

Abstract
Heterotrimeric G protein signaling is involved in many pathways essential to development including those controlling cell migration, proliferation, differentiation and apoptosis. One key developmental event known to rely on proper heterotrimeric G protein signaling is primordial germ cell (PGC) migration. We previously developed an in vivo PGC migration assay that identified differences in the signaling capacity of G protein gamma subunits. In this study we developed Gγ subunit chimeras to determine the regions of Gγ isoforms that are responsible for these differences. The central section of the Gγ subunit was found to be necessary for the ability of a Gγ subunit to mediate signaling involved in PGC migration. Residues found in the carboxyterminal segment of Gγ transducin (gngt1) were found to be responsible for the ability of this subunit to disrupt PGC migration. The type of prenylation did not affect the ability of a Gγ subunit to reverse prenylation-deficient-Gγ-induced PGC migration defects. However, a version of gng2, engineered to be farnesylated instead of geranylgeranylated, still lacks the ability to reverse PGC migration defects known to result from treatment of zebrafish with geranylgeranyl transferase inhibitors (GGTI), supporting the notion that Gγ subunits are one of several protein targets that need to be geranylgeranylated to orchestrate the proper long-range migration of PGCs.
Keywords: Heterotrimeric G protein, Betagamma, GPCR, Primordial germ cells, Migration
1. Introduction
Heterotrimeric G proteins are pivotal for the transduction of signals emanating from the hundreds of known G protein coupled receptors (GPCR). These signaling proteins control pathways as diverse as neurotransmission, odorant reception, migration and apoptosis [1]. Heterotrimeric G proteins are composed of a beta (β), gamma (γ) and a GDP-bound alpha (α) subunit. Ligand binding to a GPCR results in a conformational switch in the heterotrimeric G protein, which causes the release of GDP from the Gα subunit in exchange for GTP, and the activation of both the Gα subunit and the Gβγ dimer. Once activated, the Gα subunit and Gβγ dimer can each activate their own effectors. Heterotrimeric G proteins are believed to process the input from a multitude of receptors with specificity due in part to the large number of isoforms that exist for each of these subunits. There are 16 α, 5 β and 12 γ subunit gene loci that have been identified in humans [2]. The zebrafish has at least 26 α, 9 β and 17 γ subunits [3,4].
The diversity of the signaling capabilities of heterotrimeric G proteins has increased through vertebrate evolution in the temporal and spatial restrictions of the expression profiles of subunit isoforms and their ability to selectively form heterotrimers. Gβ and Gγ isoforms exhibit selectivity in their obligate heterodimerization [5–11]. The binding efficiencies of βγ dimers with certain Gα subunits also differ [12–14]. Furthermore, the individual isoforms that make up a heterotrimer influence a heterotrimer's affinity and activation by certain receptors [15–19]. Similar to Gα, Gβγ dimers composed of different subunit isoforms exhibit heterogeneity in their efficiency in binding and activating effectors [7,20,21].
Gγ consists of two alpha helices separated by a kink (alpha helix #1: residues 10–25; kink: 26–29; alpha helix #2: 30–46; residue numbers correspond to gng2 [22,23]). While many residues along Gγ make hydrophobic contact with the Gβ subunit, growing evidence suggests that specific regions of the Gγ subunit are required for the efficient interaction of Gγ with Gβ, Gβγ with GPCRs, and Gβγ with effectors. Among Gγ isoforms, the N-terminus is the most variable region, but is largely conserved when compared across species, which has led to suggestions that it contains functional relevance [24]. Recently, the N-terminal region of Gγ was found to influence the efficiency with which the Gβγ dimer activates type II adenylyl cyclase [14]. The central region of the Gγ subunit has been shown to influence the ability of Gγ isoforms to selectively bind Gβ subunits to form heterodimers [25–27]. Regions in the carboxy terminus of Gγ influence the ability of a Gβγ dimer to bind receptors [14,28–31] and to translocate to intracellular membranes upon activation [32,33].
The carboxy-terminus of all Gγ subunits contains a CaaX motif which directs them to be post-translationally modified with the covalent addition of either a 15-carbon farnesyl or a 20-carbon geranylgeranyl lipid through a process called prenylation. The affinity of the prenyl transferases that perform this reaction is largely determined by the amino acids represented in the CaaX motif (where C = cysteine, a = an aliphatic amino acid, and X = a variable amino acid). This reaction results in the added hydrophobicity necessary for these proteins to interact with membranes and other hydrophobic proteins. While the formation of the Gβγ dimer occurs prior to prenylation [34], Gγ subunits must be prenylated in order for the Gβγ dimer to interact efficiently with the Gα subunit [5,31,34,35] and receptors [30,36] and to activate effectors [37–40]. The type of prenylation, farnesylation versus geranylgeranylation, affects the efficiency of the interaction of Gβγ dimers with Gα, membranes and receptors and the ability of Gβγ dimers to activate effectors [20,30,31,38,41].
When introduced to a cellular environment, prenylation-deficient forms of Gγ (where the cysteine of the CaaX motif has been mutated to a serine) result in Gβγ dimers that are cytosolically localized instead of being membrane bound [42,43]. These prenylation-deficient Gγ subunits have the ability to disrupt GPCR signaling, presumably by sequestering Gβ, and perhaps other proteins, into inactive cytosolically-localized complexes [44–46]. Zebrafish injected with mRNA transcripts encoding the majority of these prenylation-deficient Gγ subunits exhibit PGCs that lack directional migration [3].
PGCs are believed to migrate directionally as a result of random membrane protrusions that are stabilized in the direction of increasing amounts of the chemokine stromal cell-derived factor 1 alpha (sdf1a; recently renamed cxcl12a). This chemokine is recognized by the GPCR cxcr4b, which is expressed in a number of cells in the developing zebrafish including migrating PGCs [47,48]. The exact signaling molecules that respond to the extracellular chemokine, however, remain unknown. Signal transduction is known to result in an increase in intracellular calcium in the protrusions of migrating PGCs [49].
When zebrafish embryos are injected with gng2-SaaX(nos) mRNA, which encodes a PGC-driven, prenylation-deficient version of the Gγ2 subunit, PGCs fail to accumulate calcium in protrusions and thus lose the ability to migrate directionally. Subsequent injection of mRNA encoding gng2, gng3, gng4, gng7, gng8, gng12a, gng12b or gng13 has the ability to overcome the prenylation-deficient gng-SaaX-induced defects and restore proper PGC migration [3]. While overexpression of the majority of Gγ subunits has no detrimental effects on PGC migration, orthologs of the transducin Gγ subunits (gngt1 and gngt2a) disrupt PGC migration when injected into embryos as mRNA in their wild type form. In the present study, we constructed a number of Gγ subunit chimeras to investigate which sections of the Gγ subunit are responsible for the observed differences in their capacity to participate in the signaling necessary for directional PGC migration. Analysis of these chimeras reveals that the central domain and multiple motifs in the C-terminal domain of Gγ subunits influence the functional diversity of heterotrimeric G protein signaling in vivo.
