

Abstract
Cardiac AT2R expression is upregulated in the normal process of aging. In this study we determined the contribution of AT2R to chronic antihypertensive and remodelling effects of AT1R blockade in aged hypertensive rats. Adult (20 weeks) and senescent (20 months) spontaneously hypertensive rats (SHRs) were treated with either the AT1R antagonist, candesartan cilexetil (2 mg/kg/day), the AT2R antagonist, PD123319 (10 mg/kg/day), or a combination of the 2 compounds. Mean arterial pressure (MAP) and left ventricular volume were markedly decreased by candesartan cilexetil, however, simultaneous treatment with PD123319 had no additional effect on either parameter. Perivascular fibrosis was significantly reduced by candesartan cilexetil in aged animals only, and this effect was reversed by concomitant PD123319 administration. Vascular hypertrophy was reduced by candesartan cilexetil, and these effects were reversed by simultaneous PD123319. These results suggest that AT2R stimulation does not significantly influence the antihypertensive effect of chronic AT1R blockade, but plays a role in the regulation of vascular structure. The severe degree of cardiac perivascular fibrosis in senescent animals was regressed by AT1R blockade and this effect was reversed by simultaneous AT2R inhibition, demonstrating an antifibrotic role of AT2R stimulation in the aging hypertensive heart.
1. Introduction
The incidence of hypertension, cardiac hypertrophy, and heart failure increases significantly with aging [1], and age-related structural adaptations may contribute to deteriorating function of the cardiovascular system. The aging heart is characterised by myocyte loss, hypertrophy of remaining cells, and exaggerated accumulation of extracellular (ECM) proteins [1, 2], which is associated with increased incidence of both contractile and conductile dysfunction of senescent hearts [2]. In addition, structural modifications of the aorta and coronary vasculature, particularly involving hypertrophy/hyperplasia of smooth muscle cells and increased collagen deposition within and surrounding the media of vessels [3], result in arterial stiffening, alterations in vascular permeability, and deterioration of coronary haemodynamics [4].
Ang II is known to promote cardiovascular hypertrophy and fibrosis via AT1R stimulation [5, 6], whereas the role of AT2R has been less conclusively defined [7]. AT2R activation is thought to oppose AT1R-mediated hypertrophic and fibrotic effects; however, studies in transgenic mouse models of targeted deletion [8, 9] or overexpression [10] of AT2R have reported contrasting effects on cardiovascular structure, emphasising the need for further pharmacological investigation and elucidation of AT2R function.
AT1R antagonists increase circulating levels of Ang II, which may stimulate unopposed AT2R and potentially contribute to the effects of AT1R blockade [11]. We have previously shown that impaired in vitro AT2R-mediated relaxation in SHRs was restored by antihypertensive treatment [12]. Furthermore, AT2R stimulation may influence cardiovascular function and structure during chronic AT1R blockade [13–15]. These studies have been performed in animal models of genetic hypertension or following cardiovascular infarct and have deduced various degrees of AT2R-mediated antihypertrophic and antifibrotic effects, depending on the study.
Importantly, although cardiac AT2R expression is relatively low in the adult rat heart [16], expression may be upregulated in certain disease states and has been particularly associated with conditions of increased fibrosis [17], cardiac hypertrophy [18], heart failure [19], and also with increasing age [20, 21]. Moreover, increased myocardial angiotensinogen and ACE indicate that intracardiac production of Ang II may also be potentiated with senescence [22].
Given the possibility of augmented cardiac RAS activity with increased age, and also the fact that chronic AT1R blockade increased longevity in rodent models of aging and was associated with cardiovascular protective effects [23, 24], we reasoned that a greater AT2R contribution to AT1R inhibition may be manifest in the aged hypertensive state. Therefore, the aims of this study were to determine the contribution of the AT2R to the antihypertensive and cardiovascular remodelling effects of chronic AT1R blockade in aged SHRs.
2. Materials and Methods
2.1. Animals and Treatment
Male SHRs (12 weeks) were obtained from the Animal Resource Centre, Western Australia and were maintained on a 12-hour day/night cycle with free access to food and water until animals were either 20 weeks or 20 months of age. Senescent animals were used at 20 months, as at this age, SHRs display many of the features of hypertensive and age-related cardiac remodelling (including cardiovascular hypertrophy and fibrosis) but are yet to complete the transition to heart failure [25].
Radiotelemetry transmitters (TA11PA-C40, Data Sciences) were inserted into the abdominal aorta of SHRs under isoflurane anaesthesia (2–4%, O2), as previously described [26]. Animals were allowed to recover for 1 week, after which time a continuous baseline recording of MAP and HR was made for a further week. Animals were then given the AT1R antagonist, candesartan cilexetil (2 mg/kg/day), its vehicle, or the nonangiotensin antihypertensive, hydralazine (30 mg/day), in drinking water. At the same time, senescent SHRs were also briefly anaesthetised with isoflurane, and osmotic mini pumps containing either PD123319 (10 mg/kg/day) or saline vehicle were inserted into a subcutaneous pocket formed between the scapulae. Doses of candesartan cilexetil and PD123319 were based on previous studies performed in senescent Wistar Kyoto rats [26]. Adult SHRs were treated for 2 weeks with candesartan cilexetil (2 mg/kg/day), before implantation of osmotic mini pumps, such that animals received the combination of 6-week candesartan cilexetil and 4-week PD123319 treatment. In senescent SHRs, all drug treatments were initiated simultaneously and continued for 4 weeks duration. MAP and HR were recorded continuously during the entire 4- or 6-week treatment period. Treatment groups were as follows:
Adult (20 weeks) SHRs
control (n = 6),
candesartan cilexetil alone (n = 7),
candesartan cilexetil + PD123319 (n = 7),
PD123319 alone (n = 7).
