

Abstract
Microfabricated thermoelectric controllers can be employed to investigate mechanisms underlying myosin-driven sliding of Ca2+-regulated actin and disease-associated mutations in myofilament proteins. Specifically, we examined actin filament sliding—with or without human cardiac troponin (Tn) and α-tropomyosin (Tm)—propelled by rabbit skeletal heavy meromyosin, when temperature was varied continuously over a wide range (∼20–63°C). At the upper end of this temperature range, reversible dysregulation of thin filaments occurred at pCa 9 and 5; actomyosin function was unaffected. Tn-Tm enhanced sliding speed at pCa 5 and increased a transition temperature (Tt) between a high activation energy (Ea) but low temperature regime and a low Ea but high temperature regime. This was modulated by factors that alter cross-bridge number and kinetics. Three familial hypertrophic cardiomyopathy (FHC) mutations, cTnI R145G, cTnI K206Q, and cTnT R278C, cause dysregulation at temperatures ∼5–8°C lower; the latter two increased speed at pCa 5 at all temperatures.
1. Introduction
Biomolecular motors have clear promise for transport applications in micro- and nanofabricated devices although control of motion remains a major challenge [1–4]. Toward that end, we constructed a microfabricated thermoelectric controller for rapid and reversible actuation using myosin, the biomolecular motor of muscle, and its partner filament, actin [5, 6]. Thermal control of kinesin-microtubule motility using thermally responsive polymers has also been demonstrated as a possible temperature-dependent control mechanism for biomolecular motors [7]. We present here novel applications of our thermoelectric control system to investigate changes in actin-myosin motility when Ca2+-regulated thin (actin) filaments are used, and when thin-filament Ca2+-regulatory protein mutations are introduced that underlie an inherited cardiovascular disease, familial hypertrophic cardiomyopathy (FHC) [8–10]. Results provide insights into mechanisms of altered function when thin filaments are reconstituted with human cardiac Ca2+-regulatory proteins troponin (Tn) and tropomyosin (Tm), and further modulated by FHC-related mutations in cardiac Tn.
In the heart—and in vertebrate striated muscles in general—contraction is physiologically controlled by binding of cytoplasmic Ca2+ to thin filaments. Thin filaments are comprised of three major proteins: actin, Tm, and Tn that is itself a complex of three polypeptides [11]. Ca2+ activates thin filaments by binding to Tn, leading to structural changes that ultimately expose myosin-binding sites on actin subunits [11]. Our previous work on the thermoelectric controller utilized only myosin and unregulated F-actin, that is, Ca2+-independent motility achieved using actin filaments without Tn or Tm [5, 6]. This study was initiated because Ca2+-regulated thin filaments could have several advantages over unregulated F-actin in a thermoelectric nanoactuator. First, Ca2+ would provide a secondary control mechanism that is separate from temperature. An additional benefit is that the Ca2+-regulatory proteins Tn and Tm at saturating Ca2+ can enhance function of the actomyosin system [12–20]. These effects of Tn and Tm on actomyosin function, however, appear to vary markedly over the limited temperature ranges studied previously [16, 18, 21] and thus could introduce nonlinearities to a thermoelectric nano-actuator.
Temperature has obvious physiological relevance as a determinant of mammalian striated muscle contraction. It affects several fundamental aspects of muscle structure and function [22–24] although temperature effects on muscle function appear to be greater at low temperatures used in many experiments than at physiological temperature; examples are isometric tetanic tension [25–27] and maximum mechanical power output [28]. Temperature also modulates the control of striated muscle contraction by Ca2+ regulatory proteins Tn and Tm, although there are both qualitative and quantitative discrepancies on this point in the striated muscle literature [22, 26, 29, 30]. Interestingly, mild hypothermia has been reported to be a positive inotropic effector in living cardiac muscle due to interplay between temperature and cardiac thin filament Ca2+ regulation [31]. Our thermo-electric controller allows us to address these issues and more in a single experiment through rapid exploration of broad temperature regimes surrounding physiological temperature.
Some mutations in Tn and Tm alter thin-filament function in ways that might make the mutant proteins useful in biomolecular motor-based transport applications. In particular, mutations associated with familial hypertrophic cardiomyopathy (FHC)—one of a growing number of diseases caused by mutations in the genes for sarcomere proteins including thin-filament Ca2+-regulatory proteins [8–10]—may be especially useful in bionanomechanical systems even though those mutations are devastating for patients [32–41]. The molecular basis by which these mutations alter thin-filament function and bring about cardiovascular disease is unknown [8, 10, 42]. Our thermoelectric controller could provide important insight into mechanisms by which these mutations in myofilament proteins alter contractile function, and directly or indirectly signal changes in gene expression that underlie the progression to pathological cardiac hypertrophy in FHC.
2. Materials and Methods
2.1. Protein Preparations: Myosin, Heavy Meromyosin, Actin, Troponin, and Tropomyosin
Florida State University's Institutional Animal Care and Use Committee approved all procedures and protocols involving vertebrate animals. Adult New Zealand White rabbits were housed in Florida State University's Laboratory Animal Resources facility and were handled in accordance with the current National Institutes of Health/National Research Council Guide for the Care and Use of Laboratory Animals. Briefly, rabbits were injected IM with 3 mg Acepromazine + 10 mg/kg Xylazine + 50 mg/kg Ketamine. Following verification of appropriate surgical depth of anesthesia, the rabbits were exsanguinated via laceration of the carotid artery. The euthanized animals were skinned, eviscerated, and chilled on ice. Back and leg muscles were removed for acetone powder, or back muscles only for myosin preparation [12, 13, 43]. Myosin and the soluble chymotryptic digest fragment heavy meromyosin (HMM) were prepared from fast skeletal muscle as described [12, 13, 43, 44]. HMM was made from myosin stored <5 weeks in glycerol (1 : 1) at −20°C; HMM was used within 3 days after preparation. Rabbit muscle actin was purified from muscle acetone powder as described [12, 13, 43, 45]. F-actin was labeled with rhodamine phalloidin (RhPh; Molecular Probes) for fluorescence microscopy (Figure 1(a)). F-actin (33 μg/mL) and RhPh (1 μM) were incubated in actin buffer (AB) with 1 mM DTT at 4°C overnight; fluorescently labeled F-actin was stable at 4°C for at least 2 weeks.
Figure 1.
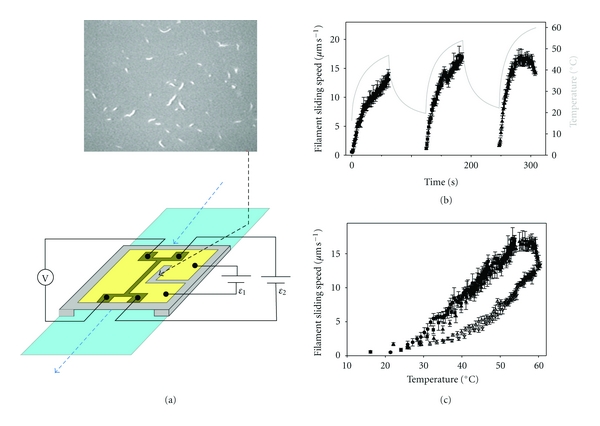
Temperature dependence of HMM-driven sliding of RhPh-labeled actin filaments determined using a microfabricated thermoelectric heater. (a) Modified flow cells were constructed as described [5]. Flow cells consisted of a microscope slide (light blue) and a modified cover slip (gray and gold), separated by two, thin spacers and held together by nonfluorescent vacuum grease. Solutions (Section 2.2) are pipetted through the flow cell as indicated by the blue, dashed arrows. To warm the flow cell, current is injected via circuit ε1 into a microfabricated thermoelectric heater (gold) on the coverslip's outer surface; flow cells are cooled by circulating chilled water through a copper coil wrapped around the microscope objective. Temperature is monitored via a linear, resistive thermometer (dark gold structure) that was microfabricated on the inner face of the coverslip (i.e., on the same side where actomyosin motility occurred, and opposite to the side with the thermoelectric heater); temperature is obtained from a calibrated voltage readout (V) with bias current set by circuit ε2. Motion of fluorescently labeled filaments on the inner side of the coverslip (top micrograph) is observed and recorded through a window (black, dashed arrow) located immediately adjacent to the thermometer. (b) Time course of temperature (gray line; right ordinate) and sliding speed (solid symbols; left ordinate) for regulated thin filaments (F-actin plus native cTn and α-Tm) at pCa 5 during the heating phase of three cycles of heating and cooling (Section 2). Points represent mean ± S.D. of the sliding speed for 3–8 filaments during 1 s. Note that the upper limit of the temperature-dependent increase in sliding speed occurs at ∼54°C (t > 4.5 min). (c) The data from (b) were replotted to eliminate time: cycle 1 (⬤), cycle 2 (■), and cycle 3 (▲). An additional dataset for unregulated F-actin sliding is shown: cycle 1 (▵) and cycle 2 (▿). Note that the sliding speed of regulated thin filaments at the highest temperature is almost the same as for unregulated F-actin.
