table 3.
Combination therapy approaches in the treatment of EBV malignancies.
| Lytic replication inducer | Drug | Target cells | In vivo | Reference |
|---|---|---|---|---|
| DNA methylase transferase inhibitors | ||||
| 5-Azacytidine | GCV and 5-bromodeoxyuridine | EBV+ and EBV- BL cells | None | Moore et al. [75] |
|
| ||||
| HDAC inhibitors | ||||
| Arg-Butyrate | GCV | LCL from lung transplant recipient | Single human patient | Mentzer et al. [70] |
| Arg-Butyrate | GCV | 10 human patients | Mentzer et al. [76] | |
| Arg-Butyrate | GCV | 15 human patients | Perrine et al. [74] | |
| Valproic Acid* | Cisplatin, 5-FU, Gemcitabine, Doxorubicin | LCL, gastric carcinoma-EBV, NPC | SCID mice | Feng et al. [77] |
| Na-Butyrate | GCV | P3HR1 | None | Ghosh et al. [78] |
|
| ||||
| Radiation | ||||
| γ-Radiation + Na-butyrate | GCV & AZT | LCL and BL cell lines | SCID mice | Westphal et al. [71] |
| γ-Radiation | AZT+GCV | LCL-4A1A | Nude rats | Roychowdhury et al. [79] |
|
| ||||
| B-cell receptor blockade | ||||
| Rituximab+Dexamethasone | GCV | AKATA | Nude mice | Daibata et al. [80] |
|
| ||||
| Proteasome inhibitor | ||||
| Bortezomib | 131I-FIAU | BL cell line | SCID xenograft | Fu et al. [81] |
|
| ||||
| Other | ||||
| Cis-platinum, 5-fluorouracil, Taxol | GCV | Gastric carcinoma, NPC | Nude mice | Feng et al. [82] |
| Gemcitabine and Doxorubicin | GCV | LCL, and BL cell lines | SCID mice | Feng et al. [83] |
While the basic concept of combination therapy has been tested in multiple in vitro studies and animal studies, and proof-of principle has been documented in the clinical trials, the optimal pharmacological inducer of EBV lytic-phase gene expression has not yet been determined. Ideally, this inducer should be highly active in inducing EBV lytic-phase genes and should have excellent bioavailability. In our clinical trial, we demonstrated that butyrate acts as an effective inducer of EBV lytic replication, and, together with an anti-herpesvirus drug, inhibits tumor growth and induces tumor regression [74, 76]. Because of its very short half-life in vivo, however, butyrate required continuous infusion over several days for therapeutic synergy with antivirals. In a recent study, we demonstrated that multiple short 6–8 hr exposures of P3HR1 cells to butyrate also efficiently induced EBV TK, and in the presence of GCV inhibited tumor cell growth [78]. This observation was translated into a clinical protocol to treat a rituximab-refractory EBV-positive PTLD patient who had received a cord-blood stem cell transplant for Hodgkin's disease [85]. A 5-day infusion of arginine butyrate and 21 days of GCV/valganciclovir treatment resulted in complete resolution of 4 out of 6 lesions in this patient, and decrease in the size of two other lesions. It is also noteworthy that this patient was symptomatic from EBV, CMV, and HHV-6 viremia, with very high plasma viral loads, despite ongoing administration of GCV/valganciclovir. After 1 cycle of combination therapy, these viral levels decreased by several logs or fell to undetectable levels, and her symptoms resolved, suggesting the applicability of this combination approach to multiple types of viruses in addition to EBV.
In vitro studies have demonstrated that HDACi are potent antiproliferative agents that cause cell-cycle arrest, apoptosis and/or differentiation of tumors [86, 87]. Furthermore, their preferential cytotoxic activity on tumor cells over normal cells suggested potential anticancer therapeutic application. In recent years, a number of HDACi have been tested in clinical trials. In addition, efforts have been made to develop more potent, or more HDAC class selective, HDACi. Although HDACi as a class are well-known inducers of EBV lytic-phase gene expression [88], only butyrate and valproic acid have been tested for their activity in treating EBV malignancies [70, 76, 77]. We have recently completed studies to test a variety of HDACi of different chemical classes, including some new and highly potent compounds, for their ability to sensitize EBV-lymphoma cells to anti-herpesvirus drugs. The HDACi studied included short-chain fatty acids (butyrate, valproate), hydroxamic acids (SAHA, oxamflatin, LBH589, scriptaid, PDX101), benzamide (MS275), a cyclic tetrapeptide (apicidin), and largazoles (originally isolated from marine cyanobacterium). With the exception of SAHA and PXD101, all of the other HDACi produced sensitization to GCV, and the combination caused cytotoxicity in EBV+ lymphoma cells (when used as a single agent, PDX101 itself exerted a strong cytotoxic effect on the cells [89, 90]). LBH589, MS275, and synthetic largazole derivatives were 104 to 105 times more potent in killing EBV+ lymphoma cells in presence of GCV, compared to sodium butyrate (Figure 2). The effective concentration of LBH589 was in the range of 50–100 nM, MS275 at 200–500 nM, and largazoles at 100–200 nM. The effectiveness of these HDAC-inhibitory (HDACi) compounds at such low concentrations makes them potentially applicable as sensitizers to antiviral therapeutics for the treatment of EBV-associated lymphomas. Furthermore, butyrate and LBH589 were also found to potently sensitize another BL cell line (Daudi) and a lymphoblastoid cell line (JY) (Figure 3). These findings therefore suggest that that these new and potent HDACi may provide alternative therapeutic options to butyrate, in combination with nucleoside antiviral agents, for the treatment of EBV-associated tumors.