Senescent (20 months) SHRs
2.2. Determination of Plasma Ang II Levels
At the end of the treatment period, a sample of blood was collected directly from the catheterised aorta of each animal into chilled, heparinised tubes, and then centrifuged at 4000 rpm at 4°C for 10 minutes to isolate plasma. The resultant plasma sample was stored at −80°C for later analysis. Ang II concentrations were analysed in duplicate by RIA as described previously [27]. Briefly, plasma (100 μL) was equilibrated with antibody raised in rabbit against Ang II, which was N-terminally conjugated to bovine thyroglobulin. Monoiodinated 125I- Ang II tracer (10 000 cpm in 100 μL) was added and allowed to equilibrate for 16 hrs at 4°C, whereupon bound and free phase was separated using Dextran 10-coated charcoal and centrifugation. Sensitivity was 3.5 pg/mL. Intra- and interassay variabilities were 6.4 and 12.0%. Cross reactivity to other angiotensins were Ang I = 0.52%, Ang (1–7) = 0.01%, and to all other pertinent hormones less than 0.10%.
2.3. Perfusion Fixation
After 4 or 6 weeks treatment, animals were anaesthetised (ketamine/xylazine; 100 mg/10 mg per kg), and the abdominal aorta briefly ligated to enable removal of the radiotelemetry probe. A catheter was inserted into the abdominal aorta, and a sample of blood was collected into a heparinised tube. Heparin sodium (1 IU/g body weight), papaverine hydrochloride (1.2 mg/rat), and potassium chloride (60 mM in 0.1 mL) were administered via the catheterized aorta to prevent blood from clotting, maximally dilate blood vessels, and arrest the heart in diastole, respectively. Organs were cleared of blood with physiological saline, and then perfusion fixed with 4% paraformaldehyde in 0.1 M phosphate buffer. Perfusion pressure was maintained at a pressure corresponding to the in vivo systolic pressure of adult and aged SHRs by use of a perfusion apparatus attached to a sphygmomanometer. Hearts and blood vessels were then excised and stored immersed in paraformaldehyde at 4°C for later processing.
2.4. Cardiac Remodelling
Both left and right atria were removed from fixed hearts, and the remaining left ventricle (LV), right ventricle (RV), and septum were weighed. Hearts were then cut into approximately twelve 1.5 mm thick slices using a razor blade slicing device. Each slice was then placed on a light table, images were captured using a video camera module (Sony, XC-77CE CCD, Japan) displayed on a monitor, and analysed using imaging computer software (Microscope Computed Imaging Device M4 (MCID), Imaging Research, Canada). Sampled cross-sectional areas of the LV, RV, and both LV and RV chambers were then multiplied by slice thickness to calculate the volume of each sampled area. Total volumes of LV, RV, LV chamber and RV chamber of each heart were determined by adding measurements taken from heart slices throughout the entire heart. Ventricular weight and volume measurements were normalized to body weight for each animal.
2.5. Interstitial and Perivascular Fibrosis in the Heart
After heart volumes had been determined, five 1.5 mm heart slices from each animal were embedded in paraffin, sectioned at 5 μm and stained for collagen with 0.001% Picrosirius Red. Each section was viewed under a light microscope (Olympus, BH-2, Japan) with a video camera module interfaced to a computer. Images were displayed onto a monitor and analysed using imaging computer software (MCID). All sections were examined under ×200 magnification.
The area of interstitial fibrosis in 6 fields of view of the LV per section and 2 fields of view of the RV per section were sampled, and the percentage of fibrosis within each sampled area was averaged for each animal. Collagen volume fraction (%) was calculated by determining the area stained for collagen as a percentage of the total area of sampled tissue, per field of view. Perivascular fibrosis was investigated in the LV only. Two intramyocardial arterioles (measuring 100–200 μm in diameter) were randomly selected per section. The cross-sectional area (CSA) of adventitia (representing perivascular fibrosis), media, and lumen were determined and averaged for each animal. Perivascular fibrosis was normalised to lumen area. As an index of microvascular remodelling, media-to-lumen ratio of intramyocardial vessels was determined from CSA measurements and averaged for each animal.
2.6. Aortic Hypertrophy
Segments of fixed vessel were dissected from the thoracic portion of the aorta of each animal, embedded in epon-araldite, cut at 1 μm, and stained with toluidine blue. Each section was viewed under a light microscope and analysed using imaging computer software (MCID). All sections were examined under ×100 magnification. CSA of the media was determined for each vessel and normalised to lumen CSA.
2.7. Statistics
The effect of drug treatments on MAP and HR over time was assessed by one- or two-way analysis of variance (ANOVA) with repeated measures, as appropriate. Differences in morphometric data between treatments were determined using one-way ANOVA, followed by a Bonferroni post hoc test. Results are expressed as mean ± standard error of the mean (SEM). Statistical significance was accepted as a probability of P < 0.05.
3. Results
3.1. Body Weight and Plasma Ang II
Body weight was not affected by any drug treatments (Table 1). Compared to age-matched untreated animals, plasma Ang II levels were increased more than 7- and 3-fold by candesartan cilexetil treatment, either alone or in combination with PD123319, in adult and senescent SHRs, respectively (Table 1). Both PD1231319 alone and hydralazine had no effect on plasma Ang II levels (Table 1).
Table 1.
Effect of drug treatments on body weight, ventricular weight, and plasma Ang II of adult and senescent SHRs.
| Control | Candesartan cilexetil | Candesartan cilexetil + PD123319 | PD123319 | Hydralazine | |
|---|---|---|---|---|---|
| Body weight (g) | |||||
| Adult SHRs | 411 ± 11 | 415 ± 5 | 410 ± 5 | 414 ± 3 | — |
| Senescent SHRs | 417 ± 9 | 427 ± 7 | 428 ± 10 | 399 ± 9 | 419 ± 11 |
| Ventricular weight (mg) | |||||
| Adult SHRs | 1423 ± 23 | 1229 ± 39* | 1207 ± 30* | 1474 ± 48 | — |
| Senescent SHRs | 2114 ± 74 | 1796 ± 85* | 1962 ± 86 | 2136 ± 163 | 1952 ± 49 |
| Plasma Ang II (pg/mL) | |||||
| Adult SHRs | 220 ± 100 | 1597 ± 234* | 2785 ± 817* | 68 ± 18 | — |
| Senescent SHRs | 80 ± 10 | 383 ± 89* | 271 ± 51* | 64 ± 23 | 91 ± 16 |
Values are mean ± SEM. *P < 0.05 versus age-matched control (1-way ANOVA).