Recombinant human α-Tm was expressed in E. coli as a homodimeric fusion protein with maltose binding protein (MBP); α-Tm was purified following removal of the MBP affinity tag via thrombin cleavage [13, 33]. After removal of the MBP tag, recombinant α-Tm has two extra amino acids (GS-) at the N-terminus; GS- is a conservative alternative to the AS-dipeptide in bacterially expressed Tm that substitutes functionally for acetylation of Tm's N-terminus [46, 47]. Purified Tn from human cardiac muscle (cTn) was obtained from Research Diagnostics (Flanders, NJ), or coexpressed recombinantly (rHcTn) in E. coli as a fusion protein with glutathione S-transferase (GST); the ternary rHcTn complex was purified following removal of the GST affinity tag via cleavage with TEV protease [13]. FHC mutations of rHcTn were introduced via site-directed mutagenesis to the bacterial coexpression plasmid; changes were verified by DNA sequencing. Proteins were assessed by coomassie-stained Tricine-SDS PAGE [48] and quantitative analysis with a Kodak EDAS 290 digital imaging system.
2.2. In Vitro Motility Assays
The speed of RhPh F-actin over HMM-coated surfaces was measured to assess the kinetics of actomyosin interactions and, for reconstituted thin filaments, their regulation by Ca2+. All aspects of the motility experiments with unregulated F-actin, such as flow cell assembly, solution preparation, assay procedures, and data collection were conducted as described [5, 13, 43, 49]. [HMM] applied to flow cells was 250 μg mL−1 in the majority of assays to achieve high density of HMM (ρ) on the nitrocellulose-coated motility surface; to reduce ρ in a limited set of experiments, 75 μg mL−1 HMM was applied to the flow cell.
The standard motility buffer (MB) contained 10 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 20 mM 3-(N-Morpholino)propanesulfonic acid (MOPS), [K+] = 50 mM, [Na+] = 15 mM, pMg 3 (pMg = −log10[Mg2+], where [Mg2+] is in molar units), and appropriate amounts of Tris+, and acetate (anion) to obtain an ionic strength of 0.085 M at pH 7. Concentrations of Ca2+, Mg2+, Na+, K+ were determined taking into account known metal ion binding constants and their enthalpies [32, 50]; solution composition was chosen to minimize enthalpic changes in pH in flow cells when temperature was varied. Glucose (3 mg/mL), 0.1 mg/mL glucose oxidase, 0.018 mg/mL catalase (Boeringher-Mannheim), and 40 mM dithiothreitol (DTT) were added immediately prior to an assay to minimize photooxidation and photobleaching [49]. Experiments were conducted at saturating [Ca2+] (pCa 5, where pCa = −log10[Ca2+], with [Ca2+] in molar units) or, where indicated, at low [Ca2+] (pCa 9); note that [Ca2+] does not affect sliding speed of unregulated F-actin [12, 14]. Methylcellulose (final concentration 0.3%), dialyzed against sodium azide (0.02%), was added to MB to prevent actin filament diffusion away from the assay surface [12, 49, 51]. Solutions for assays with reconstituted thin filaments were the same as for unregulated F-actin, except for the last two solutions: the buffer applied to the flow cell before MB contained 75 nM each of α-Tm and either native cTn or recombinant hcTn and was incubated for three minutes before infusing MB containing the same concentrations of Tn and Tm [12, 13, 32, 50]. The minimum concentrations of native or WT Tn and Tm added to MB to obtain well-regulated filaments at 30°C were determined by titrations and applying the following criteria: filament sliding was inhibited at pCa 9, while motility was fast and uniform at pCa 5 and standard temperature of 30°C [12, 13, 32, 50]. In a limited set of motility experiments, standard MB was modified to vary metabolite concentrations (Table 1) as follows: either 5.3 μg mL−1 sucrose phosphorylase and 10 mM sucrose were added to reduce inorganic phosphate (Pi) [52] or acetate was partially replaced by Pi to achieve the high Pi condition (sucrose phosphorylase was not added); contaminating ADP was reduced to <1 μM by addition of creatine phosphate (CP) and creatine phosphokinase (CK) [53].
Table 1.
Metabolite concentrations in standard and modified motility buffers.
| Metabolites | Standard1 | Modified solutions for motility assays2 | ||
|---|---|---|---|---|
| Motility buffer | Control | High Pi | Low ATP | |
| [ATP] | 2 mM | 2 mM | 2 mM | 0.2 mM |
| Sucrose phosphorylase | — | 5.3 μg/mL | — | 5.3 μg/mL |
| Sucrose | — | 10 mM | 10 mM | 10 mM |
| [Pi] | ∼10 μM | ∼1 μM | 4 mM | ~1 μM |
|
| ||||
| Creatine kinase (CK) | — | 0.1 mg/mL | 0.1 mg/mL | 0.1 mg/mL |
| Creatine (Cr) | — | 30 μM | 30 μM | 12 μM |
| Creatine phosphate (CP) | — | 1 mM | 1 mM | 1 mM |
| [ADP] | ~10 μM | 0.42–0.57 μM | 0.42–0.57 μM | ~0.02 μM |
Values for [ATP], [Pi], and [ADP] represent final concentrations in motility buffers. [Pi] in columns 3 and 5 were estimated using Keq = 0.06 for the reaction sucrose + Pi ↔α-D-glucose-1-phosphate + D-fructose [52, 54]. [ADP] estimates are based on the equilibrium constant (Keq) for the reaction ATP + Cr ↔ ADP + CP, and are given in columns 3-4 with lowest and highest value corresponding to the lowest (27°C) and highest (42°C) temperatures employed, respectively. The estimate of [ADP] in column 2 was based on Chase and Kushmerick [53].
1Motility buffer (MB) employed in the majority of experiments (data in Figures 1–4) did not include sucrose phosphorylase and creatine kinase.
2Modified motility buffers were used for experiments shown in Figure 5.
Motility data were collected while varying temperature by employing modified flow cells containing microfabricated Au heater and thermometer elements (Figure 1(a)), where the thermometer is located immediately adjacent to the viewing region [5]. In experiments using the thermoelectric heater, cooling was achieved by circulating chilled water through a copper coil wrapped around the microscope objective; in a limited set of experiments, assays at constant temperature were achieved by circulating temperature-controlled water through the coil surrounding the objective [13, 43]. Motility speed was analyzed using MetaMorph software (Universal Imaging) as described [5]. Stacks of frames (one stack for each second of temperature transient data, or 10–12 stacks for each constant temperature experiment from one flow cell) were created from digitized movies.
2.3. Statistical Analyses
Averages and error estimates were calculated with Microsoft Excel 2000. Averages are given as unweighted mean ± SD. Linear regression analyses were performed using Microsoft Excel 2000 or SigmaPlot (Windows versions 8.0 and 11.2; SPSS Inc., Chicago, IL); nonlinear regression analyses were performed using SigmaPlot.
A transition temperature (Tt) was estimated from Arrhenius transformed speed versus temperature data that exhibited a change in slope with temperature. To obtain Tt, the data were divided into two parts (high- and low-temperature regimes) that were separated by two adjacent points on the Arrhenius plot's temperature axis; several such divisions of each dataset were processed using the following algorithm. Linear regressions were obtained for each regime, and the intersection point determined for those regressions. In general, the intersection point does not fall between the two data points that separate the high and low temperature regimes. The small number of intersection points that did fall between the high- and low-temperature regimes were considered to be candidates for Tt; in the few instances when there was more than one candidate point, the median value was chosen for Tt.
3. Results
3.1. Temperature Dependence of Unregulated F-Actin and Regulated Thin-Filament Sliding
To examine sliding of Ca2+-regulated thin filaments over a broad range of temperatures, we employed a thermoelectric controller (Figure 1(a)) that enables both rapid and continuous variation of a flow cell's temperature [5]. Figure 1(b) shows sliding speed (solid symbols) as a function of time for thin filaments reconstituted with native cTn·α-Tm at pCa 5 during three heating phases of cyclic heating and cooling (~20–60°C). Note that the peak temperature (gray line) increased with each cycle. Speed increased with temperature during the heating phase of each cycle over most of the range examined. As temperature increased during the third cycle, however, sliding speed of regulated actin reached a maximum at ∼54°C, and then speed declined above ∼58°C (Figure 1(b)). This anomalous decline in speed for regulated thin filaments is particularly evident in Figure 1(c), solid symbols where data from all three heating cycles of Figure 1(b) were combined to show the temperature dependence of sliding speed. Superposition of the data from three cycles suggests that brief exposure to elevated temperature over at least the first two cycles was fully reversible.
Comparison of regulated versus unregulated F-actin sliding speed at high temperature suggests that the anomalous decline for regulated thin-filament speed is more likely due to an effect of temperature on Tn·Tm rather than on actin or HMM. Regulated filament speed at pCa 5 (solid symbols) was at least as fast as that for unregulated F-actin (open symbols) at each temperature over the range examined (∼20–60°C) (Figure 1(c)), consistent with previous reports over more limited temperature ranges [12, 13, 16, 20]. The speed of unregulated F-actin, however, increased continuously over the entire temperature range—even at the highest temperatures examined, where regulated thin-filament speed declined. To test the hypothesis that Tn·Tm is responsible for the anomalous decline in speed (Figure 1), we examined the effect of temperature on regulated thin-filament motility at pCa 9, that is, in the absence of activating Ca2+. Figure 2(a) shows sliding speed (red symbols) as a function of time for thin filaments reconstituted with rHcTn·α-Tm at pCa 9 during five heating phases of cyclic heating and cooling. Note that the peak temperature (gray line) increased with each cycle, as in Figure 1(b). Data from all five heating cycles of Figure 2(a) were combined to show the temperature dependence of sliding speed in Figure 2(b). As expected in the absence of Ca2+, regulated thin filaments exhibited little or no motion (note red symbols at speed ~0) over a broad range of temperatures (~20–43°C). Above ~43°C, filament sliding was observed indicating loss of Ca2+ regulation (red symbols; Figure 2) although the average speed was generally less than or similar to that of unregulated F-actin (open symbols; Figure 2(b)). As for native cTn (Figure 1(c), solid symbols), rHcTn WT regulated thin filaments at pCa 5 (Figure 2(b), solid symbols) were faster than unregulated F-actin (Figure 2(b), open symbols); an anomalous decline in speed with WT rHcTn (Figure 2(b), solid black circles) occurred at slightly a higher temperature (by ∼4–5°C) than for native cTn (Figure 1(c), solid symbols), suggesting increased stability of the recombinantly expressed protein or its interactions with F-actin-α-Tm. The apparent loss of Ca2+-regulation, indicated in Figure 2 by thin filament sliding (nonzero speeds) at pCa 9 for temperatures above ∼43°C, is completely reversible because thin-filaments stopped moving after temperature was lowered during the cooling phase of each cycle at pCa 9 (Figure 2(a)).