2. Methods
2.1. Construction of chimeric and domain-swapped Gγ subunits
gngt1, gng2 and gng15 were each PCR amplified in three segments with the primers outlined in Table S1. Primers were designed to amplify the regions flanking amino acids that are highly conserved throughout the Gγ subunit family which correspond to the residues present at position 21–23 (QLK) and position 48–50 (DPL) of gngt1. The gngt1(T3TS), gng2(T3TS), and gng15(T3TS) constructs previously described [3] were used as templates for this PCR. Amplified segments were cloned into the nos1-3′UTR (nos) vector according to the scheme shown in Fig. 1. The nos1-3′UTR vector directs mRNA in-vitro transcribed from it to be stabilized in PGCs and degraded in somatic cells [50]. Chimeras were named according to the makeup of their three segments as outlined in Fig. 1 (Gγ2-2-2 = Gγ2-WT; Gγ15-15–15 = Gγ15-WT; Gγt1-t1-t1 =Gγt1-WT; Gγ2-15–2 = a Gγ2 subunit chimera with the central region of Gγ15).
Fig. 1.

Strategy for constructing Gγ subunit chimeras. The gng2 and gng15 (A) or gng2 and gngt1 (B) subunits were aligned with Clustal to show their similarities and differences at the amino acid level. Gγ subunits were split at the conserved sites boxed in red (QLK which corresponds to residues 18–20 in gng2 and DPL, which corresponds to residues 48050 of gng2). The residues that make up the CaaX motif (blue box), and the N-terminal, middle and C-terminal sections have been indicated. The three sections of each gamma subunit were individually PCR amplified and recombined to form the chimeras outlined in (C). Chimeras were named according to the makeup of their three sections as indicated to the left of each subunit Swapping the ‘translocation to endomembranes’ motif (*) involved mutating the five amino acids indicated in an alignment of their C-terminal sequences (D). Subunits harboring these point mutations were named according to whether their new motif should (trans+) or should not (trans-) be consistent with the ability to translocate upon receptor activation. Subunits with swapped CaaX motifs have been named with the amino acids that make up their new motif (E) (gng2-WT terminates with -CAIL; after swap = gng2-CVIC). The prenyl lipid that will be post-translationally added to these Gγ chimeras is indicated to the right.(For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To create the Gγt1trans−, Gγt2atrans− and Gγ2trans + mutants in which the “translocates to endomembranes motif” was swapped between gngt1 and gng2, or gngt2a and gng2, PCR was performed on the gngt1(nos), gngt2a(nos) and gng2(nos) constructs using two sets of mutagenic primers as outlined in Table S1. Swapping the CaaX motifs of gng2 and gngt1 so that they are alternatively farnesylated (gng2-CVIC) or geranylgeranylated (gngt1-CAIL) was accomplished by PCR mutagenesis of the FLAG-gng2(nos) construct and PCR amplification of gngt1 using the mutagenic primers outlined in Table S1. All constructs were confirmed by DNA sequencing prior to in vitro transcription.
2.2. In vitro transcription and injection of mRNA
5′ capped, 3′ polyadenylated mRNAs were in vitro transcribed from the nos1-3′UTR vector using the SP6 polymerase mMessage kit (Ambion: AM1340) as previously described [3]. Each mRNA was synthesized on at least two separate occasions to confirm reproducibility. 1–2 cell embryos were injected using a N2-based micro injector (Harvard Apparatus: PLI-100). Injection needles (Narishige: #NGD1) were pulled from capillaries with filament using a micropipette puller (Flaming/Brown Model P97). Injection needles were calibrated by injecting sample into 1 μl capillary tubes (Drummond: #10000010), counting the number of pulses necessary to fill the capillary to the 100 nl line and adjusting the pulse time so that a single pulse resulted in ejection of the desired volume (0.5–3 nl).
2.3. Immunoprecipitation of FLAG-tagged Gγ subunits
1–2 cell embryos injected with in vitro transcribed mRNA (100– 300 pg/embryo) were isolated and immunoprecipitated as previously described [3]. FLAG-tagged proteins were immunoprecipitated from 24 hours post fertilization (hpf) larvae lysate by incubation with anti-FLAG m2 affinity gel (Sigma-Aldrich: A2220) at 4 °C overnight. Proteins were separated on a gradient 10–20% SDS-PAGE gel, transferred to a PVDF membrane and subjected to Western blotting using rabbit anti-FLAG Antibody (F7425, Sigma) and ECL-HRP-linked donkey anti-rabbit secondary (Amersham: NA934).
2.4. PGC migration assays
PGC migration was assayed at 24 hpf in larvae injected at the 1–2 cell stage with a mixture of the in vitro transcribed mRNA and GFPnos1-3′ UTR mRNA (150 pg/embryo). Embryos were scored as described previously [3]. Briefly, at 24 hpf, ectopic PGCs were counted and assigned an ectopic PGC migration score based on the percentage of PGCs that were found outside the wildtype location (the anterior of the yolk extension; 0=0–5% ectopic; 1=6–20% ectopic; 2=21–40% ectopic; 3=41–60% ectopic; 4=61–80% ectopic; 5=81–100% ectopic). Only embryos with a minimum of 25 fluorescent PGCs were scored. The ability of native, prenylated Gγ subunits to disrupt PGC migration was assayed by co-injecting 100, 200 or 300 pg of the indicated Gγ-WT(nos) subunit mRNA with 150 pg GFP-nos1 -3′UTR and scoring larvae at 24 hpf. The ability of Gγ subunits and chimeras to reverse the gng2-SaaX(nos)-induced PGC migration defects was assayed by injecting a batch of embryos with a mixture of 50 pg gng2-SaaX(nos) and 150 pgGFP-nos1-3′UTR. The batch was then split in half, with half of the embryos receiving a phenol red control injection and the other half receiving 100, 200 or 300 pg of the indicated Gγ mRNA. All injected embryos were incubated at 28.5 °C and scored at 24 hpf.
2.5. Geranylgeranyl transferase inhibition
2.5.1. Soaking approach
1–2 cell embryos were injected with a mixture of 150 pg GFP-CVLL (nos) mRNA, which encodes a geranylgeranylated version of mGFP that is predominantly plasma membrane bound (as previously described [3]) and 200 pg of either gng2-WT(nos) or gng2-CVIC(nos) mRNA. GGTI-2166 was resuspended in ethanol at a stock concentration of 10 mM. Embryos were immediately soaked for 24 h in a 12-7well plate in 2 mL of embryo medium supplemented with either a final concentration of 20 μM GGTI-2166 (ethanol added to a final concentration of 2%) or 2% ethanol (control). PGC migration was assayed at the end of the incubation period.