3.2. Blood Pressure and Heart Rate in Senescent SHRs
Candesartan cilexetil caused a marked reduction in MAP compared to control animals, and administration of PD123319 did not reverse this antihypertensive effect, in either adult or senescent animals (Figures 1(a) and 1(c)). MAP was unaffected by PD123319 alone. In senescent SHRs, hydralazine caused a reduction in MAP that was similar in magnitude to that caused by AT1R blockade (Figure 1(c)). Candesartan cilexetil and hydralazine increased HR at the initiation of antihypertensive treatment in both age groups, which most likely represents a reflex tachycardia which persisted for 2-3 days until resetting of the baroreflex occurred. HR after this initial period was unaffected by treatments (Figures 1(b) and 1(d)).
Figure 1.
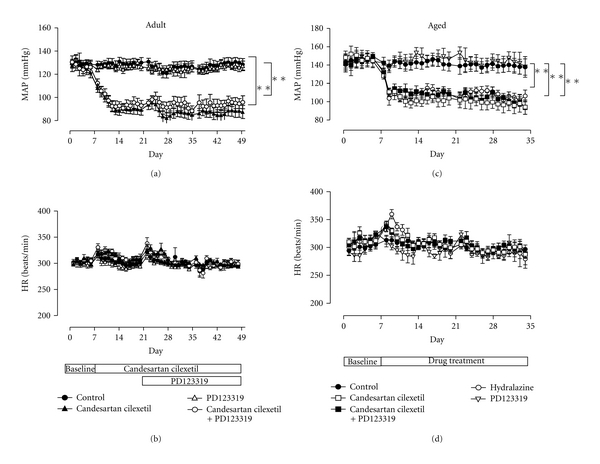
Radiotelemetry recordings of (a) MAP and (b) HR of adult SHRs, at baseline and during treatment with vehicle (control, n = 6), candesartan cilexetil (2 mg/kg/day) alone (n = 7) or in combination with PD123319 (10 mg/kg/day, n = 7), or PD123319 alone. Analogous radiotelemetry recordings of (c) MAP and (d) HR of senescent SHRs, at baseline and during treatment with vehicle (control, n = 10), candesartan cilexetil alone (n = 9) or in combination with PD123319 (n = 9), PD123319 alone (n = 4), or hydralazine (30 mg/kg/day, n = 7). **P < 0.01 versus control (2-way ANOVA).
3.3. Cardiac Remodelling
Ventricular weight (Table 1), ventricular weight to body weight ratio (Figure 2(a)) and LV volume to body weight ratio (Figure 2(b)) of adult SHRs were reduced by candesartan cilexetil; however, this antihypertrophic action was not further influenced by simultaneous AT2R inhibition. Similarly, ventricular weight (Table 1), ventricular weight to body weight ratio (Figure 2(c)), and LV volume to body weight ratio (Figure 2(d)) of senescent SHRs were also decreased by AT1R blockade. Furthermore, the regression of both ventricular weight and ventricular weight to body weight ratio were partially reversed by concurrent PD123319 treatment, such that these indices were not significantly different from control values. There were no effects of drug treatments on RV, LV chamber or RV chamber, volume to body weight ratios (data not shown).
Figure 2.
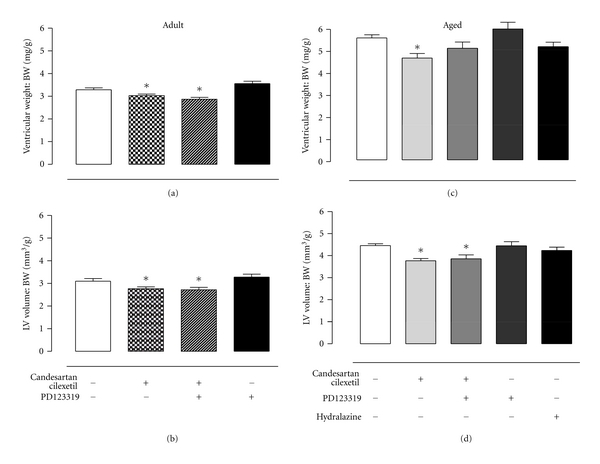
(a) Ventricular weight to body weight (BW) ratio and (b) left ventricular (LV) volume to body weight ratio of adult SHRs, at baseline and during treatment with vehicle (control, n = 4), candesartan cilexetil (2 mg/kg/day) alone (n = 4), or in combination with PD123319 (10 mg/kg/day, n = 4), and PD123319 alone (n = 7). (c) Ventricular weight to BW ratio and (d) LV volume to BW ratio of senescent SHRs, at baseline and during treatment with vehicle (control, n = 10), candesartan cilexetil (2 mg/kg/day) alone (n = 9), or in combination with PD123319 (10 mg/kg/day, n = 9), PD123319 alone (n = 4), or hydralazine (30 mg/kg/day, n = 7). *P < 0.05 versus control (1-way ANOVA).
3.4. Interstitial and Perivascular Fibrosis
Representative light micrographs of perivascular and interstitial fibrosis of senescent SHRs are shown in Figure 3. Group data shows that neither left (Figures 4(a) and 4(c)) nor right (Figures 4(b) and 4(d)) ventricular interstitial fibrosis of adult and senescent SHRs were altered by any drug treatments. Likewise, perivascular fibrosis of adult SHRs was not influenced by AT1 or AT2R inhibition (Figure 5(a)). In contrast, perivascular fibrosis was significantly decreased by ~28% in senescent SHRs receiving candesartan cilexetil, and this effect was completely reversed by simultaneous AT2R blockade (Figure 5(b)).
Figure 3.