Figure 2.
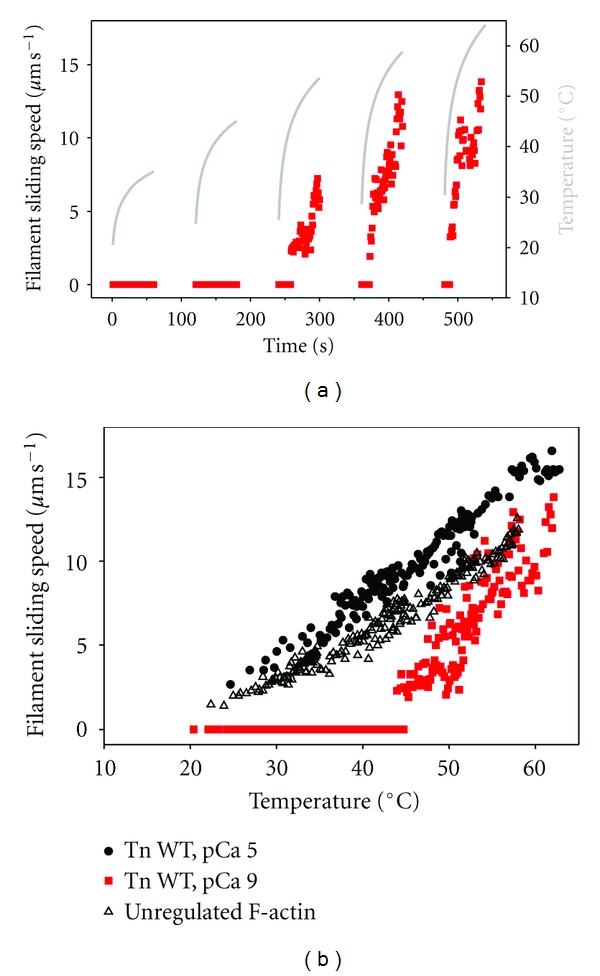
Thin filaments remain motile but lose Ca2+-regulation at high temperature. (a) Time course of temperature (gray line; right ordinate) and sliding speed (solid squares, red fill; left ordinate) for regulated thin filaments (F-actin plus rhcTn and α-Tm) at pCa 9 during the heating phase of five cycles of heating and cooling. Points represent the mean sliding speed for 3–5 filaments during 1 s. At pCa 9, nonzero sliding speeds in heating cycles 3–5 indicate loss of Ca2+-regulation (dysregulation); note that temperature-dependent loss of Ca2+-regulation is fully reversible following cooling (beginning of cycles 4-5). (b) Data from (a) (pCa 9; solid squares, red fill) were replotted to eliminate time. Additional data for regulated thin filaments (F-actin plus rhcTn and α-Tm) at pCa 5 (⬤) and unregulated F-actin (▵) from the same series of experiments are also shown for comparison.
Arrhenius analysis of the data in Figure 1(c) reveals that the activation energy (Ea) for regulated thin-filament speed at pCa 5 exhibited two distinct values, with a change in the slope at Tt ~ 38°C (Figure 3, solid symbols and black solid lines). For regulated thin filaments (Figure 3, solid symbols and black solid lines), Ea at temperatures below physiological (100.4 kJ/mol) was >2-fold greater than Ea for the higher-temperature regime (41.8 kJ/mol), while the latter value was closer to that of unregulated F-actin (61.9 kJ/mol) (Figure 3, open symbols and black dashed line). Temperature has a dramatic, nonlinear effect on the viscosity of water [55], and the speed of filament sliding varies inversely with solvent viscosity [43, 56]; we therefore asked whether temperature-dependent changes in solvent viscosity could explain the nonlinear Arrhenius relation (Figure 3). The data in Figure 3 were processed to remove the temperature dependence of solvent viscosity by first normalizing viscosity with respect to that of water at 37°C (i.e., body temperature), and second assuming that speed varies inversely with solvent viscosity [43, 56]. After removing the effects of viscosity, slopes of the Arrhenius plots were reduced (Figure 3, blue lines). Ea for unregulated F-actin decreased from 61.9 kJ mol−1 to 47.0 kJ mol−1 (Figure 3, blue dashed line). For regulated thin filaments, Ea decreased from 100.4 to 83.9 kJ mol−1 for T < 38°C, and from 41.8 to 26.6 kJ mol−1 for T > 38°C (Figure 3, black versus blue solid lines). The latter value of Ea is suggestive of a diffusion-limited process, in accord with the previously observed inverse relationship between speed and viscosity [43, 56]. Despite the reduction of slopes for regulated thin-filaments, the ratio of Ea's between the two temperature regimes increased from 2.4 to 3.1. Below 38°C, the ratio of Ea's for regulated thin-filament sliding speed with respect to that of unregulated F-actin increased from 1.6 to 1.8. It is clear from this analysis that it is important to consider temperature-dependent changes in solvent viscosity when evaluating actin filament sliding speed. Nonlinearities in the Arrhenius plot (Figure 3), however, cannot be explained by temperature-dependent changes in solvent viscosity.
Figure 3.
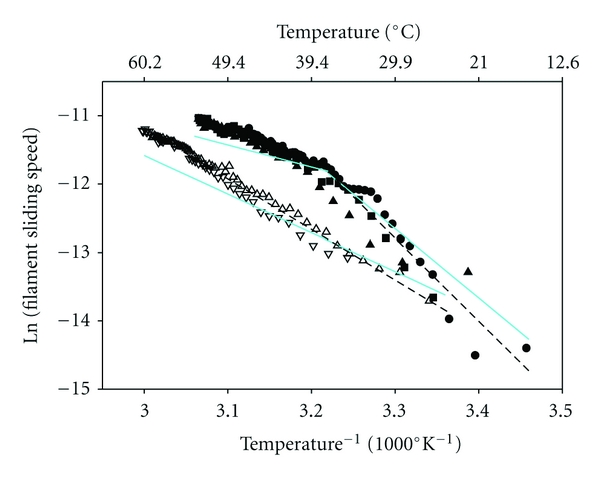
Correction for temperature-dependent effects of solvent viscosity (solid, cyan lines) on filament sliding speed of regulated thin filaments (solid symbols) and unregulated F-actin (open symbols). Data from Figure 1(c) were replotted in Arrhenius form (symbols as in Figure 1), with data omitted for regulated thin filaments above ∼54°C and filament sliding speeds expressed in μm s−1. Dashed lines (black) are linear least squares regressions on the data before correction for temperature-dependent changes in solvent viscosity. Note the change in slope at ∼38°C (Tt) for regulated thin filaments. Effects of temperature-dependent changes in solvent viscosity (normalized to that at body temperature, 37°C) were removed by assuming that speed varies inversely with viscosity, as demonstrated experimentally [43]; the net result of this correction in all instances is a decrease in apparent Ea.
3.2. Influence of FHC Mutations in Troponin on the Temperature Dependence of Thin-Filament Sliding
Because the temperature dependence of maximum filament sliding speed is influenced by the presence and function of Tn·Tm (Figures 1–3), and FHC-related mutations in troponin profoundly influence the Ca2+ dependence of myofilament function [8–10, 57], we utilized the thermoelectric controller to investigate temperature effects on thin filaments reconstituted with rHcTn·α-Tm when one of three FHC-related mutations—TnI R145G, TnI K206Q, or TnT R278C—was incorporated into the troponin complex (Section 2). At pCa 5, filament sliding speed for two mutants (TnI K206Q and TnT R278C) was faster than WT over the range of temperatures examined, consistent with previously reported data for TnI K206Q using recombinant rat cardiac Tn [32]. In contrast, speed with TnI R145G was similar to WT (Figure 4).
Figure 4.