2.5.2. Injection approach
GGTI-2166 was resuspended in DMSO at a stock concentration of 10 mM.1–2cell embryoswere injected with2 nLofamixtureof2.5 mM GGTI-2166 (25% DMSO) and 150 pg GFP-CVLL(nos) and subsequently injected with either phenol red (control), gng2-WT(nos) or gng2-CVIC (nos) mRNA. PGC migration was assayed at 24 hpf. DMSO and ethanol caused no significant disruption of PGC migration on their own when injected or soaked at the indicated concentrations (data not shown).
2.6. Zebrafish strains and fish maintenance
Danio rerio of the laboratory AB strain were outcrossed once to petstore zebrafish to reintroduce hybrid vigor, inbred several generations and designated WT fish. WT fish were raised and maintained as previously described [51,52]. Zebrafish care and experimental procedures were carried out as specified in the Institutional Animal Care and Use Committee Protocol (#647A).
3. Results
3.1. Construction of Gγ subunit chimeras
To determine which section(s) of the Gγ subunit are mediating their differences in signaling capacity, we generated chimeras using a Gγ subunit that belongs to each of three representative groups (can reverse Gγ-SaaX-induced defects: gng2; fails to reverse Gγ-SaaX-induced defects: gng15; disrupts PGC migration when injected in the wildtype form: gngt1). gngt1, gng2 and gng15, which are 73, 71, and 70 amino acids-long respectively, were split approximately into thirds at sites where the residues are conserved in the Gγ subunit family (divided at the amino acids corresponding to position 21–23 (QLK) and 51–53 (DPL) of Gγt1 as shown in Fig. 1). Each possible combination of the N-terminal, middle and C-terminal sections of gngt1 and gng2 or gng15 and gng2 were generated, resulting in 12 chimeric constructs. While both Gγ2 and Gγ15 subunits are post-translationally modified with a geranylgeranyl moiety, they differ in primary sequence in that they are only 46% identical (33 of 71 amino acids; Fig. 1A). Gγt1 shares only 36% identity to Gγ2 (27 of 73 amino acids; Fig. 1B).
3.2. Residues in the central domain of gng2 are necessary for proper PGC migration
Only Gγ chimeras that have the ability to 1) form a dimer with the appropriate Gβ subunit, 2) form a heterotrimer with the relevant Gα subunit, 3) interact as a heterotrimer with the necessary receptors and 4) become activated to allow transduction of the signal by an activated Gα subunit or Gβγ dimer through the desired effectors, should be able to reverse the phenotype induced by gng2-SaaX and result in wildtype PGC migration. To assess the ability of chimeras to transduce the signals necessary for PGC migration, chimeras were assayed for their ability to reverse PGC migration defects induced by gng2-SaaX(nos) mRNA injection. 1–2 cell zebrafish embryos were injected with a mixture of in vitro transcribed gng2-SaaX(nos) mRNA and GFP-CVLL(nos) mRNA followed by an injection of either phenol red or one of the chimeric Gγ subunit mRNAs. All of the gng2/gng15 chimeras that contained the central region of Gγ2 (Gγ2-2-2 (Gγ2-WT), Gγ15-2-2, Gγ2-2-15, and Gγ15-2–15) were able to reverse the SaaX-induced defects, suggesting that this region is responsible for mediating the interactions necessary for proper PGC migration (Fig. 2A). Consistent with this finding, the Gγ2-15–2 chimera, which has only the central region of the Gγ subunits replaced, failed to reverse the gng-SaaX-induced defect. None of the Gγ2/15 chimeras induced migration defects when injected alone (Fig. 2B).
Fig. 2.

Ability of the Gγ2/Gγ15 subunit chimeras to reverse gng2-SaaX-induced PGC migration defects. Embryos were injected with 50 pg of gng2-SaaX(nos) mRNA (scored when injected alone on the leftmost in red) and received a subsequent injection with 100, 200, or 300 pg of the gng15/gng2 chimeric Gγ mRNA (A). The ectopic PGC score of larvae injected with 100–300 pg/embryo of the chimeric Gγ-WT(nos) mRNA alone is shown in (B). Each concentration is summarized from 2 to 5 experiments with a minimum of 20 larvae scored for the number of ectopic PGCs at 24 hpf. (Ectopic PGC migration score is expressed as mean +/− S.D.) An ectopic score of 0 represents wild type migration; 1=5–20% ectopic; 2=21–40% ectopic; 3=41–60% ectopic; 4=61– 80% ectopic; 5=81–100% ectopic PGCs. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3. The C-terminus of gngt1 is responsible for its ability to disrupt PGC migration
To determine which section of gngt1 is responsible for its ability to disrupt PGC migration, we assayed the abilityof gngt1/gng2 chimeras to perturb PGC migration when injected as mRNA in their native –CaaX motif form. Injection of mRNA in vitro transcribed from the Gγ2-2–2 (Gγ2-WT), Gγt1-t1-2, Gγt1-2–2, or Gγ2-t1-2 chimeras failed to disrupt PGC migration (Fig. 3A). All of the gngt1/gng2 chimeras that contain the C-terminal segment of gngt1 (Gγt1-t1-t1 (Gγt1-WT), Gγ2-t1-t1, Gγt1-2-t1, or Gγ2-2-t1) disrupted PGC migration when injected in their native -CaaX form suggesting that the C-terminal segment is key to mediating this disruption. The inability of the Gγt1-t1-2 chimera, which contains the N-terminal two thirds of gngt1 and only the C-terminal region of gng2, to disrupt PGC migration when injected, corroborates the C-terminal region as being responsible for disrupting migration.
Fig. 3.