Figure 2.
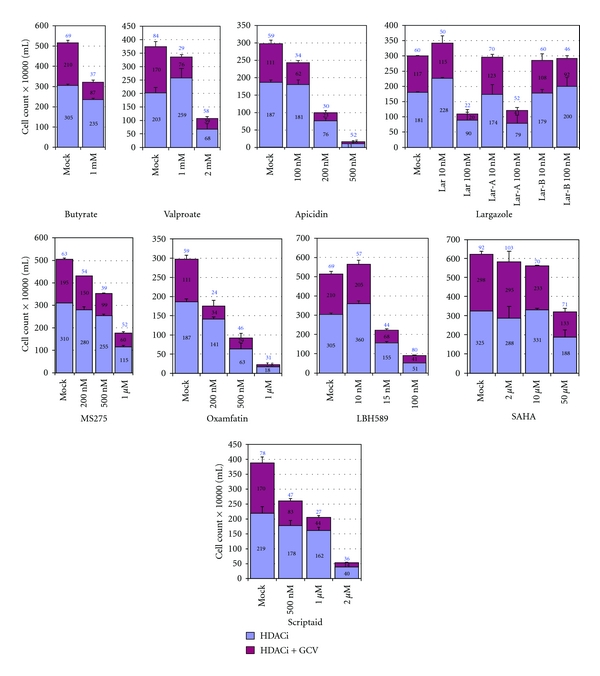
Cytotoxic activity of various HDACi in the presence of an anti-herpesvirus nucleoside analog prodrug, GCV. Three hundred thousand P3HR1 cells were exposed to either 40 μM GCV or vehicle, and the indicated concentrations of individual HDACi, in a 1 mL volume in 24-well plates, in triplicate. Seventy-two hrs later, 800 μL of the media was removed without disturbing the settled cells and 1 mL of fresh growth media containing GCV (40 μM) was added and the cells, which were cultured for another 72 hrs. HDACi studied included butyrate, valproate, apicidin, largazole and its analogs, MS275, oxamflatin, LBH589, SAHA, and Scriptaid. The number above the HDAC+GCV bar represents the percentage of cells surviving, relative to the cultures exposed to that particular HDAC inhibitor alone (assigned a value of 100%). Error bars represent standard deviation.
Figure 3.
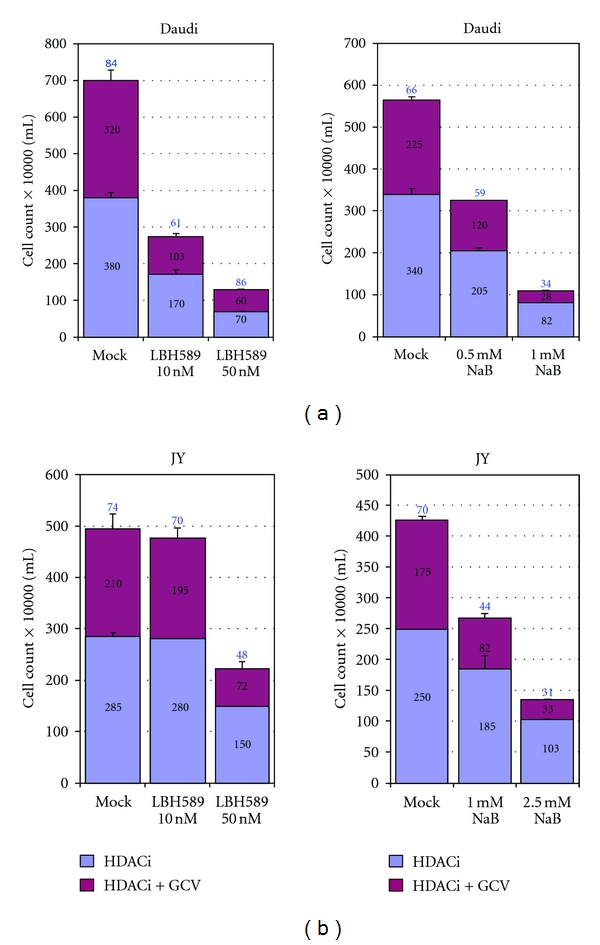
Effect of HDACi and GCV combination treatment on other EBV-positive lymphoma cells. (a) Effect of combination treatment on the BL line Daudi. Four hundred thousand cells per ml per well were cultured with 60 μM GCV in the appropriate wells. Assay parameters were as described in the legend to Figure 2. (b) Effect of combination treatment on the EBV-transformed lymphoblastoid cell line JY. In this case, 200,000 cells per well were plated in the presence of 60 μM GCV, or vehicle, as indicated. Experiments were repeated a minimum of three times and error bars represent standard deviation.