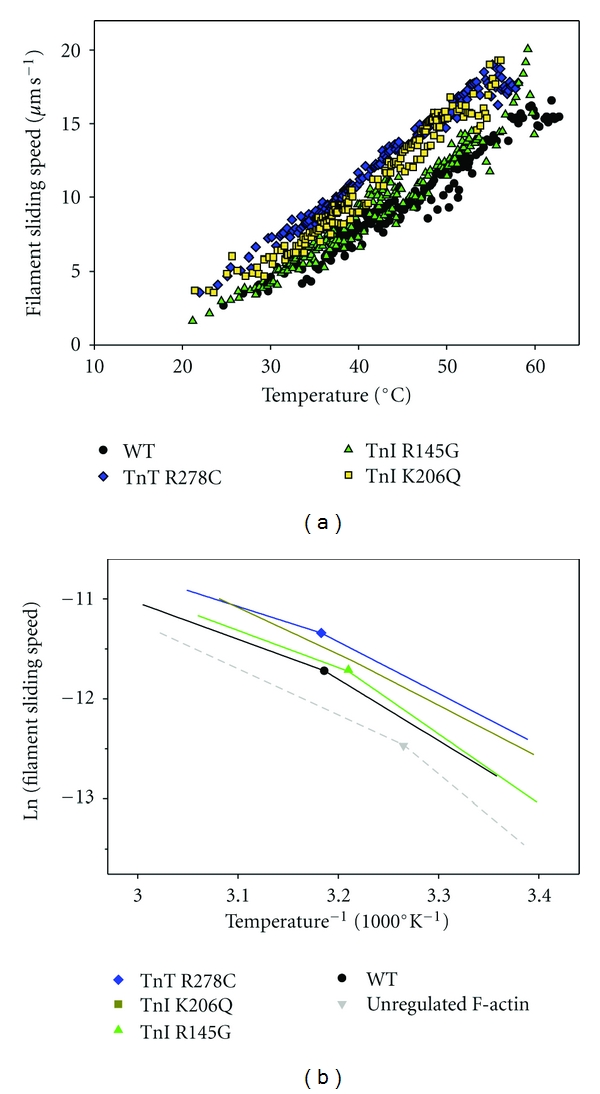
Influence of FHC-linked mutations in human cardiac troponin on temperature dependence of thin-filament sliding at pCa 5. Thin filaments were reconstituted with α-Tm and either rhcTn WT (circles, black), rhcTn TnI,R145G (triangles, green), rhcTn TnI,K206Q (squares, yellow), or rhcTn TnT, R278C (diamonds, blue). Data were obtained during the heating phases of multiple heating/cooling cycles, as in Figure 1; note that WT data were replotted from Figure 2(b). (a) Points represent the mean sliding speed for 3–8 filaments during 1 s. Note that speeds with TnI mutant K206Q [32] and TnT mutant R278C were generally elevated over WT. (b) Arrhenius analysis of data from (a). Symbols and colors as in (a); unregulated F-actin from the same series of experiments (Figure 2(b)) was also included in (b) (inverted triangle and dashed lines; light gray). Each dataset was fit by two linear regressions after omitting data above ∼60°C (WT), ∼54°C (cTnI R145G), ∼51.5°C (cTnI K206Q), or ∼55°C (cTnT R278C). Regression lines are shown without correction for temperature-dependent changes in solvent viscosity; points correspond to Tt, the intersection of each pair of lines. Filament sliding speeds are expressed in μm s−1.
The micromechanical thermal assays with thin filaments containing FHC mutations in rHcTn suggest that either the mutant troponins are intrinsically less thermally stable, or the regulatory interaction of mutant troponin with F-actin-α-Tm is less stable, or both (Figure 4(a)). All three of the FHC mutants exhibited anomalous declines in speed at lower temperatures than WT rHcTn, with the cTnT R278C and cTnI R145G mutations resulting in ∼4-5°C decreases, and the cTnI K206Q mutation leading to a larger, ∼8-9°C decrease in the temperature at which the anomalous decline in speed occurred as temperature continued to rise (Figure 4(a)).
Arhennius analysis suggests that there was little effect of the FHC mutations on Tt for thin filaments reconstituted with WT or mutant rHcTn, with values of 38–41°C where they could be unambiguously determined (Figure 4(b)). This was similar to the value of Tt = ~38°C obtained with native cTn (Figure 3). The slopes of the high- and low-temperature regimes were more similar for the two fastest mutants (cTnT R278C and cTnI K206Q) compared with WT or the cTnI R145G mutant (Figure 4(b)); in fact, it was not possible to unambiguously identify a value of Tt from data with TnI K206Q by the algorithm used (Section 2). When the data for unregulated F-actin from Figure 2(b) (open symbols) were plotted in Arrhenius form (Figure 4(b), gray dashed lines), a value of Tt ~ 33°C was obtained in contrast to the dataset in Figures 1 and 3, where Tt for unregulated F-actin could not be determined because there were not sufficient points below Tt to define a low-temperature regime.
3.3. Influence of Factors that Affect Cross-Bridge Number and Kinetics on Temperature Dependence of Unregulated F-Actin and Regulated Thin-Filament Sliding Speed
To address whether changes in the number of cross-bridges and distribution of cross-bridge states could be responsible for differences in sliding speed and Ea between unregulated F-actin and regulated thin filaments at saturating Ca2+, we studied the effects of altered HMM density, and metabolite concentrations which are known to affect the fraction of weakly versus strongly bound cross-bridges. Solutions were modified in the following ways, and compared with a control condition that was itself modified in comparison with assays reported in Figures 1–4 (Section 2): decreased [ATP] from 2 mM (Control) to 200 μM, increased [Pi] from ~1 μM (Control) to 4 mM, or reduced HMM density (ρ) from 0.25 mg/mL (Control) to 0.075 mg/mL (Figure 5). In these experiments only, motility assays were performed at steady-state temperatures of 27, 32, 37, or 42°C so that we could calculate metabolite concentrations in the presence of CP/CK to buffer ATP/ADP and reduce ADP (Section 2), conditions that did not apply to the nonsteady state temperature assays reported in Figures 1–4.
Figure 5.
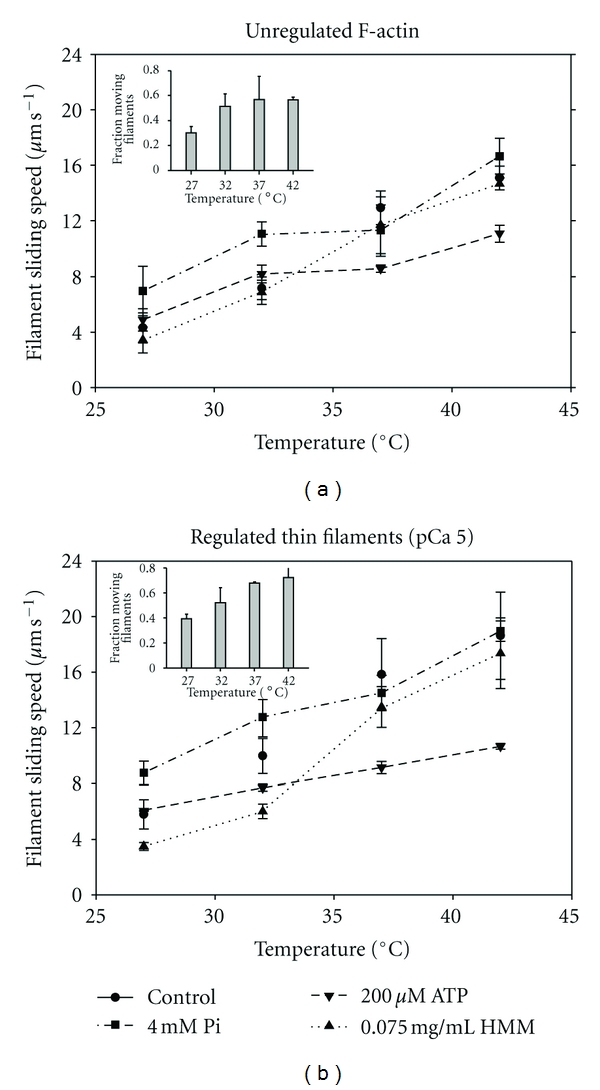
Influence of motility conditions on temperature dependence of filament sliding speeds for (a) unregulated F-actin and (b) regulated thin filaments (F-actin plus native cTn and α-Tm) at pCa 5. Points are the mean of means ± S.D. from 45–75 filaments in each of ≥3 individual flow cells and are plotted against steady-state temperature (27– 42°C) for four conditions. For this series of experiments only, control conditions (circles, solid lines) were [Pi] = ∼1 μM (achieved by removal of contaminating Pi; Section 2); [ATP] = 2 mM (buffered by CP and CK; Section 2), and high ρ achieved by infusing 0.25 mg/mL [HMM] into the flow cell. Conditions were modified to test low ATP (200 μM ATP; inverted triangles, short dashed lines); high Pi (4 mM Pi; squares, long dash dotted lines); or low ρ (0.075 mg/mL HMM; triangles, dash dotted line). Lines were drawn to connect the points. Insets: temperature dependence of fraction of filaments moving under low ρ condition plotted for (a) unregulated F-actin and (b) regulated thin filaments at pCa 5. Note that the fraction of regulated thin filaments, but not unregulated F-actin, increases continuously over this temperature range.
Sliding speed increased with temperature for each condition tested for both regulated thin filaments at pCa 5 and unregulated F-actin (Figure 5) as expected for the temperature range examined. Sliding speed with respect to the Control condition was reduced significantly for the low ATP condition at the two highest temperatures for unregulated F-actin (Figure 5(a)) and at the three highest temperature points for regulated thin filaments (Figure 5(b)). Sliding speed at high Pi (Figure 5) was higher than in Control conditions at the two lower temperatures for both regulated thin filaments and unregulated F-actin. Decreasing ρ had little effect on unregulated F-actin sliding speed (Figure 5(a)) but decreased regulated thin-filament speed significantly at the three lowest temperatures (27, 32, and 37°C, Figure 5(b)). Addition of Tn·Tm was typically associated with increased speed, but not at low ATP, which is in good agreement with the results of Homsher et al. [16]. At reduced ρ, the enhancing effect of Tn·Tm was greater at higher temperatures. The fraction of motile filaments at reduced ρ increased over the three lowest temperatures (27, 32, and 37°C) for unregulated F-actin (inset, Figure 5(a)), but over the entire temperature range for regulated thin filaments (inset Figure 5(b)).