Ability of the Gγ2/Gγt1 subunit chimeras to reverse the gng2-SaaX-induced PGC migration defects. Embryos were injected with 50 pg of gng2-SaaX(nos) mRNA (scored when injected alone on the leftmost in red) and received a subsequent injection with 100, 200, or 300 pg of the gngt1/gng2 chimeric Gγ mRNA (A). The ectopic PGC score of larvae injected with 100–300 pg/embryo of the chimeric Gγ-WT(nos) mRNA alone is shown in (B). Each concentration is summarized from 2 to 5 experiments with a minimum of 20 larvae scored for the number of ectopic PGCs at 24 hpf. (Ectopic PGC migration score is expressed as mean +/− S.D.) An ectopic score of 0 represents wildtype migration and a score of 5=81–100% ectopic PGCs. The amino acids swapped in the Gγ translocation motif chimeras (*) are listed in Figs. 4–1. Wildtype Gγ subunits and chimeras with swapped CaaX motifs have been indicated with whether they are to be post translationally modified with the 15-carbon farnesyl or 20-carbon geranylgeranyl lipid. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To determine whether the Gγt1 subunit would be able to mediate PGC migration if it were not masked by the disruptive effects of its C-terminal region, gngt1/gng2 chimeras were assayed for their ability to reverse the gng2-SaaX(nos) phenotype. Consistent with the findings from the gng2/gng15 chimeras, only the Gγt1-2–2 chimera, which has the central region of gng2 and lacks the C-terminal region of gngt1, was able to reverse the gng2-SaaX-induced migration defects (Fig. 3B). When the C-terminal segment of gngt1 was replaced with the C-terminus of gng2, the gngt1 subunit behaved similarly to the gng15 subunit as evidenced by the inability of either the Gγ2-t1-2 or Gγt1-t1-2 chimera to reverse the gng2--SaaX-induced migration defects. This suggests that in addition to disrupting PGC migration, Gγt1 is incapable of forming the interactions necessary to properly transduce the sdf1a/cxcr4b signal, presumably through either mislocalization of signaling partners or the interaction with inappropriate signaling partners.
3.4. The ability of Gγt1 to disrupt PGC migration is due in part to a translocation motif found in its C-terminal segment
Alignment of the C-terminal segments of representative Gγ subunits that disrupt (gngt1 and gngt2a) with those that do not disrupt (gng2) PGC migration when injected in their native pre-nylatable form highlights several residues that differ (Fig. 1B; gngt2a has only 2 conservative amino acid substitutions outside of the CaaX motif when compared to gngt1: V53I and D56E). One particular motif present in the C-terminal segment that differs between these subclasses of Gγ subunits is the translocation motif outlined by [32]. The residues in this motif were previously found to influence whether a Gγ subunit remains at the plasma membrane or translocates to the ER or golgi membranes upon GPCR activation in transfected M2-CHO cells. The amino acid makeup of the translocation motif present in zebrafish Gγt1 and Gγt2a subunits suggests that they would have the ability to translocate upon receptor activation (amino acids corresponding to position 56–57 = ‘EK’ and 64–65 = ‘GG’ of gngt1; ‘E/DKxxxxx-KGG’). With the exception of gng13, all Gγ subunits capable of reversing gng2-SaaX-induced defects (Gγ2, 3,4, 7, 8,12a, and 12b) have a motif suggesting that they do not translocate, but rather, remain at the plasma membrane in the presence of agonists (amino acids corresponding to position 57–58 = SE and 64–67 = KKFF of gng2: ‘SExxxxxKKFF’). To determine if these residues could confer either the gain or loss of a Gγ subunit's ability to disrupt PGC migration when expressed in the native prenylatable state, chimeras were made with the translocation motifs swapped between gngt1 and gng2 (Fig. 1D). The Gγt1trans− chimera mRNA, which has the translocation motif of gng2 (consistent with a subunit remaining at the plasma membrane) had a significantly attenuated ability to disrupt PGC migration (Fig. 3A). This decrease in disruption was not due to a lack or decrease of protein being made from the chimeric mRNA in vivo as determined by immunoprecipitation of a FLAG-tagged version of these proteins from 24 hpf larvae lysate (data not shown). Similar to Gγt1-WT, Gγt1trans− mRNA was incapable of reversing the gng2-SaaX-induced defects, which supports the hypothesis that the central section of Gγ2 is necessary for the ability to reverse Gγ-SaaX-induced PGC migration defects (Fig. 3B). Injection of the Gγ2trans + chimera, that contains the residues supporting translocation to endomembranes (DKxxxxx-KGG), resulted in a mild increase in ectopic PGCs. Gγ2trans+chimera mRNA showed no significant difference in its ability to reverse the gng2-SaaX-induced PGC migration phenotype (Fig. 3B). These results suggest that while the presence of the motif supporting translocation to intracellular membranes does influence the ability of Gγt1 to disrupt PGC migration, it is not sufficient to account for the extent of disruption shown in larvae overexpressing Gγt1-WT.
3.5. Farnesylation of Gγt1 is necessary for the ability of Gγt1 to disrupt PGC migration
To determine if the makeup of the carboxy-terminal CaaX motif (which primarily determines whether a protein will be post translationally modified with a farnesyl or geranylgeranyl lipid moiety) also accounts for the differencesin the ability of Gγt1 and Gγ2 todisrupt PGC migration, chimeras were made by swapping the CaaX motif of Gγt1 (CVIC), a farnesylated subunit, with the CaaX motif of Gγ2 (CAIL), a geranylgeranylated subunit (Fig. 1E). When injected alone, the geranylgeranylated version of Gγt1-CAIL exhibited a significant decrease in its ability to perturb PGC migration, suggesting that farnesylation of gngt1 influences the interactions mediating disruption. Despite the altered prenylation status of the proteins they encode, Gγt1-CAIL and Gγ2-CVIC mRNAs behaved identically to their wildtype counterparts in their inability or ability to reverse gng2-SaaX-induced defects, respectively. Farnesylation of Gγt1 is therefore also necessary but not sufficient to account for the migration defects induced by Gγt1 overexpression.
3.6. pGGT1 acts through multiple pathways to mediate PGC migration
Injection of zebrafish embryos with geranylgeranyl transferase inhibitors (GGTI) disrupts PGC migration. If unprenylated Gγ subunits are responsible for the PGC migration defects in the presence of GGTIs, we expect that injection of a farnesylated version of a Gγ subunit that normally mediates PGC migration would reverse the phenotype induced by a GGTI. To test this hypothesis we injected the farnesylated form of Gγ2, Gγ2-CVIC, in the presence of GGTI-2166 (an inhibitor previously shown to result in PGC migration defects [53]). Injecting embryos with Gγ2-CVIC mRNA was insufficient to reverse the PGC migration defects induced by GGTI-2166, suggesting that the geranylgeranylation of additional protein targets needs to occur for PGCs to migrate properly (Fig. 4).
Fig. 4.