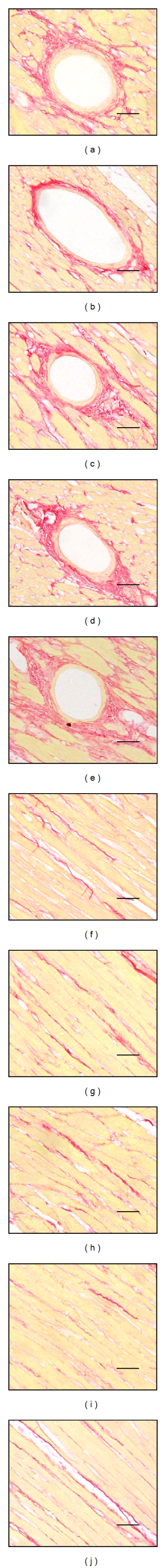
Representative light micrographs of cardiac (a–e) perivascular and (f–j) interstitial fibrosis in senescent SHRs treated with (a, f) vehicle (control), (b, g) candesartan cilexetil (2 mg/kg/day), (c, h) candesartan cilexetil in combination with PD123319 (10 mg/kg/day), (d, i) PD123319 alone, or (e, j) hydralazine (30 mg/kg/day). Scale bar = 50 μm.
Figure 4.
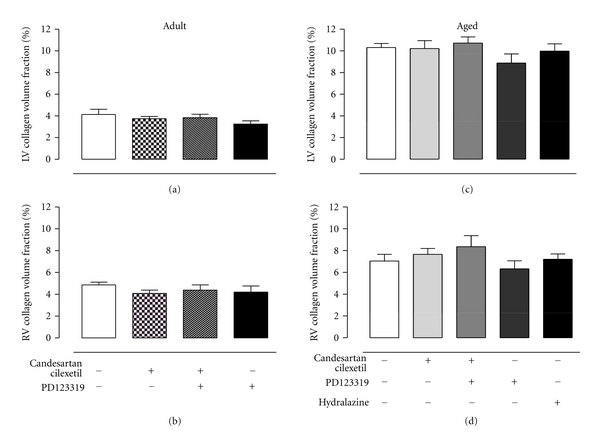
Mean data of interstitial collagen volume fraction of (a) left and (b) right ventricles of adult SHRs treated with vehicle (control, n = 6), candesartan cilexetil (2 mg/kg/day) alone (n = 7) or in combination with PD123319 (10 mg/kg/day, n = 7) or PD123319 alone. Interstitial collagen volume fraction of (c) left and (d) right ventricles of senescent SHRs treated with vehicle (control, n = 10), candesartan cilexetil alone (n = 9) or in combination with PD123319 (n = 9), PD123319 alone (n = 4), or hydralazine (30 mg/kg/day, n = 7).
Figure 5.
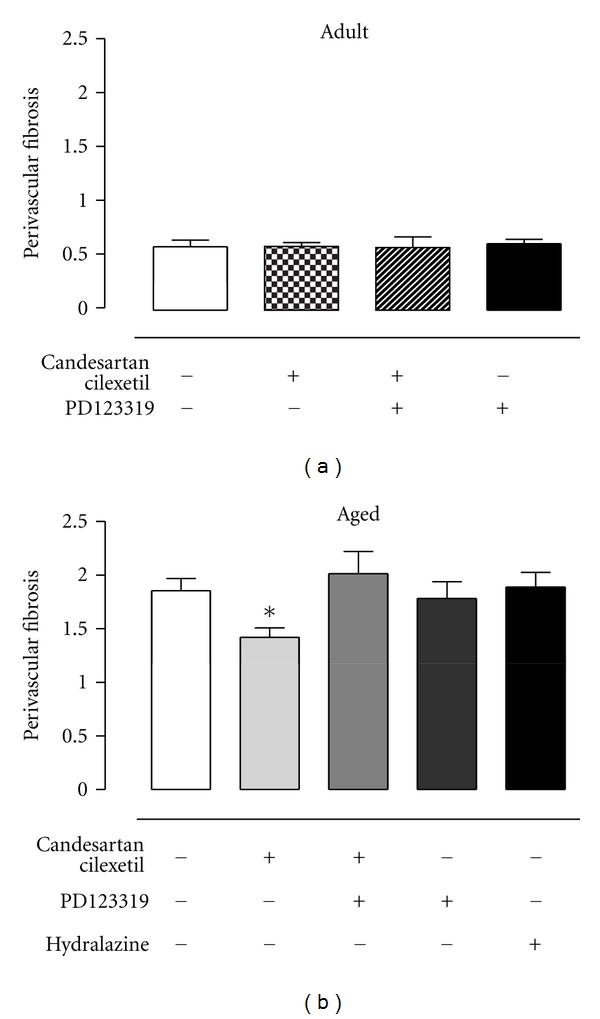
(a) Perivascular fibrosis of adult SHRs treated with vehicle (control, n = 6), candesartan cilexetil (2 mg/kg/day) alone (n = 7) or in combination with PD123319 (10 mg/kg/day, n = 7), or PD123319 alone (n = 7). (b) Perivascular fibrosis of senescent SHRs treated with vehicle (control, n = 10), candesartan cilexetil alone (n = 9) or in combination with PD123319 (n = 9), PD123319 alone (n = 4), or hydralazine (30 mg/kg/day, n = 7). Perivascular fibrosis of intramyocardial arterioles, calculated as cross-sectional area of adventitia to lumen ratio. *P < 0.05 versus control (1-way ANOVA).
3.5. Vascular Hypertrophy
Media to lumen ratios of aortic vessels in both adult and senescent SHRs (Figures 6(a) and 6(c)) and intramyocardial vessels of senescent SHRs (Figure 6(d)) were decreased by candesartan cilexetil, and this antihypertrophic effect of AT1R blockade was reversed by concomitant PD123319 administration. Hydralazine also caused a significant reduction in media-to-lumen ratios of aortic (Figure 6(b)) and intramyocardial (Figure 6(d)) vessels of senescent SHRs.
Figure 6.