4. Discussion
In this study, we utilized microfabricated thermoelectric devices to rapidly and reversibly investigate the influence of temperature on myosin-driven sliding of Ca2+-regulated, muscle thin filaments over a wide temperature range (∼20–60°C). To investigate molecular mechanisms of normal cardiac function and pathophysiology, thin filaments were reconstituted with human cardiac Tn and Tm, or with FHC-related mutants of human cardiac Tn. The major results of this study are as follows.
At pCa 5, sliding speed of regulated thin filaments increased reversibly with increasing temperature (∼20–∼54°C) as expected and was faster than unregulated F-actin over that range.
Reversible dysregulation—loss of Ca2+-regulation—occurred at both pCa 5 and pCa 9 at the upper end of the temperature range examined; this suggests thermal unfolding of the cardiac regulatory proteins and/or their dissociation from actin.
Arrhenius analysis indicates two temperature regimes with different Ea; the transition temperature between those regimes was higher for regulated thin filaments than unregulated F-actin, and this is explained by a simple model.
Factors that alter cross-bridge number and kinetics—[ATP], [Pi], and ρHMM—alter the temperature dependence of sliding speed of both regulated thin filaments and unregulated F-actin in qualitatively similar ways that provide insight into rate limiting factors.
Three FHC mutants in cTn (cTnI R145G, cTnI K206Q, and cTnT R278C) destabilize regulatory protein structure and/or interactions with F-actin-Tm and may also alter the kinetics of thin filament sliding at pCa 5.
4.1. Functional Assays for Thermal Stability of Regulated Thin Filaments and Ca2+-Regulatory Proteins
A custom thermoelectric controller [5] that was used in this study (Figure 1(a)), and miniaturized versions thereof [6], provide a novel approach to evaluate contractile protein function over a wide range of temperatures and multiple temperature cycles. The multiple datasets collected this way with native and WT regulatory proteins (Figures 1–3) complement and expand the range of information that can be obtained using conventional motility assays [16, 58, 59] and traditional approaches to studying protein structure in solution [60–63]. Our device provides a simpler readout of temperature than relying on intensity of the fluorescent label [64]. Speed of unregulated F-actin increased monotonically over the temperature range used (Figures 1-2, open symbols), indicating that HMM and actin are not affected detrimentally at temperatures below 60°C; this is consistent with previous functional measurements [5, 64] and structural assays showing that the structure of Ph-stabilized F-actin in solution is thermally stable up to 78°C [62], while unfolding of subfragment 1 (S1) was reported for temperatures greater than 58°C in the presence of analogs of ATP or ADP·Pi [63].
Dysregulation of thin filaments at elevated temperatures was observed at both pCa 5 (Figures 1-2) and pCa 9 (Figure 2), suggesting that altered function was likely due to dissociation of Tn·Tm from actin or denaturation of Tn and/or Tm. At pCa 5, the anomalous decline in sliding speed for regulated thin filaments above ∼54°C suggests a loss of regulation (dysregulation) because as temperature increased, speed decreased toward that of unregulated F-actin (Figure 1(c)). At pCa 9, filament sliding was observed at temperatures above ∼43°C; speed increased with temperature and was generally lower than, but approached that of unregulated F-actin (Figure 2), indicating that cTn stabilizes actin·Tm at pCa 5. Taken together, these results from microthermal heater assays provide functional correlates that are consistent with previous solution studies showing half-maximal dissociation of Tm from Ph-stabilized F-actin·Tm complex (no Tn) at 46–48°C, and unfolding of Tm alone in solution that was resolved into two separate calorimetric domains with maxima of 42 and 51°C [61, 62]. More generally, our results demonstrate that microthermal heater assays can provide unique, molecular level insights into muscle thin-filament function, particularly when the maximum temperature is limited such that the effects of temperature on Ca2+ regulation are fully reversible (Figure 2(a)).
4.2. Temperature Dependence of Ca2+-Regulated Thin-Filament Sliding at pCa 5
Comparison of temperature-dependence of regulated thin-filament sliding at pCa 5 with that of unregulated F-actin provides insights into the roles of cardiac troponin and tropomyosin. This is important in light of the observation that not only do Tn-Tm modulate the thin filament side of actomyosin interactions but cTn can also directly accelerate myosin ATPase activity even in the absence of actin and Tm [20]. This direct effect of cTn on myosin kinetics is presumed to at least partially underlie Tn·Tm's enhancement of maximum unloaded sliding speed under a wide variety of conditions (e.g., Figures 1(c), 2(b), 3, 4(b), and 5), although it is also possible that there are differences in the myosin binding interface due to Tm·S1 interactions [65] or regulatory protein-induced structural changes in actin itself [15].
While it is clear that Tn·Tm modulate filament sliding speed primarily by affecting the number of cross-bridges at submaximal Ca2+ activation [12, 14, 50], the most straightforward mechanism by which Tn·Tm could directly enhance actomyosin kinetics at saturating Ca2+ is through a reduction in the time that cross-bridges spend in the strongly bound state (ts) under the unloaded conditions of a motility assay, as observed experimentally [33]. The maximum sliding speed (sm) for regulated thin filaments at saturating Ca2+ or for unregulated F-actin is simply related to myosin step size (dx) and ts as proposed by Uyeda et al. [51]:
| (1) |
To accommodate the observed, biphasic Arrhenius plots (Figures 3 and 4(b)), we assume that dx = 0.0055 μm [66] for all conditions examined and rewrite (1) to express ts as the sum of two exponential terms:
| (2) |
which, when fit to the data for regulated thin filaments in Figure 3, yields sm = 0.0055/(0.301e−0.1855T + 0.00145e−0.0277T) with R2 = 0.97. Separating the two terms and plotting them as rates, we can see that one varies little with temperature (black solid line; Figure 6) and while the other is highly temperature sensitive (red solid line; Figure 6). Assuming that the more highly temperature-dependent term is similar for both regulated thin filaments and unregulated F-actin, regression estimates were obtained for unregulated F-actin sliding in Figure 3, c = 0.0225 and d = 0.0676 (R2 = 0.97), and the corresponding rate prediction was plotted in Figure 6 (black dashed line).
Figure 6.
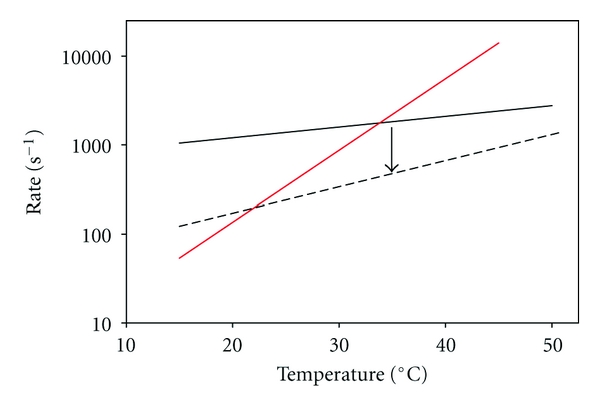
Temperature dependence of two apparent rate constants that may limit actin filament sliding speed at pCa 5. Data in Figure 3 (T < ~54°C) for sliding speed of regulated thin filaments were fitted to (2). The inverse of each of the exponential rate terms in the denominator of (2) is plotted: for WT regulated thin filaments, one rate exhibits only slight temperature sensitivity (solid black line) while the other is markedly temperature sensitive (red line). The rate-limiting step for filament sliding is the smaller of the two rates, and Tt is the intersection of the two lines. The less temperature sensitive rate is also shown for unregulated F-actin (dashed black line; data from Figure 3). Note that by assuming the temperature-sensitive rate (red line) is similar for both instances, decreasing the less temperature sensitive rate (black arrow, as observed experimentally with unregulated F-actin) decreases Tt.
If the intersections of the red line with the two black lines in Figure 6 are related to discontinuities in the Arrhenius plots (Figures 3 and 4(b)) and thus values of Tt, then the simple analysis of (1), (2) is in accord with observations that the transition from high to low Ea will occur at higher temperature for regulated thin filaments (red and solid black lines) than for unregulated F-actin (red and dashed black lines). In this model, the rate limitation for filament sliding at a given temperature would be the lower of the two rates from the red line and one black line: at low temperatures, the steeper, red value is limiting, while the shallower, black value is limited at elevated temperatures (Figure 6). The temperature dependence of unloaded filament sliding speed has been reported to vary as a function of temperature in some but not all conditions [16, 58, 59]. For unregulated F-actin, Arrhenius plots have been previously reported to be linear within 20–60°C [5] and 7–25°C [16], but nonlinear within 3–42°C with a transition temperature (Tt) of 15.4°C [59], and 10–35°C with a Tt ~ 25°C [58, 67]. Tt depends not only on the experimental conditions but also on whether Ca2+-regulatory proteins are present. The Arrhenius plot of fish myofibrillar ATPase at saturating Ca2+ exhibited a discontinuity [68], while this was not the case for in vitro motility sliding speed and actin-activated MgATPase activity with fish myosin and unregulated F-actin [69]. For regulated thin filaments, Homsher et al. [16] reported a break in the Arrhenius plot around 12.5°C that was not observed for unregulated F-actin within the 7–25°C temperature range. In this study using micromechanical thermal assays, discontinuities in Arrhenius plots for regulated thin filaments (Figures 3 and 4(b)) occurred at a higher temperature than when Tt could be identified for unregulated F-actin (Figure 4(b)). While there is no universal agreement in the limited data available on absolute values for Tt for filament sliding, there does appear to be general agreement that Tn·Tm modulates the temperature dependence of unloaded filament sliding. Our data suggest that this modulation of Tt by human cardiac Tn·Tm occurs around physiological temperature.