Ability of Gγ subunit chimeras to reverse the PGC migration defects caused by GGTI-2166. (GGTI-2166 injection) Embryos were injected with 2 nL of a mixture of 2.5 mM GGTI-2166 and 150 pg GFP-CVLL(nos). Injected embryos were split into three groups and received a 1 nL injection with either 1.) phenol red (injection control), 2.) 200 pg gng2-WT(nos) mRNA or 3.) 200 pg gng2-CVIC(nos) mRNA. PGC migration was assayed at 24 hpf. (GGTI-2166 soaking) Embryos were injected with 150 pg GFP-CVLL (nos) mRNA either alone or in a mixture with 200 pg gng2-WT(nos) mRNA or 200 pg gng2-CVIC(nos) mRNA. Injected embryos were soaked for 24 h in 2% ethanol (control) or 20 μM GGTI-2166 (final ethanol concentration=2%). PGC migration was assayed at the end of the incubation period. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
In vitro studies have shown that heterotrimeric G protein γ subunits have both overlapping and distinct affinities for signaling partners and that the primary sequence and lipid modification of Gγ influence the interactions of Gβγ with Gα subunits, receptors and effectors. How these differences translate into the capacity of a Gγ subunit to transduce signals in vivo remains unclear. We had previously shown that Gγ subunit isoforms differ in their ability to transduce the signals necessary for PGC migration [3]. To ascertain the regions of Gγ subunit domains responsible for these differences, we constructed chimeras and assayed their capacity to restore proper signaling to a disrupted, GPCR-mediated signaling pathway. The results of this study indicate that several Gγ subunit domains influence the functionality of an in vivo signaling cascade. Analysis of Gγ chimeras suggests that both the central section and the C-terminal section of the Gγ subunit influence the ability of a heterotrimeric G protein to transduce the cellular signals necessary for PGC migration.
4.1. Residues in the C-terminal segment of Gγt1 determine its ability to disrupt PGC migration
In this study, we define a region in the C-terminus of Gγ that is sufficient to confer upon a Gγ subunit the ability to disrupt PGC migration. The C-terminal region encompasses two motifs that are both necessary but not sufficient for Gγt1 to disrupt PGC migration. These motifs include the translocation motif, implicated in influencing the ability of Gγ to translocate to intracellular endomembranes, and the CaaX motif which directs the type of prenylation. When the residues that make up the C-terminal region of gng2 and gngt1 are compared, only 9 of the 24 amino acids exhibit non-conservative changes (Fig. 5A). Of these nine, the lysine and glycine at positions 55 and 56 and glutamic acid at position 59 of gngt1 are the only residues that differ in this region that were not swapped in this study. The glycine at position 56 is found in many of the Gγ subunits that do not disrupt PGC migration and in Gγ subunits that have the ability to reverse gng-SaaX-induced defects, suggesting that this residue is not crucial for the ability to disrupt. The lysine at position 55 and glutamic acid at position 59, however, are conserved among the zebrafish, mouse and human transducin orthologs and not found in the other Gγ subunits (Fig. S1). These positions may also represent residues key to conferring a Gγ subunit with the ability to disrupt PGC migration.
Fig. 5.
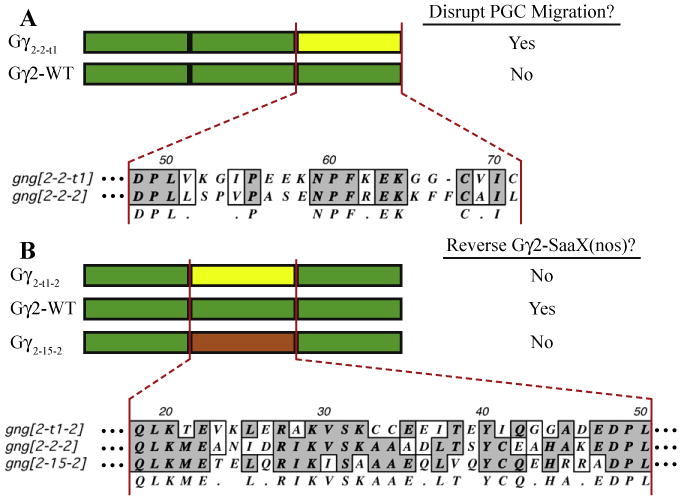
Alignment of the Gγ subunit regions responsible for differences in signaling capabilities. The C-terminal region of gngt1, which was found to confer upon the gamma subunit the ability to disrupt PGC migration when injected in its native-CaaX form, is aligned to the C-terminus of gng2 (A). The central region of the gamma subunit confers a difference in ability to reverse gng2-SaaX-induced defects. Clustal alignment of this region highlights the sequence differences among the gngt1, gng2 and gng15 subunits (B). Shown to the right of each of the cartoon schematics of the Gγ subunit chimeras is their ability to disrupt PGC migration (A) or reverse the gng2-SaaX-induced defects (B).
One explanation for the ability of chimeras containing the C-terminus of the transducin orthologs, Gγt1 and Gγt2a, to disrupt PGC migration is that residues in the C-terminal region could influence interactions of Gγt or the Gβγt dimer with other signaling molecules. Several studies have shown that mammalian Gγ chimeras (split at the ‘QLK’ and the ‘DPL’ residues) form functional proteins in vitro that heterodimerize with Gβ, form heterotrimers with Gα, are activated by receptors, and can activate downstream effectors [14,26,31]. It is therefore likely that the Gγ chimeras constructed in this study are capable of these basic functions in vivo. The C-terminal region of gngt1 could determine the Gγ subunit's ability to activate an unintended pathway that disrupts directional cell migration.
The C-terminal region could alternatively influence the ability of Gγ to sequester endogenously acting signaling molecules into complexes incapable of transducing the signal from Cxcr4b due to either altered subcellular localization or incompatible signaling components, thereby acting as a competitive inhibitor. In vitro studies have shown that residues in the C-terminus of Gγ, including but not limited to those that dictate prenylation type, can affect interactions of Gβγ dimers with Ga subunits and effectors, and interactions of Gαβγ with receptors [14,29,31,33,54-56]. The findings that swapping either the translocation motif or the CaaX motif of gngt1 with those of gng2 results in the loss of the ability to disrupt PGC migration (gngt1trans− and gngt1-CAIL do not disrupt), and that neither of these motifs when swapped alone confer upon gng2 the gain-of-function of disrupting PGC migration or the loss-of-function of no longer being able to reverse gng2-SaaX-induced defects (gng2trans + and gng2-CVIC maintain the ability to reverse gng2-SaaX), suggest that both these motifs, perhaps in conjunction with K55 and E59 (Gγ2-2-t1 does disrupt), confer gngt1 with the selectivity needed to maintain this inappropriate signaling interaction or localization. Further analysis of these C-terminal residues could lead to a better understanding of endogenous functions of this region of Gγt1 that may have been overlooked using traditional assays.
4.2. The complexes formed by prenylation-deficient Gγ subunits in vivo remain unknown
Prenylation-deficient Gγ subunits are believed to disrupt GPCR-signaling through the sequestration of endogenous Gβ subunits to the cytosol where they can no longer interact with Gα or the receptors necessary for signal transduction. Whether prenylation-deficient Gγ-SaaX subunits form larger nonfunctional complexes in vivo remains unknown due to controversy over the degree of interaction prenylation-deficient Gγ subunits have with their signaling partners. Unprenylated Gγ subunits are deficient in their ability to bind Gtα, Giα1 and Goα as assayed by either the ability of the Gγ subunits to support the pertussis toxin-induced ADP ribosylation of Gα in reconstituted membranes or transfected cells [5,31,34,38]. Gβγ dimers with unprenylated Gγ are also deficient in binding GDP-bound, Gα agarose columns [5]. Unprenylated Gγ fails to interact with the receptors rhodopsin and metarhodopsin II, as measured by receptor-catalyzed GTPγS binding to Ga [31,38]. Unprenylated Gγ fails to stimulate phospholipase C β2 [39] or to inhibit adenylyl cyclase type I or II properly [5,38], presumably due to a lack of interaction with these effectors in the absence of the prenyl moiety.