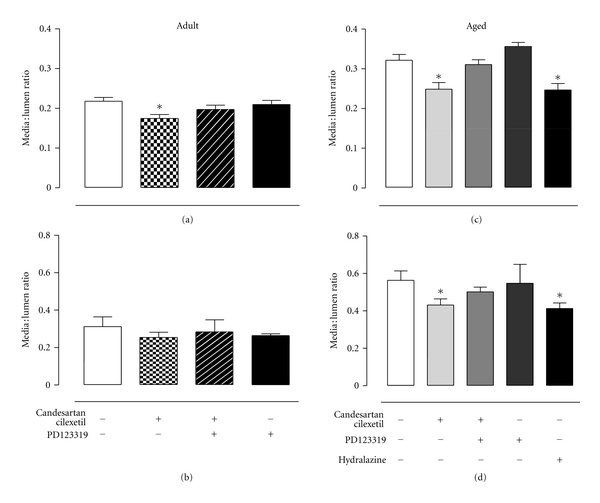
Media to lumen ratio of (a) aortic and (b) intramyocardial vessels of adult SHRs treated with vehicle (control, n = 6), candesartan cilexetil (2 mg/kg/day) alone (n = 7) or in combination with PD123319 (10 mg/kg/day, n = 7) or PD123319 alone (n = 7). Media to lumen ratio of (c) aortic and (d) intramyocardial vessels of senescent SHRs treated with vehicle (control, n = 10), candesartan cilexetil alone (n = 9) or in combination with PD123319 (n = 9), PD123319 alone (n = 4), or hydralazine (30 mg/kg/day, n = 7). *P < 0.05 versus control (1-way ANOVA).
4. Discussion
We have shown for the first time, a role for AT2R in cardiac and vascular remodelling in a clinically relevant animal model of aging and hypertension. Notably, AT2R stimulation by endogenously raised Ang II levels contributed to the cardiac antifibrotic and vascular antihypertrophic effects of chronic AT1R blockade. Thus, this study highlights the importance of AT2R in the chronic regulation of cardiovascular structure in the aging hypertensive heart and vasculature.
Candesartan cilexetil caused a marked reduction in MAP in both adult and senescent SHRs, which was not further affected by AT2R blockade. These results imply that stimulation of the AT2R does not significantly influence chronic blood pressure regulation and is consistent with other long-term studies that showed either no [13, 14, 28] or minimal [15] reversal of AT1R-blocker- (ARB-) mediated blood pressure-lowering by simultaneous AT2R blockade in SHRs. These findings are in direct contrast to the acute setting, in which the antihypertensive effect of ARB compounds was reversed by simultaneous AT2R blockade with PD123319 [29–31]. In addition, acute stimulation of AT2R has also been shown to lower blood pressure in rats, supporting a role for AT2R in acute blood pressure regulation [32–35].
Since both human and animal studies have shown circulating Ang II and renin levels to be reduced with increasing age [22, 36, 37], it is possible that the absence of AT2R-mediated actions on blood pressure in aged SHRs is due to depressed systemic RAS activity in senescence. However, in this study we have shown a similar inability of PD123319 to reverse the ARB-induced reduction in blood pressure in both adult and senescent rats. Moreover, even though baseline levels of Ang II are relatively low in aged SHRs compared to adult SHRs (~3-fold lower than adult SHRs, Table 1), AT1R inhibition caused an increase in plasma Ang II of 3-4-fold, suggesting that the RAS is still sensitive to perturbation in aged SHRs. Moreover, local tissue production of Ang II has been shown to be elevated in aged humans [6] and rodents [38]. Thus it is more likely that the inability of PD123319 administration to reverse ARB-induced antihypertensive effects in the current context reflects a subtle influence of AT2R stimulation on blood pressure regulation being masked by the dominant impact of AT1R blockade.
Candesartan cilexetil decreased indices of cardiac growth of adult rats (ventricular weight, and LV volume to body weight ratios), and PD123319 administration had no further influence on these parameters, suggesting no major role for AT2R in cardiac hypertrophy. Other studies in hypertensive models have also reported that PD123319 administration did not significantly reverse cardiac hypertrophy [15, 19, 28], and additionally, AT2R were deduced to have no major function in the regulation of cardiac mass from studies in transgenic mice models of targeted deletion or cardiac-specific over expression of AT2R [10, 39, 40]. In contrast, a dependence on AT2R for ARB-mediated cardiac remodelling following MI has been demonstrated in rats [13] and AT2R knock out mice [40]. These mismatches in reported AT2R influence on cardiac hypertrophy most likely reflect the gross measures of cardiac hypertrophy made in the majority of studies, as heart mass is commonly employed as a surrogate marker for cardiac hypertrophy (i.e., increased cardiomyocyte size) but is unable to distinguish between changes in proportion of specific components within the heart.
Indeed, ventricular weight and LV volume to body weight ratios were also reduced by AT1R blockade in senescent SHRs; however, in these aged animals, simultaneous AT2R inhibition caused a partial reversal of ventricular weight to body weight ratio. Given that LV volume is heavily influenced by changes in cardiomyocyte area [2], and that LV volume was not influenced by AT2R blockade, PD123319-mediated reversal of heart weight to body weight ratio most likely reflects changes in the nonmyocyte components of the heart, rather than a true effect on cardiac hypertrophy. Indeed, we have shown that perivascular fibrosis of coronary microvessels is decreased by AT1R blockade and that this effect is reversed by concomitant AT2R blockade, but only in senescent hearts. We have previously shown a similar mismatch between LV volume and ventricular weight following AT1R blockade in senescent normotensive WKY rats [26], which also coincided with a cardiac AT2R-mediated antifibrotic action. Thus the PD122319-mediated increase in ventricular weight to body weight ratio during AT1R blockade may in fact be due to inhibition of an AT2R-mediated antifibrotic action in senescent SHRs.
Surprisingly, candesartan cilexetil did not reduce interstitial fibrosis in either adult or senescent SHRs. In the present study, particularly high levels of LV and RV interstitial fibrosis (collagen volume fraction ~7–10%) were seen in control senescent animals. These relatively high levels of interstitial fibrosis in aged hearts are entirely consistent with previous studies [15, 20], and contrast with the degree of fibrosis in adult hypertensive SHRs (interstitial collagen volume fraction ~4-5%). We have previously shown that an identical treatment regime markedly reduced interstitial fibrosis from similar levels in aged normotensive WKY (interstitial collagen volume fraction ~4-5%), and this effect was also reversed by PD123319 [26]. Thus in the current study, it appears that the inability of candesartan cilexetil to reduce interstitial fibrosis in both adult and senescent SHRs, results from modifications of the ECM specifically related to hypertension, rather than particularly high pretreatment basal levels of fibrosis. Indeed, collagen cross-linking has been shown to be augmented by hypertension [41, 42], and increased cross-linking is associated with diminished susceptibility of the ECM to proteolytic degradation [43]. Alteration in ECM degradation due to increased glycation cross-linking has been associated with decreased activity of proteolytic enzymes such as matrix metalloproteinase 1 and 2 (MMP-1 and MMP-2) [44], and findings of decreased activity of MMP-1 and MMP-2 by 40–45% in aged, hypertensive rats [45] further support the notion of impaired collagen degradative mechanisms in senescent hypertensive hearts.