The existence of discontinuities in Arrhenius plots suggests that different factors are rate-limiting to filament sliding above and below Tt. What factors could contribute to this difference? Prior experiments with regulated thin filaments at subsaturating levels of Ca2+ suggested that the number of cross-bridges is limiting to sliding speed at 30°C [12, 50]. The data in Figure 5 indicate that sliding speed is markedly sensitive to a reduction in the density (ρ) of HMM on the flow cell surface at 27 and 32°C for both unregulated F-actin and especially regulated thin filaments at maximal Ca2+ activation. At higher temperatures (37 and 42°C), however, there was little or no effect of the same reduction in ρ on sliding speed for either unregulated F-actin or regulated thin filaments (Figure 5). In addition, the fraction of moving filaments at low ρincreased with temperature for regulated thin filaments at pCa 5 over the entire temperature range (Figure 5(b), inset), while for unregulated F-actin, the fraction started at a lower value and plateaued (Figure 5(a), inset). Taken together, these observations suggest an increase in strong cross-bridges with temperature for both unregulated F-actin and regulated thin filaments, consistent with previous reports using in vitro assays and permeabilized muscle fibers [70–72]. Thus, it appears that Tn·Tm can modulate at least one rate-limiting factor, and also Tt, around physiological temperature: once a temperature (Tt) is attained at which the availability of cross-bridges is great enough to guarantee continuous filament sliding, further increase of speed with temperature will only depend on less temperature-sensitive kinetic rates that are associated with transitions between strongly bound cross-bridge states, and will therefore be characterized by low Ea (Figure 6, cross-over from red to black lines).
In addition to the number of cross-bridges, filament sliding speed can be altered by variations in the substrate and products of the ATPase reaction through modulation of actomyosin kinetics. According to the model implicit in (1), changes in metabolite concentration would primarily influence ts, and therefore sm, and also the number of attached cross-bridges. For example, Tn·Tm could affect the kinetics of ATP-induced cross-bridge dissociation that has been suggested to limit filament sliding speed, at least in fast skeletal muscle at low temperature [16, 73]. When [ATP] was reduced to 200 μM—a substrate concentration that is similar to the apparent Kd for sarcomere shortening velocity and filament sliding speed [16, 53, 74, 75]—inhibition of speed was greater for regulated thin filaments at pCa 5 than unregulated F-actin (Figure 5), as reported by Homsher et al. [16]. Inhibition of sm at low ATP levels is explained by slower ATP-induced detachment of cross-bridges at the end of attached phase the cycle, which increases ts (1); the fraction of strongly attached cross-bridges would also increase, although the dominant factor in determining sm in this instance is slower cross-bridge cycling. The relatively greater inhibition of regulated thin-filament sliding was accentuated at higher, more physiological temperatures (Figure 5). The net result was a generally shallow slope of speed versus temperature for both regulated thin filaments and unregulated F-actin (Figure 5) that corresponds to a regime of relatively low Ea. The expectation would then be that reduced [ATP] should shift Tt to a lower temperature (Figure 6) although it was not resolved in this experiment, likely because of the lower limit of temperature examined.
There are some interesting parallels with reduced [ATP] effects, along with significant differences, when the concentration of product Pi was increased from micromolar levels to 4 mM (Table 1), that is, the highest concentration that we could achieve without necessitating an increase in ionic strength. Temperature has previously been shown to modulate the effect of Pi on isometric force in muscle fibers, although there is disagreement about whether Pi is either more [29, 76] or less [77] inhibitory of isometric force as temperature increases. While Pi inhibits isometric force, elevated Pi increases sliding speed for both regulated thin filaments at pCa 5 and unregulated F-actin, although the effect predominates at lower temperatures and is more pronounced for regulated thin-filaments (Figure 5). Maximum velocity of sarcomere shortening [52, 70, 78, 79] and unloaded sliding of unregulated F-actin [80] have previously been reported to increase with elevated Pi. This is generally understood to be the result of reversal of cross-bridge attachment, which decreases ts as demonstrated at the single molecule level by Baker et al. [81], thus reducing the number of strongly attached cross-bridges and increasing sm (1). The relatively greater enhancement of sliding speed at lower temperatures results in a generally shallow slope of speed versus temperature for both regulated thin filaments and unregulated F-actin (Figure 5) that corresponds to a regime of relatively low Ea; in contrast to the result with low ATP, the expectation is that elevated Pi should shift Tt to a higher temperature (Figure 6). Taken together, the data in Figure 5 illustrate that the relative importance of factors which influence cross-bridge cycling kinetics and/or filament sliding speed varies with temperature around the physiological range, and which are rate-limiting also varies as implicated in Figure 6. This suggests that energetic state may impact cardiac function when core body temperature varies, such as in mild hypothermia or with exercise, in ways that may seem counterintuitive [31].
4.3. Implications for Understanding Cardiovascular Diseases
Figure 4 illustrates that the microthermal heater can be used to study effects of disease-related mutations in human cardiac Ca2+ regulatory proteins on function of individual thin filaments. This clearly goes beyond, and will ultimately help explain, prior work that examined physical properties of the mutant proteins in bulk solution, and the Ca2+-dependence of function. cTn mutations may influence not just sm (TnI K206Q and TnT R278C), but also the temperature dependence of Ea (TnI K206Q and TnT R278C) and possibly Tt (TnI K206Q). The mutations may also influence the temperature at which thin-filament structure is affected to the extent that dysregulation occurs and Ca2+ dependent function is altered, whether the disease-related change in function is most relevant in the myofilaments, or perhaps in the nucleus [82]. For all three Tn mutants examined (TnI R145G, TnI K206Q, and TnT R278C) mutations destabilized thin-filament structure and function at lower temperatures than WT; this effect was particularly noticeable for an FHC mutant of human cardiac Tm (E180G) that causes extreme shifts in Ca2+ sensitivity [33, 40]. Thus, we anticipate that the thermoelectric heater will be generally useful for characterization of clinically relevant mutants of thin filament protein structural stability through assessment of function at the level of individual thin filaments.
4.4. Conclusion
Results demonstrate the utility of our thermoelectric controller for investigating molecular mechanisms underlying biomolecular motor function, and cardiovascular diseases related to altered biomechanics of cardiac myofilament proteins. This assay provides a novel view of structure-function relationships for cardiac thin filaments.
Acknowledgments
The authors gratefully acknowledge L. A. McFadden for purifying myosin and actin; V. F. Miller for expression and M. Seavy for HPLC purification of tropomyosin; R. Dhanarajan from the Biological Science Molecular Cloning Facility for assistance with mutagenesis; Dr. S. Miller from the Biological Science DNA Sequencing Facility for assistance with DNA sequencing; L. A. Compton for assistance with gel analysis; K. Riddle from the Biological Science Imaging Resource for assistance with MetaMorph; Dr. J. R. Grubich for assistance with preliminary experiments; J. Bhuvasorakul and W. Chase for assistance with data analysis; Dr. B. Schoffstall for critical comments. This work was supported by NIH/NHLBI HL63974 (PBC), American Heart Association FL/PR Affiliate 0315097B (NMB), and NSF NIRT Grant ECS-0210332 (PX).