5. Concluding Remarks
EBV-associated malignancies remain a significant health concern worldwide, with particularly higher incidences in southeast Asia and China. In Western countries, the incidence of EBV malignancies, and particularly PTLD, is also on the rise. The increasing use of solid organ or hematopoietic transplantation, especially in situations requiring intense immunosuppression, is most likely a major contributor to this increase in EBV-lymphomas. Furthermore, as referenced above, the presence of EBV in the common NHL and HD lymphomas confers a much poorer prognosis after conventional therapy, demonstrating the need for new therapeutic approaches.
The presence of EBV in these tumors represents a potential “tumor-specific” targeting opportunity for the development of new therapeutics. Among all potential therapeutic avenues explored to date for targeting the virus in EBV-positive tumors, pharmacological induction of EBV lytic infection coupled with an anti-herpesvirus agent appears to be most promising, as a variety of potential virus-inducing agents (some of which are logs more potent than butyrate) are now available, as are active and safe antiviral prodrugs. Furthermore, because a few of the new HDACi with excellent pharmacokinetics and pharmacodynamic profiles are already “clinical-stage,” they could rapidly be employed in clinical trials.
Acknowledgments
This study was supported by grants from the V-Foundation, from the National Cancer Institute CA-153474 (D. V. Faller and S. P. Perrine), and from the Karin Grunebaum Foundation for Cancer Research (D. V. Faller).
References
- 1.Straus SE, Cohen JI, Tosato G, Meier J. Epstein-Barr virus infections: biology, pathogenesis, and management. Annals of Internal Medicine. 1993;118(1):45–58. doi: 10.7326/0003-4819-118-1-199301010-00009. [DOI] [PubMed] [Google Scholar]
- 2.Henle G, Henle W, Diehl V. Relation of Burkitt’s tumor-associated herpes-ytpe virus to infectious mononucleosis. Proceedings of the National Academy of Sciences of the United States of America. 1968;59(1):94–101. doi: 10.1073/pnas.59.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Purtilo DT. X-linked lymphoproliferative disease (XLP) as a model of Epstein-Barr virus-induced immunopathology. Springer Seminars in Immunopathology. 1991;13(2):181–197. doi: 10.1007/BF00201468. [DOI] [PubMed] [Google Scholar]
- 4.Morra M, Howie D, Grande MS, et al. X-linked lymphoproliferative disease: a progressive immunodeficiency. Annual Review of Immunology. 2001;19:657–682. doi: 10.1146/annurev.immunol.19.1.657. [DOI] [PubMed] [Google Scholar]
- 5.Liebowitz D. Nasopharyngeal carcinoma: the Epstein-Barr virus association. Seminars in Oncology. 1994;21(3):376–381. [PubMed] [Google Scholar]
- 6.Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. The Lancet. 1964;283(7335):702–703. doi: 10.1016/s0140-6736(64)91524-7. [DOI] [PubMed] [Google Scholar]
- 7.Hopwood P, Crawford DH. The role of EBV in post-transplant malignancies: a review. Journal of Clinical Pathology. 2000;53(4):248–254. doi: 10.1136/jcp.53.4.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chao NJ, Berry GJ, Advani R, Horning SJ, Weiss LM, Blume KG. Epstein-Barr virus-associated lymphoproliferative disorder following autologous bone marrow transplantation for non-Hodgkin’s lymphoma. Transplantation. 1993;55(6):1425–1428. [PubMed] [Google Scholar]
- 9.Ho M, Jaffe R, Miller G, et al. The frequency of Epstein-Barr virus infection and associated lymphoproliferative syndrome after transplantation and its manifestations in children. Transplantation. 1988;45(4):719–727. doi: 10.1097/00007890-198804000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penn I. Malignant lymphomas in organ transplant recipients. Transplantation Proceedings. 1981;13(1):736–738. [PubMed] [Google Scholar]
- 11.Weiss LM, Movahed LA, Warnke RA, Sklar J. Detection of Epstein-Barr viral genomes in Reed-Sternberg cells of Hodgkin’s disease. The New England Journal of Medicine. 1989;320(8):502–506. doi: 10.1056/NEJM198902233200806. [DOI] [PubMed] [Google Scholar]