On the other hand, the antifibrotic action of AT2R stimulation on perivascular fibrosis in senescent rats, as demonstrated in the current investigation, is in accordance with other chronic in vivo studies, which have also shown increased cardiac fibrosis during AT2R blockade [15, 19, 26]. Similarly, investigators who have used either targeted deletion [8, 40, 46] or cardiac overexpression [10] of AT2R in mice have also deduced an antifibrotic role of the AT2R. Importantly, cardiac fibrosis induced by circulating humoral factors such as Ang II, typically initiates around blood vessels and then progresses to infiltrate interstitial areas, resulting in a temporal divergence in onset (and thus conceivably also of regression) of the two types of fibrosis related to location [47]. In this context, it is possible that interstitial fibrosis may have been reduced by a longer duration treatment with an AT1R antagonist, as has been reported by other investigators following AT1R blockade for 12 weeks [15].
In the current study, media-to-lumen ratio of both aortae and coronary vessels was decreased by candesartan cilexetil treatment and also by hydralazine in aged SHRs. This vascular antihypertrophic effect is consistent with previous reports that increased medial thickness due to hypertrophy/hyperplasia of smooth muscle cells is closely related to pressure [48, 49]. However, the other major modification of vascular structure that occurs in hypertension and senescence is an increase in vascular collagen content, the levels of which have been shown to be poorly associated with MAP, but sensitive to AT1R inhibition [48]. As the vascular antihypertrophic effect of candesartan cilexetil was reversed, but MAP was unchanged by PD123319, it is reasonable to suggest that the effect of AT2R inhibition on vascular remodelling was pressure-independent and thus may be via a reduction in vascular collagen. Furthermore, such a pressure-independent influence of AT2R on collagen accumulation in aged SHRs is consistent with effects on perivascular fibrosis in this study, which were decreased by AT1R blockade but unaffected by hydralazine, despite both treatments resulting in similar reductions in MAP.
A limitation of this study was that we did not confirm that the reversal of ARB-mediated antifibrotic effects by PD123319 is solely via AT2R mechanisms. Indeed, we [50] and others have shown that in certain situations, PD123319 may inhibit the effects of Ang 1–7, which is considered the endogenous ligand for the Mas receptor (MasR). However, we have also recently reported that Ang 1–7 shows significant AT2R binding [51], which is consistent with PD123319-mediated reversal of Ang 1–7 effects being due to inhibition of AT2R rather than a nonselective action at MasR. Nevertheless, definitive elucidation of this issue regarding selectivity of PD123319 requires future determination of MasR binding.
The present study demonstrates an important role for AT2R in cardiovascular remodelling in senescent SHRs, as evidenced by the fact that AT2R inhibition with PD123319 reversed ARB-mediated regression of perivascular fibrosis in aged SHRs only. Furthermore, we have shown an inhibitory influence of AT2R in vascular remodelling, which was apparent in both adult and senescent SHRs, and occurred despite a lack of AT2R-mediated effects on blood pressure. Given that our population is aging and that AT1R antagonists are commonly used antihypertensives in this demographic, this study provides information regarding the functional relevance of AT2R in the physiologically relevant setting of hypertension and senescence, which may have important implications for optimising cardiovascular therapeutics in the elderly.
Acknowledgments
These studies were supported in part by the National Health and Medical Research Council of Australia. The authors would like to thank D. Casley for performing the plasma Ang II assay for these experiments. At the time that these studies were performed, E. Jones was a recipient of a National Heart Foundation of Australia Postgraduate Research Scholarship.
References
- 1.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation. 2003;107(2):346–354. doi: 10.1161/01.cir.0000048893.62841.f7. [DOI] [PubMed] [Google Scholar]
- 2.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiological Reviews. 1999;79(1):215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 3.Gaballa MA, Jacob CT, Raya TE, Liu J, Simon B, Goldman S. Large artery remodeling during aging: biaxial passive and active stiffness. Hypertension. 1998;32(3):437–443. doi: 10.1161/01.hyp.32.3.437. [DOI] [PubMed] [Google Scholar]
- 4.Susic D, Nunez E, Hosoya K, Frohlich ED. Coronary hemodynamics in aging spontaneously hypertensive and normotensive Wistar-Kyoto rats. Journal of Hypertension. 1998;16(2):231–237. doi: 10.1097/00004872-199816020-00014. [DOI] [PubMed] [Google Scholar]
- 5.de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacological Reviews. 2000;52(3):415–472. [PubMed] [Google Scholar]
- 6.Wang M, Khazan B, Lakatta E. Central arterial aging and angiotensin II signaling. Current Hypertension Reviews. 2010;6(4):266–281. doi: 10.2174/157340210793611668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones ES, Vinh A, McCarthy CA, Gaspari TA, Widdop RE. AT2 receptors: functional relevance in cardiovascular disease. Pharmacology and Therapeutics. 2008;120(3):292–316. doi: 10.1016/j.pharmthera.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akishita M, Iwai M, Wu L, et al. Inhibitory effect of angiotensin II type 2 receptor on coronary arterial remodeling after aortic banding in mice. Circulation. 2000;102(14):1684–1689. doi: 10.1161/01.cir.102.14.1684. [DOI] [PubMed] [Google Scholar]