The preponderance of experiments showing that Gγ must be prenylated in order for Gβγ to bind Gα, interact with receptors and activate effectors efficiently makes it unlikely that Gγ-SaaX forms large nonfunctional complexes. The possibility that nonfunctional heterotrimers form in vivo cannot be ruled out, however, since Gγt1 and Gγ2 subunits that lack prenyl modification still form heterotrimers in vitro as evidenced by crystallographic study of the heterotrimers Giα1β1γ2 and Giα1/Giα-β1γt1 [22,57]. Studies in transfected cells have also shown that it is possible to immunoprecipitate Gαs with Gγ2-SaaX [58]. Recent studies analyzing Gβγ interactions in cell lines using BRET (bioluminescence resonance energy transfer) and BiFC (bimolecular fluorescence complementation) have suggested that the initial interactions of Gβγ dimers with receptors and effectors occur in the ER prior to plasma membrane localization and prior to Gβγ interacting with Gα [59,60]. The ability of wild type Gγ mRNA to reverse gng2-SaaX-induced defects may, therefore, rely on the ability of the Gγ subunit to not only outcompete gng2-SaaX for the appropriate Gβ subunit but also for the proper Gα, receptor and effectors.
4.3. Residues in the central region of the Gγ2 subunit are necessary for the ability to reverse gng2-SaaX-induced PGC migration defects
Residues that are present in the central region of gng2, corresponding to amino acids 21–47, were found to be necessary and sufficient to confer upon gng15 and necessary but not sufficient to confer upon gngt1 the ability to reverse gng2-SaaX-induced defects. The Gγ15-2–15 chimera that contains only the central region of Gγ2 was able to reverse gng2-SaaX-induced defects. The C-terminus of gngt1, present in the Gγt1-2-t1 chimera, also had to be swapped in order for gngt1 to reverse gng2-SaaX-induced migration defects, presumably due to the disruptive influence of the C-terminal region (Gγt1-2–2 can reverse). When the sequences of the central region of gng2 are aligned to the Gγ subunits that fail to reverse the gng2-SaaX-induced defects (gng15 and gngt1), 18 of the 27 amino acid positions harbor non-conservative changes in one of the subunits (Fig. 5B). Gng2 is identical to gngt1 or gng15 at 9 of these 18 positions and similar at 5 positions. The remaining 4 positions have non-conservative changes in both of the non-reversing subunits and make likely candidates for further analysis ofthe residues responsible for determining the ability of a Gγ subunit to mediate the signaling necessary for PGC migration.
The ability of a Gγ isoform to reverse gng2-SaaX-induced defects could be determined by its ability to form a heterodimer with the appropriate Gβ. The central region of Gγ, which is primarily known to mediate its interaction with the Gβ subunit and was found here to determine the ability of a Gγ subunit to reverse gng2-SaaX, imparts Gγ2 but not Gγt1 with the specificity to bind Gβ2. The region that confers selectivity in Gβγ dimer formation was originally defined to be contained in the 14 amino acids between the relatively conserved ‘VSK’ and ‘EDPL’ sites of Gγ2 (which corresponds to positions 33–46 of Gγ2) [25]. The selectivity to bind Gβ2 was later shown to be conferred by the residues that correspond to positions 35–37 [26,61] or positions 33–35 of Gγ2 (AAA) [27]. When compared to positions 33–35 of gng2, the gng15 subunit has 2 nonconservative changes, and all 5 residues exhibit nonconservative changes in gngt1 suggesting that the affinity of these Gγ isoforms for Gβ isoforms may not entirely overlap (Fig. 5B). It remains to be determined whether the Gγ isoforms that reverse gng2-SaaX bind distinct Gβ isoforms and whether this selective binding primarily determines their ability to reverse gng2-SaaX-induced defects. Sections of Gγ that overlap the central region also have been reported to influence their efficiency of effector activation. Residues 35–71 of Gγ2 are required for Gβ1γ2 to fully activate the GIRK4 channel [56].
The fact that no single residue is conserved among all the Gγ subunits that can reverse gng2-SaaX-induced defects or not conserved in subunits that fail to reverse suggests that this ability is multifactorial. If the ability of a Gγ subunit to reverse prenylation-deficient Gγ subunits is determined by binding to the proper Gβ subunit, the contribution of multiple residues would be consistent given the number of residues in Gγ that are purported to make contact with the Gβ subunit [23]. It is also plausible that there are separate functions that are influenced by the residues in the central region of Gγ. Swapping the central region ofthe Gγ subunits could result in a functional difference in binding the endogenously acting relevant Gβ subunits, or in the affinity ofthe Gβγ dimer for other signaling partners such as Gα, the Cxcr4b receptor, or necessary effectors.
4.4. Multiple protein targets must be prenylated for PGCs to properly migrate
The inability of farnesylated gng2-CVIC subunits to reverse the effects of geranylgeranyl transferase I inhibition suggests that proteins other than the heterotrimeric G protein -y subunits need to be geranylgeranylated for proper PGC migration. In Drosophila, a cell-autonomous role for geranylgeranylated G protein γ subunits has been inferred from the function of the PGC-expressed GPCR, trapped in endoderm-1 (Tre1), in the transepithelial migration of PGCs [62]. However, ectopic expression of HMGCR is sufficient to attract PGCs in flies, suggesting that geranylgeranylated proteins are also needed in somatic cells for the attraction of PGCs [63]. The ability of globally applied mevalonate to rescue HMGCR inhibition in the zebrafish [53] indicates that other geranylgeranylated proteins in addition to Gγ could be acting in somatic cells, PGCs, or both. A recent report of potential cell autonomous contributions of the geranylgeranylated small GTPases Rho and Rac [64] and potential cell-non-autonomous contributions of unidentified geranylgeranylated proteins involved in adhesion or attraction are important issues to resolve in future research.
5. Conclusion
The findings of this study provide evidence that multiple domains present in Gγ subunits influence the functionality of heterotrimeric G protein signaling in vivo. Regions in the C-terminus of Gγ, including but not limited to those dictating the prenylation type and the ability of a Gγ subunit to translocate upon receptor activation determine the ability of transducin Gγ subunits to disrupt PGC migration. The central region of the Gγ2 subunit was found to be necessary for the ability of a Gγ subunit to reverse prenylation-deficient Gγ-induced PGC migration defects. Further dissection of these subunits and the influences they have on the localization and interactions of the Gβγ heterodimer will be key to understanding signaling complexes pivotal to cellular behaviors as diverse as migration and apoptosis.