- 12.Ott G, Kirchner TH, Muller-Hermelink HK. Monoclonal Epstein-Barr virus genomes but lack of EBV-related protein expression in different types of gastric carcinoma. Histopathology. 1994;25(4):323–329. doi: 10.1111/j.1365-2559.1994.tb01350.x. [DOI] [PubMed] [Google Scholar]
- 13.Yuen ST, Chung LP, Leung SY, Luk ISC, Chan SY, Ho J. In situ detection of Epstein-Barr virus in gastric and colorectal adenocarcinomas. American Journal of Surgical Pathology. 1994;18(11):1158–1163. doi: 10.1097/00000478-199411000-00010. [DOI] [PubMed] [Google Scholar]
- 14.Bonnet M, Guinebretiere JM, Kremmer E, et al. Detection of Epstein-Barr virus in invasive breast cancers. Journal of the National Cancer Institute. 1999;91(16):1376–1381. doi: 10.1093/jnci/91.16.1376. [DOI] [PubMed] [Google Scholar]
- 15.Arbach H, Viglasky V, Lefeu F, et al. Epstein-Barr virus (EBV) genome and expression in breast cancer tissue: effect of EBV infection of breast cancer cells on resistance to paclitaxel (taxol) Journal of Virology. 2006;80(2):845–853. doi: 10.1128/JVI.80.2.845-853.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glaser SL, Hsu JL, Gulley ML. Epstein-Barr Virus and breast cancer: state of the evidence for viral carcinogenesis. Cancer Epidemiology Biomarkers and Prevention. 2004;13(5):688–697. [PubMed] [Google Scholar]
- 17.Diepstra A, Van Imhoff GW, Schaapveld M, et al. Latent Epstein-Barr virus infection of tumor cells in classical Hodgkin’s lymphoma predicts adverse outcome in older adult patients. Journal of Clinical Oncology. 2009;27(23):3815–3821. doi: 10.1200/JCO.2008.20.5138. [DOI] [PubMed] [Google Scholar]
- 18.Tsimberidou AM, Keating MJ, Bueso-Ramos CE, Kurzrock R. Epstein-Barr virus in patients with chronic lymphocytic leukemia: a pilot study. Leukemia and Lymphoma. 2006;47(5):827–836. doi: 10.1080/10428190500398856. [DOI] [PubMed] [Google Scholar]
- 19.Park S, Lee J, Young HK, et al. The impact of Epstein-Barr virus status on clinical outcome in diffuse large B-cell lymphoma. Blood. 2007;110(3):972–978. doi: 10.1182/blood-2007-01-067769. [DOI] [PubMed] [Google Scholar]
- 20.Paydas S, Ergin M, Erdogan S, Seydaoglu G. Prognostic significance of EBV-LMP1 and VEGF-A expressions in non-Hodgkin’s lymphomas. Leukemia Research. 2008;32(9):1424–1430. doi: 10.1016/j.leukres.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 21.Sasikala PS, Nirmala K, Sundersingh S, Mahji U, Rajkumar T. Frequency and distribution of Epstein-Barr virus infection and its association with P53 expression in a series of primary nodal non-Hodgkin lymphoma patients from South India. International Journal of Laboratory Hematology. 2010;32(1):56–64. doi: 10.1111/j.1751-553X.2008.01125.x. [DOI] [PubMed] [Google Scholar]
- 22.Carbone A, Cesarman E, Spina M, Gloghini A, Schulz TF. HIV-associated lymphomas and gamma-herpesviruses. Blood. 2009;113(6):1213–1224. doi: 10.1182/blood-2008-09-180315. [DOI] [PubMed] [Google Scholar]
- 23.Raab-Traub N, Flynn K. The structure of the termini of the Epstein-Barr virus as a marker of clonal cellular proliferation. Cell. 1986;47(6):883–889. doi: 10.1016/0092-8674(86)90803-2. [DOI] [PubMed] [Google Scholar]
- 24.Brown NA, Liu C, Garcia CR. Clonal origins of lymphoproliferative disease induced by Epstein-Barr virus. Journal of Virology. 1986;58(3):975–978. doi: 10.1128/jvi.58.3.975-978.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hammerschmidt W, Sugden B. Identification and characterization of oriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell. 1988;55(3):427–433. doi: 10.1016/0092-8674(88)90028-1. [DOI] [PubMed] [Google Scholar]
- 26.Neri A, Barriga F, Inghirami G, et al. Epstein-Barr virus infection precedes clonal expansion in Burkitt’s and acquired immunodeficiency syndrome-associated lymphoma. Blood. 1991;77(5):1092–1095. [PubMed] [Google Scholar]