References
- 1.Hess H. Toward devices powered by biomolecular motors. Science. 2006;312(5775):860–861. doi: 10.1126/science.1126399. [DOI] [PubMed] [Google Scholar]
- 2.Hess H, Vogel V. Molecular shuttles based on motor proteins: active transport in synthetic environments. Journal of Biotechnology. 2001;82(1):67–85. doi: 10.1016/s1389-0352(01)00029-0. [DOI] [PubMed] [Google Scholar]
- 3.Sundberg M, Bunk R, Albet-Torres N, et al. Actin filament guidance on a chip: toward high-throughput assays and lab-on-a-chip applications. Langmuir. 2006;22(17):7286–7295. doi: 10.1021/la060854i. [DOI] [PubMed] [Google Scholar]
- 4.Jaber JA, Chase PB, Schlenoff JB. Actomyosin-driven motility on patterned polyelectrolyte mono-and multilayers. Nano Letters. 2003;3(11):1505–1509. [Google Scholar]
- 5.Mihajlović G, Brunet NM, Trbović J, Xiong P, Von Molnár S, Chase PB. All-electrical switching and control mechanism for actomyosin-powered nanoactuators. Applied Physics Letters. 2004;85(6):1060–1062. [Google Scholar]
- 6.Grove TJ, Puckett KA, Brunet NM, et al. Packaging actomyosin-based biomolecular motor-driven devices for nanoactuator applications. IEEE Transactions on Advanced Packaging. 2005;28(4):556–563. [Google Scholar]
- 7.Ionov L, Stamm M, Diez S. Reversible switching of microtubule motility using thermoresponsive polymer surfaces. Nano Letters. 2006;6(9):1982–1987. doi: 10.1021/nl0611539. [DOI] [PubMed] [Google Scholar]
- 8.Parmacek MS, Solaro RJ. Biology of the troponin complex in cardiac myocytes. Progress in Cardiovascular Diseases. 2004;47(3):159–176. doi: 10.1016/j.pcad.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circulation Research. 2011;108(6):765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annual Review of Genomics and Human Genetics. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- 11.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiological Reviews. 2000;80(2):853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 12.Gordon AM, LaMadrid MA, Chen Y, Luo Z, Chase PB. Calcium regulation of skeletal muscle thin filament motility in vitro. Biophysical Journal. 1997;72(3):1295–1307. doi: 10.1016/S0006-3495(97)78776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schoffstall B, Brunet NM, Williams S, et al. Ca2+ sensitivity of regulated cardiac thin filament sliding does not depend on myosin isoform. Journal of Physiology. 2006;577(3):935–944. doi: 10.1113/jphysiol.2006.120105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Homsher E, Kim B, Bobkova A, Tobacman LS. Calcium regulation of thin filament movement in an in vitro motility assay. Biophysical Journal. 1996;70(4):1881–1892. doi: 10.1016/S0006-3495(96)79753-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Homsher E, Lee DM, Morris C, Pavlov D, Tobacman LS. Regulation of force and unloaded sliding speed in single thin filaments: effects of regulatory proteins and calcium. Journal of Physiology. 2000;524(1):233–243. doi: 10.1111/j.1469-7793.2000.00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Homsher E, Nili M, Chen IY, Tobacman LS. Regulatory proteins alter nucleotide binding to acto-myosin of sliding filaments in motility assays. Biophysical Journal. 2003;85(2):1046–1052. doi: 10.1016/S0006-3495(03)74543-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gorga JA, Fishbaugher DE, VanBuren P. Activation of the calcium-regulated thin filament by myosin strong binding. Biophysical Journal. 2003;85(4):2484–2491. doi: 10.1016/s0006-3495(03)74671-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clemmens EW, Regnier M. Skeletal regulatory proteins enhance thin filament sliding speed and force by skeletal HMM. Journal of Muscle Research and Cell Motility. 2004;25(7):515–525. doi: 10.1007/s10974-004-3787-0. [DOI] [PubMed] [Google Scholar]
- 19.Fujita H, Sasaki D, Ishiwata S, Kawai M. Elementary steps of the cross-bridge cycle in bovine myocardium with and without regulatory proteins. Biophysical Journal. 2002;82(2):915–928. doi: 10.1016/S0006-3495(02)75453-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoffstall B, Labarbera VA, Brunet NM, et al. Interaction between troponin and myosin enhances contractile activity of myosin in cardiac muscle. DNA and Cell Biology. 2011;30(9):653–659. doi: 10.1089/dna.2010.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujita H, Kawai M. Temperature effect on isometric tension is mediated by regulatory proteins tropomyosin and troponin in bovine myocardium. Journal of Physiology. 2002;539(1):267–276. doi: 10.1113/jphysiol.2001.013220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Tombe PP, Stienen GJM. Impact of temperature on cross-bridge cycling kinetics in rat myocardium. Journal of Physiology. 2007;584(2):591–600. doi: 10.1113/jphysiol.2007.138693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janssen PML, Stull LB, Marbán E. Myofilament properties comprise the rate-limiting step for cardiac relaxation at body temperature in the rat. American Journal of Physiology. 2002;282(2):H499–H507. doi: 10.1152/ajpheart.00595.2001. [DOI] [PubMed] [Google Scholar]
- 24.Martyn DA, Adhikari BB, Regnier M, Gu J, Xu S, Yu LC. Response of equatorial X-ray reflections and stiffness to altered sarcomere length and myofilament lattice spacing in relaxed skinned cardiac muscle. Biophysical Journal. 2004;86(2):1002–1011. doi: 10.1016/S0006-3495(04)74175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coupland ME, Ranatunga KW. Force generation induced by rapid temperature jumps in intact mammalian (rat) skeletal muscle fibres. Journal of Physiology. 2003;548(2):439–449. doi: 10.1113/jphysiol.2002.037143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stephenson DG, Williams DA. Temperature-dependent calcium sensitivity changes in skinned muscle fibres of rat and toad. Journal of Physiology. 1985;360:1–12. doi: 10.1113/jphysiol.1985.sp015600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ranatunga KW, Wylie SR. Temperature-dependent transitions in isometric contractions of rat muscle. Journal of Physiology. 1983;339:87–95. doi: 10.1113/jphysiol.1983.sp014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ranatunga KW. Temperature dependence of mechanical power output in mammalian (rat) skeletal muscle. Experimental Physiology. 1998;83(3):371–376. doi: 10.1113/expphysiol.1998.sp004120. [DOI] [PubMed] [Google Scholar]
- 29.Debold EP, Romatowski J, Fitts RH. The depressive effect of Pi on the force-pCa relationship in skinned single muscle fibers is temperature dependent. American Journal of Physiology. 2006;290(4):C1041–C1050. doi: 10.1152/ajpcell.00342.2005. [DOI] [PubMed] [Google Scholar]
- 30.MacIntosh BR. Role of calcium sensitivity modulation in skeletal muscle performance. News in Physiological Sciences. 2003;18(6):222–225. doi: 10.1152/nips.01456.2003. [DOI] [PubMed] [Google Scholar]
- 31.Weisser J, Martin J, Bisping E, et al. Influence of mild hypothermia on myocardial contractility and circulatory function. Basic Research in Cardiology. 2001;96(2):198–205. doi: 10.1007/s003950170071. [DOI] [PubMed] [Google Scholar]
- 32.Köhler J, Chen Y, Brenner B, et al. Familial hypertrophic cardiomyopathy mutations in troponin I (K183Δ, G203S, K206Q) enhance filament sliding. Physiological Genomics. 2003;14:117–128. doi: 10.1152/physiolgenomics.00101.2002. [DOI] [PubMed] [Google Scholar]
- 33.Wang F, Brunet NM, Grubich JR, et al. Facilitated cross-bridge interactions with thin filaments by familial hypertrophic cardiomyopathy mutations in α-tropomyosin. Journal of Biomedicine and Biotechnology. 2011;2011:12 pages. doi: 10.1155/2011/435271. Article ID 435271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gafurov B, Fredricksen S, Cai A, Brenner B, Chase PB, Chalovich JM. The Δ14 mutation of human cardiac troponin T enhances ATPase activity and alters the cooperative binding of S1-ADP to regulated actin. Biochemistry. 2004;43(48):15276–15285. doi: 10.1021/bi048646h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi-Yanaga F, Morimoto S, Harada K, et al. Functional consequences of the mutations in human cardiac troponin I gene found in familial hypertrophic cardiomyopathy. Journal of Molecular and Cellular Cardiology. 2001;33(12):2095–2107. doi: 10.1006/jmcc.2001.1473. [DOI] [PubMed] [Google Scholar]
- 36.Lang R, Gomes AV, Zhao J, Housmans PR, Miller T, Potter JD. Functional analysis of a troponin I (R145G) mutation associated with familial hypertrophic cardiomyopathy. Journal of Biological Chemistry. 2002;277(14):11670–11678. doi: 10.1074/jbc.M108912200. [DOI] [PubMed] [Google Scholar]
- 37.Szczesna D, Zhang R, Zhao J, Jones M, Guzman G, Potter JD. Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. Journal of Biological Chemistry. 2000;275(1):624–630. doi: 10.1074/jbc.275.1.624. [DOI] [PubMed] [Google Scholar]
- 38.Yanaga F, Morimoto S, Ohtsuki I. Ca2+ sensitization and potentiation of the maximum level of myofibrillar ATPase activity caused by mutations of troponin T found in familial hypertrophic cardiomyopathy. Journal of Biological Chemistry. 1999;274(13):8806–8812. doi: 10.1074/jbc.274.13.8806. [DOI] [PubMed] [Google Scholar]
- 39.Elliott K, Watkins H, Redwood CS. Altered regulatory properties of human cardiac troponin I mutants that cause hypertrophic cardiomyopathy. Journal of Biological Chemistry. 2000;275(29):22069–22074. doi: 10.1074/jbc.M002502200. [DOI] [PubMed] [Google Scholar]