Supplementary Material
Acknowledgments
The authors would like to thank Amy Kowalski for technical assistance and Jennifer Anderson for editorial assistance. We would also like to thank Marnie Halpern, Haiqing Zhao, Alex Bortvin, Mark Van Doren and Narasimhan Gautam for critical reading and advice.
Footnotes
Supplementary materials related to this article can be found online at doi: 10.1016/j.cellsig.2011.05.015.
References
- 1.Wettschureck N, Offermanns S. Physiol Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 2.Hurowitz EH, Melnyk JM, Chen YJ, Kouros-Mehr H, Simon MI, Shizuya H. DNA Res. 2000;7:111–120. doi: 10.1093/dnares/7.2.111. [DOI] [PubMed] [Google Scholar]
- 3.Mulligan T, Blaser H, Raz E, Farber SA. Cell Signal. 2010;22:221–233. doi: 10.1016/j.cellsig.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oka Y, Saraiva LR, Kwan YY, Korsching SI. Proc Natl Acad Sci U S A. 2009;106:1484–1489. doi: 10.1073/pnas.0809420106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iniguez-Lluhi JA, Simon MI, Robishaw JD, Gilman AG. J Biol Chem. 1992;267:23409–23417. [PubMed] [Google Scholar]
- 6.Pronin AN, Gautam N. Proc Natl Acad Sci U S A. 1992;89:6220–6224. doi: 10.1073/pnas.89.13.6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dingus J, Wells CA, Campbell L, Cleator JH, Robinson K, Hildebrandt JD. Biochemistry. 2005;44:11882–11890. doi: 10.1021/bi0504254. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt CJ, Thomas TC, Levine MA, Neer EJ. J Biol Chem. 1992;267:13807–13810. [PubMed] [Google Scholar]
- 9.Mervine SM, Yost EA, Sabo JL, Hynes TR, Berlot CH. Mol Pharmacol. 2006;70:194–205. doi: 10.1124/mol.106.022616. [DOI] [PubMed] [Google Scholar]
- 10.Yost EA, Mervine SM, Sabo JL, Hynes TR, Berlot CH. Mol Pharmacol. 2007;72:812–825. doi: 10.1124/mol.107.038075. [DOI] [PubMed] [Google Scholar]
- 11.Ray K, Kunsch C, Bonner LM, Robishaw JD. J Biol Chem. 1995;270:21765–21771. doi: 10.1074/jbc.270.37.21765. [DOI] [PubMed] [Google Scholar]
- 12.Ohguro H, Fukada Y, Yoshizawa T, Saito T, Akino T. Biochem Biophys Res Commun. 1990;167:1235–1241. doi: 10.1016/0006-291x(90)90656-8. [DOI] [PubMed] [Google Scholar]
- 13.Casey PJ, Graziano MP, Gilman AG. Biochemistry. 1989;28:611–616. doi: 10.1021/bi00428a029. [DOI] [PubMed] [Google Scholar]
- 14.Myung CS, Lim WK, DeFilippo JM, Yasuda H, Neubig RR, Garrison JC. Mol Pharmacol. 2006;69:877–887. doi: 10.1124/mol.105.018994. [DOI] [PubMed] [Google Scholar]
- 15.Jian X, Sainz E, Clark WA, Jensen RT, Battey JF, Northup JK. J Biol Chem. 1999;274:11573–11581. doi: 10.1074/jbc.274.17.11573. [DOI] [PubMed] [Google Scholar]
- 16.Richardson M, Robishaw JD. J Biol Chem. 1999;274:13525–13533. doi: 10.1074/jbc.274.19.13525. [DOI] [PubMed] [Google Scholar]
- 17.McIntire WE, MacCleery G, Murphree LJ, Kerchner KR, Linden J, Garrison JC. Biochemistry. 2006;45:11616–11631. doi: 10.1021/bi0604882. [DOI] [PubMed] [Google Scholar]
- 18.Kleuss C, Scherubl H, Hescheler J, Schultz G, Wittig B. Science. 1993;259:832–834. doi: 10.1126/science.8094261. [DOI] [PubMed] [Google Scholar]
- 19.Hou Y, Azpiazu I, Smrcka A, Gautam N. J Biol Chem. 2000;275:38961–38964. doi: 10.1074/jbc.C000604200. [DOI] [PubMed] [Google Scholar]
- 20.Myung CS, Yasuda H, Liu WW, Harden TK, Garrison JC. J Biol Chem. 1999;274:16595–16603. doi: 10.1074/jbc.274.23.16595. [DOI] [PubMed] [Google Scholar]
- 21.Bommakanti RK, Vinayak S, Simonds WF. J Biol Chem. 2000;275:38870–38876. doi: 10.1074/jbc.M007403200. [DOI] [PubMed] [Google Scholar]
- 22.Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, Sprang SR. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 23.Sondek J, Bohm A, Lambright DG, Hamm HE, Sigler PB. Nature. 1996;379:369–374. doi: 10.1038/379369a0. [DOI] [PubMed] [Google Scholar]
- 24.Cook LA, Schey KL, Cleator JH, Wilcox MD, Dingus J, Hildebrandt JD. Protein Sci. 2001;10:2548–2555. doi: 10.1110/ps.ps.26401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spring DJ, Neer EJ. J Biol Chem. 1994;269:22882–22886. [PubMed] [Google Scholar]
- 26.Lee C, Murakami T, Simonds WF. J Biol Chem. 1995;270:8779–8784. doi: 10.1074/jbc.270.15.8779. [DOI] [PubMed] [Google Scholar]
- 27.Meister M, Dietrich A, Gierschik P. Eur J Biochem. 1995;234:171–177. doi: 10.1111/j.1432-1033.1995.171_c.x. [DOI] [PubMed] [Google Scholar]
- 28.Azpiazu I, Gautam N. J Biol Chem. 2001;276:41742–41747. doi: 10.1074/jbc.M104034200. [DOI] [PubMed] [Google Scholar]
- 29.Azpiazu I, Cruzblanca H, Li P, Linder M, Zhuo M, Gautam N. J Biol Chem. 1999;274:35305–35308. doi: 10.1074/jbc.274.50.35305. [DOI] [PubMed] [Google Scholar]
- 30.Yasuda H, Lindorfer MA, Woodfork KA, Fletcher JE, Garrison JC. J Biol Chem. 1996;271:18588–18595. doi: 10.1074/jbc.271.31.18588. [DOI] [PubMed] [Google Scholar]
- 31.Jian X, Clark WA, Kowalak J, Markey SP, Simonds WF, Northup JK. J Biol Chem. 2001;276:48518–48525. doi: 10.1074/jbc.M107129200. [DOI] [PubMed] [Google Scholar]