- 27.Henle G, Henle W, Horwitz CA. Antibodies to Epstein Barr virus associated nuclear antigen in infectious mononucleosis. Journal of Infectious Diseases. 1974;130(3):231–239. doi: 10.1093/infdis/130.3.231. [DOI] [PubMed] [Google Scholar]
- 28.Henle G, Henle W, Clifford P, et al. Antibodies to Epstein-Barr virus in Burkitt’s lymphoma and control groups. Journal of the National Cancer Institute. 1969;43(5):1147–1157. [PubMed] [Google Scholar]
- 29.Henle W, Henle G. Seroepidemiology of the virus. In: Epstein M, Achong B, editors. The Epstein-Barr Virus. Berlin: Springer; 1979. pp. 61–78. [Google Scholar]
- 30.Rickinson AB, Moss DJ. Human cytotoxic T lymphocyte responses to Epstein-Barr virus infection. Annual Review of Immunology. 1997;15:405–431. doi: 10.1146/annurev.immunol.15.1.405. [DOI] [PubMed] [Google Scholar]
- 31.Tan LC, Gudgeon N, Annels NE, et al. A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. Journal of Immunology. 1999;162(3):1827–1835. [PubMed] [Google Scholar]
- 32.Sugden B, Mark W. Clonal transformation of adult human leukocytes by Epstein-Barr virus. Journal of Virology. 1977;23(3):503–508. doi: 10.1128/jvi.23.3.503-508.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen JI. Epstein-Barr virus and the immune system: hide and seek. Journal of the American Medical Association. 1997;278(6):510–513. [PubMed] [Google Scholar]
- 34.Jackman WT, Mann KA, Hoffmann HJ, Spaete RR. Expression of Epstein-Barr virus gp350 as a single chain glycoprotein for an EBV subunit vaccine. Vaccine. 1999;17(7-8):660–668. doi: 10.1016/s0264-410x(98)00248-5. [DOI] [PubMed] [Google Scholar]
- 35.Thomson SA, Burrows SR, Misko IS, Moss DJ, Coupar BEH, Khanna R. Targeting a polyepitope protein incorporating multiple class II- restricted vital epitopes to the secretory/endocytic pathway facilitates immune recognition by CD4+ cytotoxic T lymphocytes: a novel approach to vaccine design. Journal of Virology. 1998;72(3):2246–2252. doi: 10.1128/jvi.72.3.2246-2252.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rickinson AB, Kieff E. Epstein-Barr virus. In: Fields BN, Knipe DM, Howley PM, editors. Fields Virology. 3rd edition. Philadelphia, Pa, USA: Lippincott-Raven; 1996. [Google Scholar]
- 37.Sokal EM, Hoppenbrouwers K, Vandermeulen C, et al. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. Journal of Infectious Diseases. 2007;196(12):1749–1753. doi: 10.1086/523813. [DOI] [PubMed] [Google Scholar]
- 38.Moutschen M, Léonard P, Sokal EM, et al. Phase I/II studies to evaluate safety and immunogenicity of a recombinant gp350 Epstein-Barr virus vaccine in healthy adults. Vaccine. 2007;25(24):4697–4705. doi: 10.1016/j.vaccine.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Papadopoulos EB, Ladanyi M, Emanuel D, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. The New England Journal of Medicine. 1994;330(17):1185–1191. doi: 10.1056/NEJM199404283301703. [DOI] [PubMed] [Google Scholar]
- 40.Rooney CM, Smith CA, Ng CYC, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92(5):1549–1555. [PubMed] [Google Scholar]
- 41.Khanna R, Bell S, Sherritt M, et al. Activation and adoptive transfer of Epstein-Barr virus-specific cytotoxic T cells in solid organ transplant patients with posttransplant lymphoproliferative disease. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(18):10391–10396. doi: 10.1073/pnas.96.18.10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chua D, Huang J, Zheng B, et al. Adoptive transfer of autologous Epstein-Barr virus-specific cytotoxic T cells for nasopharyngeal carcinoma. International Journal of Cancer. 2001;94(1):73–80. doi: 10.1002/ijc.1430. [DOI] [PubMed] [Google Scholar]
- 43.Bollard CM, Aguilar L, Straathof KC, et al. Cytotoxic T lymphocyte therapy for epstein-barr virus+ Hodgkin’s disease. Journal of Experimental Medicine. 2004;200(12):1623–1633. doi: 10.1084/jem.20040890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Straathof KCM, Bollard CM, Popat U, et al. Treatment of nasopharyngeal carcinoma with Epstein-Barr virus-specific T lymphocytes. Blood. 2005;105(5):1898–1904. doi: 10.1182/blood-2004-07-2975. [DOI] [PubMed] [Google Scholar]