- 40.Bai F, Weis A, Takeda AK, Chase PB, Kawai M. Enhanced active cross-bridges during diastole: molecular pathogenesis of tropomyosin's HCM mutations. Biophysical Journal. 2011;100(4):1014–1023. doi: 10.1016/j.bpj.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mathur MC, Chase PB, Chalovich JM. Several cardiomyopathy causing mutations on tropomyosin either destabilize the active state of actomyosin or alter the binding properties of tropomyosin. Biochemical and Biophysical Research Communications. 2011;406(1):74–78. doi: 10.1016/j.bbrc.2011.01.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kataoka A, Hemmer C, Chase PB. Computational simulation of hypertrophic cardiomyopathy mutations in Troponin I: influence of increased myofilament calcium sensitivity on isometric force, ATPase and [Ca2+]i. Journal of Biomechanics. 2007;40(9):2044–2052. doi: 10.1016/j.jbiomech.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 43.Chase PB, Chen Y, Kulin KL, Daniel TL. Viscosity and solute dependence of F-actin translocation by rabbit skeletal heavy meromyosin. American Journal of Physiology. 2000;278(6):C1088–C1098. doi: 10.1152/ajpcell.2000.278.6.C1088. [DOI] [PubMed] [Google Scholar]
- 44.Margossian SS, Lowey S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods in Enzymology. 1982;85(C):55–71. doi: 10.1016/0076-6879(82)85009-x. [DOI] [PubMed] [Google Scholar]
- 45.Pardee JD, Aspudich J. Purification of muscle actin. Methods in Enzymology. 1982;85(C):164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- 46.Heald RW, Hitchcock-DeGregori SE. The structure of the amino terminus of tropomyosin is critical for binding to actin in the absence and presence of troponin. Journal of Biological Chemistry. 1988;263(11):5254–5259. [PubMed] [Google Scholar]
- 47.Monteiro PB, Lataro RC, Ferro JA, Reinach FDC. Functional α-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. Journal of Biological Chemistry. 1994;269(14):10461–10466. [PubMed] [Google Scholar]
- 48.Schagger H, Von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Analytical Biochemistry. 1987;166(2):368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 49.Kron SJ, Toyoshima YY, Uyeda TQP, Spudich JA. Assays for actin sliding movement over myosin-coated surfaces. Methods in Enzymology. 1991;196:399–416. doi: 10.1016/0076-6879(91)96035-p. [DOI] [PubMed] [Google Scholar]
- 50.Liang B, Chen Y, Wang CK, et al. Ca2+ regulation of rabbit skeletal muscle thin filament sliding: role of cross-bridge number. Biophysical Journal. 2003;85(3):1775–1786. doi: 10.1016/S0006-3495(03)74607-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uyeda TQP, Kron SJ, Spudich JA. Myosin step size estimation from slow sliding movement of actin over low densities of heavy meromyosin. Journal of Molecular Biology. 1990;214(3):699–710. doi: 10.1016/0022-2836(90)90287-V. [DOI] [PubMed] [Google Scholar]
- 52.Pate E, Cooke R. Addition of phosphate to active muscle fibers probes actomyosin states within the powerstroke. Pflugers Archiv. 1989;414(1):73–81. doi: 10.1007/BF00585629. [DOI] [PubMed] [Google Scholar]
- 53.Chase PB, Kushmerick MJ. Effect of physiological ADP concentrations on contraction of single skinned fibers from rabbit fast and slow muscles. American Journal of Physiology. 1995;268(2):C480–C489. doi: 10.1152/ajpcell.1995.268.2.C480. [DOI] [PubMed] [Google Scholar]
- 54.Silverstein R, Voet J, Reed D, Abeles RH. Purification and mechanism of action of sucrose phosphorylase. Journal of Biological Chemistry. 1967;242(6):1338–1346. [PubMed] [Google Scholar]
- 55.Weast RC. CRC Handbook of Chemistry and Physics. 63rd edition. Boca Raton, Fla, 1982: CRC Press; 1982. [Google Scholar]
- 56.Chase PB, Denkinger TM, Kushmerick MJ. Effect of viscosity on mechanics of single, skinned fibers from rabbit psoas muscle. Biophysical Journal. 1998;74(3):1428–1438. doi: 10.1016/S0006-3495(98)77855-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gomes AV, Potter JD. Molecular and cellular aspects of troponin cardiomyopathies. Annals of the New York Academy of Sciences. 2004;1015:214–224. doi: 10.1196/annals.1302.018. [DOI] [PubMed] [Google Scholar]
- 58.Rossi R, Maffei M, Bottinelli R, Canepari M. Temperature dependence of speed of actin filaments propelled by slow and fast skeletal myosin isoforms. Journal of Applied Physiology. 2005;99(6):2239–2245. doi: 10.1152/japplphysiol.00543.2005. [DOI] [PubMed] [Google Scholar]
- 59.Anson M. Temperature dependence and arrhenius activation energy of F-actin velocity generated in vitro by skeletal myosin. Journal of Molecular Biology. 1992;224(4):1029–1038. doi: 10.1016/0022-2836(92)90467-x. [DOI] [PubMed] [Google Scholar]
- 60.Golitsina N, An Y, Greenfield NJ, et al. Effects of two familial hypertrophic cardiomyopathy-causing mutations on α-tropomyosin structure and function. Biochemistry. 1999;38(12):p. 3850. doi: 10.1021/bi9950701. [DOI] [PubMed] [Google Scholar]
- 61.Kremneva EV, Nikolaeva OP, Gusev NB, Levitsky DI. Effects of troponin on thermal unfolding of actin-bound tropomyosin. Biochemistry. 2003;68(7):802–809. doi: 10.1023/a:1025043202615. [DOI] [PubMed] [Google Scholar]
- 62.Kremneva E, Boussouf S, Nikolaeva O, Maytum R, Geeves MA, Levitsky DI. Effects of two familial hypertrophic cardiomyopathy mutations in α-tropomyosin, Asp175Asn and Glut180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophysical Journal. 2004;87(6):3922–3933. doi: 10.1529/biophysj.104.048793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shriver JW, Kamath U. Differential scanning calorimetry of the unfolding of myosin subfragment 1, subfragment 2, and heavy meromyosin. Biochemistry. 1990;29(10):2556–2564. doi: 10.1021/bi00462a018. [DOI] [PubMed] [Google Scholar]
- 64.Kato H, Nishizaka T, Iga T, Kinosita K, Ishiwata S. Imaging of thermal activation of actomyosin motors. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(17):9602–9606. doi: 10.1073/pnas.96.17.9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. Journal of Molecular Biology. 1997;266(1):8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 66.Veigel C, Bartoo ML, White DCS, Sparrow JC, Molloy JE. The stiffness of rabbit skeletal actomyosin cross-bridges determined with an optical tweezers transducer. Biophysical Journal. 1998;75(3):1424–1438. doi: 10.1016/S0006-3495(98)74061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Höök P, Larsson L. Actomyosin interactions in a novel single muscle fiber in vitro motility assay. Journal of Muscle Research and Cell Motility. 2000;21(4):357–365. doi: 10.1023/a:1005614212575. [DOI] [PubMed] [Google Scholar]
- 68.Moerland TS, Sidell BD. Contractile responses to temperature in the locomotory musculature of striped bass Morone saxatilis. Journal of Experimental Zoology. 1986;240(1):25–33. doi: 10.1002/jez.1402380303. [DOI] [PubMed] [Google Scholar]
- 69.Grove TJ, Mcfadden LA, Chase PB, Moerland TS. Effects of temperature, ionic strength and pH on the function of skeletal muscle myosin from a eurythermal fish, Fundulus heteroclitus. Journal of Muscle Research and Cell Motility. 2005;26(4-5):191–197. doi: 10.1007/s10974-005-9010-0. [DOI] [PubMed] [Google Scholar]
- 70.Kawai M, Kawaguchi K, Saito M, Ishiwata S. Temperature change does not affect force between single actin filaments and HMM from rabbit muscles. Biophysical Journal. 2000;78(6):3112–3119. doi: 10.1016/S0006-3495(00)76848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karatzaferi C, Chinn MK, Cooke R. The force exerted by a muscle cross-bridge depends directly on the strength of the actomyosin bond. Biophysical Journal. 2004;87(4):2532–2544. doi: 10.1529/biophysj.104.039909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhao Y, Kawai M. Kinetic and thermodynamic studies of the cross-bridge cycle in rabbit psoas muscle fibers. Biophysical Journal. 1994;67(4):1655–1668. doi: 10.1016/S0006-3495(94)80638-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nyitrai M, Rossi R, Adamek N, Pellegrino MA, Bottinelli R, Geeves MA. What limits the velocity of fast-skeletal muscle contraction in mammals? Journal of Molecular Biology. 2006;355(3):432–442. doi: 10.1016/j.jmb.2005.10.063. [DOI] [PubMed] [Google Scholar]
- 74.Cooke R, Bialek W. Contraction of glycerinated muscle fibers as a function of the ATP concentration. Biophysical Journal. 1979;28(2):241–258. doi: 10.1016/S0006-3495(79)85174-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pate E, Lin M, Franks-Skiba K, Cooke R. Contraction of glycerinated rabbit slow-twitch muscle fibers as a function of MgATP concentration. American Journal of Physiology. 1992;262(4):C1039–C1046. doi: 10.1152/ajpcell.1992.262.4.C1039. [DOI] [PubMed] [Google Scholar]
- 76.Debold EP, Dave H, Fitts RH. Fiber type and temperature dependence of inorganic phosphate: implications for fatigue. American Journal of Physiology. 2004;287(3):C673–C681. doi: 10.1152/ajpcell.00044.2004. [DOI] [PubMed] [Google Scholar]
- 77.Coupland ME, Puchert E, Ranatunga KW. Temperature dependance of active tension in mammalian (rabbit psoas) muscle fibres: effect of inorganic phosphate. Journal of Physiology. 2001;536(3):879–891. doi: 10.1111/j.1469-7793.2001.00879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chase PB, Kushmerick MJ. Effects of pH on contraction of rabbit fast and slow skeletal muscle fibers. Biophysical Journal. 1988;53(6):935–946. doi: 10.1016/S0006-3495(88)83174-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karatzaferi C, Franks-Skiba K, Cooke R. Inhibition of shortening velocity of skinned skeletal muscle fibers in conditions that mimic fatigue. American Journal of Physiology. 2008;294(3):R948–R955. doi: 10.1152/ajpregu.00541.2007. [DOI] [PubMed] [Google Scholar]
- 80.Homsher E, Wang F, Sellers JR. Factors affecting movement of F-actin filaments propelled by skeletal muscle heavy meromyosin. American Journal of Physiology. 1992;262(3):C714–C723. doi: 10.1152/ajpcell.1992.262.3.C714. [DOI] [PubMed] [Google Scholar]
- 81.Baker JE, Brosseau C, Joel PB, Warshaw DM. The biochemical kinetics underlying actin movement generated by one and many skeletal muscle myosin molecules. Biophysical Journal. 2002;82(4):2134–2147. doi: 10.1016/S0006-3495(02)75560-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Asumda FZ, Chase PB. Nuclear cardiac troponin and tropomyosin are expressed early in cardiac differentiation of rat mesenchymal stem cells. Differentiation. 83(2):106–115. doi: 10.1016/j.diff.2011.10.002. [DOI] [PubMed] [Google Scholar]