- 32.Saini DK, Kalyanaraman V, Chisari M, Gautam N. J Biol Chem. 2007;282:24099–24108. doi: 10.1074/jbc.M701191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akgoz M, Kalyanaraman V, Gautam N. Cell Signal. 2006;18:1758–1768. doi: 10.1016/j.cellsig.2006.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Higgins JB, Casey PJ. J Biol Chem. 1994;269:9067–9073. [PubMed] [Google Scholar]
- 35.Dietrich A, Scheer A, Illenberger D, Kloog Y, Henis YI, Gierschik P. Biochem J. 2003;376:449–456. doi: 10.1042/BJ20030578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohguro H, Fukada Y, Takao T, Shimonishi Y, Yoshizawa T, Akino T. EMBO J. 1991;10:3669–3674. doi: 10.1002/j.1460-2075.1991.tb04934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dietrich A, Brazil D, Jensen ON, Meister M, Schrader M, Moomaw JF, Mann M, Illenberger D, Gierschik P. Biochemistry. 1996;35:15174–15182. doi: 10.1021/bi960305j. [DOI] [PubMed] [Google Scholar]
- 38.Matsuda T, Hashimoto Y, Ueda H, Asano T, Matsuura Y, Doi T, Takao T, Shimonishi Y, Fukada Y. Biochemistry. 1998;37:9843–9850. doi: 10.1021/bi973194c. [DOI] [PubMed] [Google Scholar]
- 39.Dietrich A, Meister M, Brazil D, Camps M, Gierschik P. Eur J Biochem. 1994;219:171–178. doi: 10.1111/j.1432-1033.1994.tb19927.x. [DOI] [PubMed] [Google Scholar]
- 40.Katz A, Wu D, Simon MI. Nature. 1992;360:686–689. doi: 10.1038/360686a0. [DOI] [PubMed] [Google Scholar]
- 41.Kisselev O, Ermolaeva M, Gautam N. J Biol Chem. 1995;270:25356–25358. doi: 10.1074/jbc.270.43.25356. [DOI] [PubMed] [Google Scholar]
- 42.Simonds WF, Butrynski JE, Gautam N, Unson CG, Spiegel AM. J Biol Chem. 1991;266:5363–5366. [PubMed] [Google Scholar]
- 43.Muntz KH, Sternweis PC, Gilman AG, Mumby SM. Mol Biol Cell. 1992;3:49–61. doi: 10.1091/mbc.3.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schillo S, Belusic G, Hartmann K, Franz C, Kuhl B, Brenner-Weiss G, Paulsen R, Huber A. J Biol Chem. 2004;279:36309–36316. doi: 10.1074/jbc.M404611200. [DOI] [PubMed] [Google Scholar]
- 45.Chakravorty D, Botella JR. Gene. 2007;393:163–170. doi: 10.1016/j.gene.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 46.Grishin AV, Weiner JL, Blumer KJ. Mol Cell Biol. 1994;14:4571–4578. doi: 10.1128/mcb.14.7.4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knaut H, Werz C, Geisler R, Nusslein-Volhard C. Nature. 2003;421:279–282. doi: 10.1038/nature01338. [DOI] [PubMed] [Google Scholar]
- 48.Doitsidou M, Reichman-Fried M, Stebler J, Koprunner M, Dorries J, Meyer D, Esguerra CV, Leung T, Raz E. Cell. 2002;111:647–659. doi: 10.1016/s0092-8674(02)01135-2. [DOI] [PubMed] [Google Scholar]
- 49.Blaser H, Reichman-Fried M, Castanon I, Dumstrei K, Marlow FL, Kawakami K, Solnica-Krezel L, Heisenberg CP, Raz E. Dev Cell. 2006;11:613–627. doi: 10.1016/j.devcel.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 50.Koprunner M, Thisse C, Thisse B, Raz E. Genes Dev. 2001;15:2877–2885. doi: 10.1101/gad.212401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Dev Dyn. 1995;203:253–310. doi: 10.1002/aja.1002030302. [DOI] [PubMed] [Google Scholar]
- 52.Westerfield M. A guide for the laboratory use of zebrafish (Danio rerio) 4th. University of Oregon Press; Eugene: 2000. The zebrafish book. [Google Scholar]
- 53.Thorpe JL, Doitsidou M, Ho SY, Raz E, Farber SA. Dev Cell. 2004;6:295–302. doi: 10.1016/s1534-5807(04)00032-2. [DOI] [PubMed] [Google Scholar]
- 54.Fogg VC, Azpiazu I, Linder ME, Smrcka A, Scarlata S, Gautam N. J Biol Chem. 2001;276:41797–41802. doi: 10.1074/jbc.M107661200. [DOI] [PubMed] [Google Scholar]
- 55.Akgoz M, Azpiazu I, Kalyanaraman V, Gautam N. J Biol Chem. 2002;277:19573–19578. doi: 10.1074/jbc.M201546200. [DOI] [PubMed] [Google Scholar]
- 56.Peng L, Mirshahi T, Zhang H, Hirsch JP, Logothetis DE. J Biol Chem. 2003;278:50203–50211. doi: 10.1074/jbc.M308299200. [DOI] [PubMed] [Google Scholar]
- 57.Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 58.Takida S, Wedegaertner PB. J Biol Chem. 2003;278:17284–17290. doi: 10.1074/jbc.M213239200. [DOI] [PubMed] [Google Scholar]
- 59.Dupre DJ, Baragli A, Rebois RV, Ethier N, Hebert TE. Cell Signal. 2007;19:481–489. doi: 10.1016/j.cellsig.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 60.Dupre DJ, Robitaille M, Ethier N, Villeneuve LR, Mamarbachi AM, Hebert TE. J Biol Chem. 2006;281:34561–34573. doi: 10.1074/jbc.M605012200. [DOI] [PubMed] [Google Scholar]
- 61.Adler EM, Gough NR. Sci STKE eg3 (2007) 2007 doi: 10.1126/stke.3832007eg3. [DOI] [PubMed] [Google Scholar]
- 62.Kunwar PS, Sano H, Renault AD, Barbosa V, Fuse N, Lehmann R. J Cell Biol. 2008;183:157–168. doi: 10.1083/jcb.200807049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Doren M, Broihier HT, Moore LA, Lehmann R. Nature. 1998;396:466–469. doi: 10.1038/24871. [DOI] [PubMed] [Google Scholar]
- 64.Kardash E, Reichman-Fried M, Maitre JL, Boldajipour B, Papusheva E, Messerschmidt EM, Heisenberg CP, Raz E. Nat Cell Biol. 2010;12(Suppl):47–53. 41–11. doi: 10.1038/ncb2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.