- 45.Gustafsson A, Levitsky V, Zou JZ, et al. Epstein-Barr virus (EBV) load in bone marrow transplant recipients at risk to develop posttransplant lymphoproliferative disease: prophylactic infusion of EBV-specific cytotoxic T cells. Blood. 2000;95(3):807–814. [PubMed] [Google Scholar]
- 46.Heslop HE, Slobod KS, Pule MA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115(5):925–935. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murray RJ, Kurilla MG, Brooks JM, et al. Identification of target antigens for the human cytotoxic T cell response to Epstein-Barr virus (EBV): implications for the immune control of EBV- positive malignancies. Journal of Experimental Medicine. 1992;176(1):157–168. doi: 10.1084/jem.176.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Louis CU, Straath K, Bollard CM, et al. Enhancing the in vivo expansion of adoptively transferred EBV- Specific CTL with lymphodepleting CD45 monoclonal antibodies in NPC patients. Blood. 2009;113(11):2442–2450. doi: 10.1182/blood-2008-05-157222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Merlo A, Turrini R, Dolcetti R, Zanovello P, Rosato A. Immunotherapy for EBV-associated malignancies. International Journal of Hematology. 2011;93(3):281–293. doi: 10.1007/s12185-011-0782-2. [DOI] [PubMed] [Google Scholar]
- 50.Chodosh J, Holder VP, Gan YJ, Belgaumi A, Sample J, Sixbey JW. Eradication of latent Epstein-Barr virus by hydroxyurea alters the growth-transformed cell phenotype. Journal of Infectious Diseases. 1998;177(5):1194–1201. doi: 10.1086/515290. [DOI] [PubMed] [Google Scholar]
- 51.Slobod KS, Taylor GH, Sandlund JT, Furth P, Helton KJ, Sixbey JW. Epstein-Barr virus-targeted therapy for AIDS-related primary lymphoma of the central nervous system. The Lancet. 2000;356(9240):1493–1494. doi: 10.1016/S0140-6736(00)02879-8. [DOI] [PubMed] [Google Scholar]
- 52.Kenney JL, Guinness ME, Curiel T, Lacy J. Antisense to the Epstein-Barr virus (EBV)-encoded latent membrane protein 1 (LMP-1) suppresses LMP-1 and Bcl-2 expression and promotes apoptosis in EBV-immortalized B cells. Blood. 1998;92(5):1721–1727. [PubMed] [Google Scholar]
- 53.Hirai H, Satoh E, Osawa M, et al. Use of EBV-based vector/HVJ-liposome complex vector for targeted gene therapy of EBV-associated neoplasms. Biochemical and Biophysical Research Communications. 1997;241(1):112–118. doi: 10.1006/bbrc.1997.7776. [DOI] [PubMed] [Google Scholar]
- 54.Kenney S, Ge JIQ, Westphal EM, Olsen J. Gene therapy strategies for treating Epstein-Barr virus-associated lymphomas: comparison of two different Epstein-Barr virus-based vectors. Human Gene Therapy. 1998;9(8):1131–1141. doi: 10.1089/hum.1998.9.8-1131. [DOI] [PubMed] [Google Scholar]
- 55.Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Research. 1986;46(10):5276–5281. [PubMed] [Google Scholar]
- 56.Gutiérrez MI, Judde JG, Magrath IT, Bhatia KG. Switching viral latency to viral lysis: a novel therapeutic approach for Epstein-Barr virus-associated neoplasia. Cancer Research. 1996;56(5):969–972. [PubMed] [Google Scholar]
- 57.Westphal EM, Mauser A, Swenson J, Davis MG, Talarico CL, Kenney SC. Induction of lytic Epstein-Barr virus (EBV) infection in EBV-associated malignancies using adenovirus vectors in vitro and in vivo. Cancer Research. 1999;59(7):1485–1491. [PubMed] [Google Scholar]
- 58.Feng WH, Westphal E, Mauser A, et al. Use of adenovirus vectors expressing Epstein-Barr virus (EBV) immediate-early protein BZLF1 or BRLF1 to treat EBV-positive tumors. Journal of Virology. 2002;76(21):10951–10959. doi: 10.1128/JVI.76.21.10951-10959.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Israel BF, Kenney SC. Virally targeted therapies for EBV-associated malignancies. Oncogene. 2003;22(33):5122–5130. doi: 10.1038/sj.onc.1206548. [DOI] [PubMed] [Google Scholar]