- 9.Ichihara S, Senbonmatsu T, Price E, Jr., Ichiki T, Gaffney FA, Inagami T. Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation. 2001;104(3):346–351. doi: 10.1161/01.cir.104.3.346. [DOI] [PubMed] [Google Scholar]
- 10.Kurisu S, Ozono R, Oshima T, et al. Cardiac angiotensin II type 2 receptor activates the Kinin/NO system and inhibits fibrosis. Hypertension. 2003;41(1):99–107. doi: 10.1161/01.hyp.0000050101.90932.14. [DOI] [PubMed] [Google Scholar]
- 11.Widdop RE, Jones ES, Hannan RE, Gaspari TA. Angiotensin AT2 receptors: cardiovascular hope or hype? British Journal of Pharmacology. 2003;140(5):809–824. doi: 10.1038/sj.bjp.0705448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.You D, Loufrani L, Baron C, Levy BI, Widdop RE, Henrion D. High blood pressure reduction reverses angiotensin II type 2 receptor-mediated vasoconstriction into vasodilation in spontaneously hypertensive rats. Circulation. 2005;111(8):1006–1011. doi: 10.1161/01.CIR.0000156503.62815.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu YH, Yang XP, Sharov VG, et al. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure: role of kinins and angiotensin II type 2 receptors. Journal of Clinical Investigation. 1997;99(8):1926–1935. doi: 10.1172/JCI119360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tea BS, Der Sarkissian S, Touyz RM, Hamet P, DeBlois D. Proapoptotic and growth-inhibitory role of angiotensin II type 2 receptor in vascular smooth muscle cells of spontaneously hypertensive rats in vivo. Hypertension. 2000;35(5):1069–1073. doi: 10.1161/01.hyp.35.5.1069. [DOI] [PubMed] [Google Scholar]
- 15.Varagic J, Susic D, Frohlich ED. Coronary hemodynamic and ventricular responses to angiotensin type 1 receptor inhibition in SHR: interaction with angiotensin type 2 receptors. Hypertension. 2001;37(6):1399–1403. doi: 10.1161/01.hyp.37.6.1399. [DOI] [PubMed] [Google Scholar]
- 16.Busche S, Gallinat S, Bohle RM, et al. Expression of angiotensin AT1 and AT2 receptors in adult rat cardiomyocytes after myocardial infarction: a single-cell reverse transcriptase-polymerase chain reaction study. American Journal of Pathology. 2000;157(2):605–611. doi: 10.1016/S0002-9440(10)64571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsutsumi Y, Matsubara H, Ohkubo N, et al. Angiotensin II type 2 receptor is upregulated in human heart with interstitial fibrosis, and cardiac fibroblasts are the major cell type for its expression. Circulation Research. 1998;83(10):1035–1046. doi: 10.1161/01.res.83.10.1035. [DOI] [PubMed] [Google Scholar]
- 18.Lopez JJ, Lorell BH, Ingelfinger JR, et al. Distribution and function of cardiac angiotensin AT1-and AT2-receptor subtypes in hypertrophied rat hearts. American Journal of Physiology. 1994;267(2, part 2):H844–H852. doi: 10.1152/ajpheart.1994.267.2.H844. [DOI] [PubMed] [Google Scholar]
- 19.Ohkubo N, Matsubara H, Nozawa Y, et al. Angiotensin type 2 receptors are reexpressed by cardiac fibroblasts from failing myopathic hamster hearts and inhibit cell growth and fibrillar collagen metabolism. Circulation. 1997;96(11):3954–3962. doi: 10.1161/01.cir.96.11.3954. [DOI] [PubMed] [Google Scholar]
- 20.Heymes C, Silvestre JS, Llorens-Cortes C, et al. Cardiac senescence is associated with enhanced expression of angiotensin II receptor subtypes. Endocrinology. 1998;139(5):2579–2587. doi: 10.1210/endo.139.5.6023. [DOI] [PubMed] [Google Scholar]
- 21.Widdop RE, Vinh A, Henrion D, Jones ES. Vascular angiotensin AT2 receptors in hypertension and ageing. Clinical and Experimental Pharmacology and Physiology. 2008;35(4):386–390. doi: 10.1111/j.1440-1681.2008.04883.x. [DOI] [PubMed] [Google Scholar]
- 22.Heymes C, Swynghedauw B, Chevalier B. Activation of angiotensinogen and angiotensin-converting enzyme gene expression in the left ventricle of senescent rats. Circulation. 1994;90(3):1328–1333. doi: 10.1161/01.cir.90.3.1328. [DOI] [PubMed] [Google Scholar]
- 23.Basso N, Cini R, Pietrelli A, Ferder L, Terragno NA, Inserra F. Protective effect of long-term angiotensin II inhibition. American Journal of Physiology. 2007;293(3):H1351–H1358. doi: 10.1152/ajpheart.00393.2007. [DOI] [PubMed] [Google Scholar]
- 24.Linz W, Heitsch H, Scholkens BA, Wiemer G. Long-term angiotensin II type 1 receptor blockade with fonsartan doubles lifespan of hypertensive rats. Hypertension. 2000;35(4):908–913. doi: 10.1161/01.hyp.35.4.908. [DOI] [PubMed] [Google Scholar]
- 25.Bing OH, Brooks WW, Robinson KG, et al. The spontaneously hypertensive rat as a model of the transition from compensated left ventricular hypertrophy to failure. Journal of Molecular and Cellular Cardiology. 1995;27(1):383–396. doi: 10.1016/s0022-2828(08)80035-1. [DOI] [PubMed] [Google Scholar]
- 26.Jones ES, Black MJ, Widdop RE. Angiotensin AT2 receptor contributes to cardiovascular remodelling of aged rats during chronic AT1 receptor blockade. Journal of Molecular and Cellular Cardiology. 2004;37(5):1023–1030. doi: 10.1016/j.yjmcc.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Johnston CI, Millar JA, Casley DJ, McGrath BP, Matthews PG. Hormonal responses to angiotensin blockade. Comparison between receptor antagonism and converting enzyme inhibition. Circulation Research. 1980;46(6):I128–I134. [PubMed] [Google Scholar]