- 60.Ben-Sasson SA, Klein G. Activation of the Epstein-Barr virus genome by 5-aza-cytidine in latently infected human lymphoid lines. International Journal of Cancer. 1981;28(2):131–135. doi: 10.1002/ijc.2910280204. [DOI] [PubMed] [Google Scholar]
- 61.Hudewentz J, Bornkamm GW, Zur Hausen H. Effect of the diterpene ester TPA on Epstein-Barr virus antigen- and DNA synthesis in producer and nonproducer cell lines. Virology. 1980;100(1):175–178. doi: 10.1016/0042-6822(80)90563-2. [DOI] [PubMed] [Google Scholar]
- 62.Luka J, Kallin B, Klein G. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology. 1979;94(1):228–231. doi: 10.1016/0042-6822(79)90455-0. [DOI] [PubMed] [Google Scholar]
- 63.Anisimova E, Prachova K, Roubal J, Vonka V. Effects of n-butyrate and phorbol ester (TPA) on induction of Epstein-Barr virus antigens and cell differentiation. Archives of Virology. 1984;81(3-4):223–237. doi: 10.1007/BF01309995. [DOI] [PubMed] [Google Scholar]
- 64.Saemundsen AK, Kallin B, Klein G. Effect of n-butyrate on cellular and viral DNA synthesis in cells latently infected with Epstein-Barr virus. Virology. 1980;107(2):557–561. doi: 10.1016/0042-6822(80)90326-8. [DOI] [PubMed] [Google Scholar]
- 65.Farrell PJ, Rowe DT, Rooney CM, Kouzarides T. Epstein-Barr virus BZLF1 trans-activator specifically binds to a consensus AP-1 site and is related to c-fos. The EMBO Journal. 1989;8(1):127–132. doi: 10.1002/j.1460-2075.1989.tb03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tao Q, Robertson KD. Stealth technology: how Epstein-Barr virus utilizes DNA methylation to cloak itself from immune detection. Clinical Immunology. 2003;109(1):53–63. doi: 10.1016/s1521-6616(03)00198-0. [DOI] [PubMed] [Google Scholar]
- 67.Park JH, Faller DV. Epstein-Barr virus latent membrane protein-1 induction by histone deacetylase inhibitors mediates induction of intercellular adhesion molecule-1 expression and homotypic aggregation. Virology. 2002;303(2):345–363. doi: 10.1006/viro.2002.1638. [DOI] [PubMed] [Google Scholar]
- 68.Sealy L, Chalkley R. The effect of sodium butyrate on histone modification. Cell. 1978;14(1):115–121. doi: 10.1016/0092-8674(78)90306-9. [DOI] [PubMed] [Google Scholar]
- 69.Sarkar S, Abujamra AL, Loew JE, Forman LW, Perrine SP, Faller DV. Histone deacetylase inhibitors reverse CpG methylation by regulating DNMT1 through ERK signaling. Anticancer Research. 2011;31(9):2723–2732. [PubMed] [Google Scholar]
- 70.Mentzer SJ, Fingeroth J, Reilly JJ, Perrine SP, Faller DV. Arginine butyrate-induced susceptibility to ganciclovir in an Epstein-Barr-virus-associated lymphoma. Blood Cells, Molecules and Diseases. 1998;24(2):114–123. doi: 10.1006/bcmd.1998.0178. [DOI] [PubMed] [Google Scholar]
- 71.Westphal EM, Blackstock W, Feng W, Israel B, Kenney SC. Activation of lytic Epstein-Barr virus (EBV) infection by radiation and sodium butyrate in vitro and in vivo: a potential method for treating EBV-positive malignancies. Cancer Research. 2000;60(20):5781–5788. [PubMed] [Google Scholar]
- 72.Faller DV, Mentzer SJ, Perrine SP. Induction of the Epstein-Barr virus thymidine kinase gene with concomitant nucleoside antivirals as a therapeutic strategy for Epstein-Barr virus-associated malignancies. Current Opinion in Oncology. 2001;13(5):360–367. doi: 10.1097/00001622-200109000-00008. [DOI] [PubMed] [Google Scholar]
- 73.Faller DV, Hermine O, Small T. Phase I/II trial of arginine butyrate to induce viral TK gene expression in Epstein-Barr virus (EBV)-associated lymphoma. Blood (ASH Annual Meeting Abstracts) 2000;96, abstract 577 [Google Scholar]
- 74.Perrine SP, Hermine O, Small T, et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood. 2007;109(6):2571–2578. doi: 10.1182/blood-2006-01-024703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moore SM, Cannon JS, Tanhehco YC, Hamzeh FM, Ambinder RF. Induction of Epstein-Barr virus kinases to sensitize tumor cells to nucleoside analogues. Antimicrobial Agents and Chemotherapy. 2001;45(7):2082–2091. doi: 10.1128/AAC.45.7.2082-2091.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mentzer SJ, Perrine SP, Faller DV. Epstein-Barr virus post-transplant lymphoproliferative disease and virus-specific therapy: pharmacological re-activation of viral target genes with arginine butyrate. Transplant Infectious Disease. 2001;3(3):177–185. doi: 10.1034/j.1399-3062.2001.003003177.x. [DOI] [PubMed] [Google Scholar]
- 77.Feng WH, Kenney SC. Valproic acid enhances the efficacy of chemotherapy in EBV-positive tumors by increasing lytic viral gene expression. Cancer Research. 2006;66(17):8762–8769. doi: 10.1158/0008-5472.CAN-06-1006. [DOI] [PubMed] [Google Scholar]
- 78.Ghosh SK, Forman LW, Akinsheye I, Perrine SP, Faller DV. Short, discontinuous exposure to butyrate effectively sensitizes latently EBV-infected lymphoma cells to nucleoside analogue antiviral agents. Blood Cells, Molecules, and Diseases. 2007;38(1):57–65. doi: 10.1016/j.bcmd.2006.10.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roychowdhury S, Peng R, Baiocchi RA, et al. Experimental treatment of Epstein-Barr virus-associated primary central nervous system lymphoma. Cancer Research. 2003;63(5):965–971. [PubMed] [Google Scholar]
- 80.Daibata M, Bandobashi K, Kuroda M, Imai S, Miyoshi I, Taguchi H. Induction of lytic Epstein-Barr virus (EBV) infection by synergistic action of rituximab and dexamethasone renders EBV-positive lymphoma cells more susceptible to ganciclovir cytotoxicity in vitro and in vivo. Journal of Virology. 2005;79(9):5875–5879. doi: 10.1128/JVI.79.9.5875-5879.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fu DX, Tanhehco Y, Chen J, et al. Bortezomib-induced enzyme-targeted radiation therapy in herpesvirus-associated tumors. Nature Medicine. 2008;14(10):1118–1122. doi: 10.1038/nm.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Feng WH, Israel B, Raab-Traub N, Busson P, Kenney SC. Chemotherapy induces lytic EBV replication and confers ganciclovir susceptibility to EBV-positive epithelial cell tumors. Cancer Research. 2002;62(6):1920–1926. [PubMed] [Google Scholar]
- 83.Feng WH, Hong G, Delecluse HJ, Kenney SC. Lytic induction therapy for Epstein-Barr virus-positive B-cell lymphomas. Journal of Virology. 2004;78(4):1893–1902. doi: 10.1128/JVI.78.4.1893-1902.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fu DX, Tanhehco YC, Chen J, et al. Virus-associated tumor imaging by induction of viral gene expression. Clinical Cancer Research. 2007;13(5):1453–1458. doi: 10.1158/1078-0432.CCR-06-2295. [DOI] [PubMed] [Google Scholar]
- 85.Faller DV, Ghosh S, Feldman T, et al. Short-term exposure to arginine butyrate, in combination with ganciclovir, is as effective as continuous exposure for virus-targeted therapy of EBV-positive lymphomas. Blood (ASH Annual Meeting Abstracts) 2009;114, abstract 4754 [Google Scholar]
- 86.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26(37):5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 87.Mercurio C, Minucci S, Pelicci PG. Histone deacetylases and epigenetic therapies of hematological malignancies. Pharmacological Research. 2010;62(1):18–34. doi: 10.1016/j.phrs.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 88.Ye J, Gradoville L, Daigle D, Miller G. De novo protein synthesis is required for lytic cycle reactivation of Epstein-Barr virus, but not Kaposi’s sarcoma-associated herpesvirus, in response to histone deacetylase inhibitors and protein kinase C agonists. Journal of Virology. 2007;81(17):9279–9291. doi: 10.1128/JVI.00982-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Faller DV, Ghosh S, Perrine SP, Williams RM, Berenson RJ. Histone deacetylase inhibitors: potent inducers of tumor latent EBV thymidine kinase induction. Blood (ASH Annual Meeting Abstracts) 2011;118, abstract 1831 [Google Scholar]
- 90.Ghosh SK, Perrine SP, Williams RM, Faller DV. Histone deacetylase inhibitors are potent inducers of gene expression in latent EBV and sensitize lymphoma cells to nucleoside anti-viral agents. Blood. 2012;119(4):1008–1017. doi: 10.1182/blood-2011-06-362434. [DOI] [PMC free article] [PubMed] [Google Scholar]