- 28.Makino N, Sugano M, Otsuka S, Hata T. Molecular mechanism of angiotensin II type I and type II receptors in cardiac hypertrophy of spontaneously hypertensive rats. Hypertension. 1997;30(4):796–802. doi: 10.1161/01.hyp.30.4.796. [DOI] [PubMed] [Google Scholar]
- 29.Gigante B, Piras O, De Paolis P, Porcellini A, Natale A, Volpe M. Role of the angiotensin II AT2-subtype receptors in the blood pressure-lowering effect of losartan in salt-restricted rats. Journal of Hypertension. 1998;16(12, part 2):2039–2043. doi: 10.1097/00004872-199816121-00027. [DOI] [PubMed] [Google Scholar]
- 30.Siragy HM, de Gasparo M, Carey RM. Angiotensin type 2 receptor mediates valsartan-induced hypotension in conscious rats. Hypertension. 2000;35(5):1074–1077. doi: 10.1161/01.hyp.35.5.1074. [DOI] [PubMed] [Google Scholar]
- 31.Duke LM, Evans RG, Widdop RE. AT2 receptors contribute to acute blood pressure-lowering and vasodilator effects of AT1 receptor antagonism in conscious normotensive but not hypertensive rats. American Journal of Physiology. 2005;288(5):H2289–H2297. doi: 10.1152/ajpheart.01096.2004. [DOI] [PubMed] [Google Scholar]
- 32.Barber MN, Sampey DB, Widdop RE. AT2 receptor stimulation enhances antihypertensive effect of AT1 receptor antagonist in hypertensive rats. Hypertension. 1999;34(5):1112–1116. doi: 10.1161/01.hyp.34.5.1112. [DOI] [PubMed] [Google Scholar]
- 33.Carey RM, Howell NL, Jin XH, Siragy HM. Angiotensin type 2 receptor-mediated hypotension in angiotensin type-1 receptor-blocked rats. Hypertension. 2001;38(6):1272–1277. doi: 10.1161/hy1201.096576. [DOI] [PubMed] [Google Scholar]
- 34.Li XC, Widdop RE. AT2 receptor-mediated vasodilatation is unmasked by AT 1 receptor blockade in conscious SHR. British Journal of Pharmacology. 2004;142(5):821–830. doi: 10.1038/sj.bjp.0705838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bosnyak S, Welungoda IK, Hallberg A, Alterman M, Widdop RE, Jones ES. Stimulation of angiotensin AT2 receptors by the non-peptide agonist, Compound 21, evokes vasodepressor effects in conscious spontaneously hypertensive rats. British Journal of Pharmacology. 2010;159(3):709–716. doi: 10.1111/j.1476-5381.2009.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Michel JB, Heudes D, Michel O, et al. Effect of chronic ANG I-converting enzyme inhibition on aging processes. II. Large arteries. American Journal of Physiology. 1994;267(1, part 2):R124–R135. doi: 10.1152/ajpregu.1994.267.1.R124. [DOI] [PubMed] [Google Scholar]
- 37.Thompson MM, Oyama TT, Kelly FJ, Kennefick TM, Anderson S. Activity and responsiveness of the renin-angiotensin system in the aging rat. American Journal of Physiology. 2000;279(5):R1787–R1794. doi: 10.1152/ajpregu.2000.279.5.R1787. [DOI] [PubMed] [Google Scholar]
- 38.Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging and Disease. 2011;2(2):158–173. [PMC free article] [PubMed] [Google Scholar]
- 39.Sugino H, Ozono R, Kurisu S, et al. Apoptosis is not increased in myocardium overexpressing type 2 angiotensin II receptor in transgenic mice. Hypertension. 2001;37(6):1394–1398. doi: 10.1161/01.hyp.37.6.1394. [DOI] [PubMed] [Google Scholar]
- 40.Xu J, Carretero OA, Liu YH, et al. Role of AT2 receptors in the cardioprotective effect of AT1 antagonists in mice. Hypertension. 2002;40(3):244–250. doi: 10.1161/01.hyp.0000029095.23198.ad. [DOI] [PubMed] [Google Scholar]
- 41.Tsotetsi OJ, Woodiwiss AJ, Netjhardt M, Qubu D, Brooksbank R, Norton GR. Attenuation of cardiac failure, dilatation, damage, and detrimental interstitial remodeling without regression of hypertrophy in hypertensive rats. Hypertension. 2001;38(4):846–851. doi: 10.1161/hy1001.092649. [DOI] [PubMed] [Google Scholar]
- 42.Badenhorst D, Maseko M, Tsotetsi OJ, et al. Cross-linking influences the impact of quantitative changes in myocardial collagen on cardiac stiffness and remodelling in hypertension in rats. Cardiovascular Research. 2003;57(3):632–641. doi: 10.1016/s0008-6363(02)00733-2. [DOI] [PubMed] [Google Scholar]
- 43.DeGroot J. The AGE of the matrix: chemistry, consequence and cure. Current Opinion in Pharmacology. 2004;4(3):301–305. doi: 10.1016/j.coph.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 44.Kuzuya M, Asai T, Kanda S, Maeda K, Cheng XW, Iguchi A. Glycation cross-links inhibit matrix metalloproteinase-2 activation in vascular smooth muscle cells cultured on collagen lattice. Diabetologia. 2001;44(4):433–436. doi: 10.1007/s001250051640. [DOI] [PubMed] [Google Scholar]
- 45.Robert V, Besse S, Sabri A, et al. Differential regulation of matrix metalloproteinases associated with aging and hypertension in the rat heart. Laboratory Investigation. 1997;76(5):729–738. [PubMed] [Google Scholar]
- 46.Wu L, Iwai M, Nakagami H, et al. Effect of angiotensin II type 1 receptor blockade on cardiac remodeling in angiotensin II type 2 receptor null mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22(1):49–54. doi: 10.1161/hq0102.102277. [DOI] [PubMed] [Google Scholar]
- 47.Weber KT. Angiotensin II and connective tissue: homeostasis and reciprocal regulation. Regulatory Peptides. 1999;82(1–3):1–17. doi: 10.1016/s0167-0115(99)00032-4. [DOI] [PubMed] [Google Scholar]
- 48.Benetos A, Safar ME. Aortic collagen, aortic stiffness, and AT1 receptors in experimental and human hypertension. Canadian Journal of Physiology and Pharmacology. 1996;74(7):862–866. [PubMed] [Google Scholar]
- 49.Lacolley P, Safar ME, Lucet B, Ledudal K, Labat C, Benetos A. Prevention of aortic and cardiac fibrosis by spironolactone in old normotensive rats. Journal of the American College of Cardiology. 2001;37(2):662–667. doi: 10.1016/s0735-1097(00)01129-3. [DOI] [PubMed] [Google Scholar]
- 50.Walters PE, Gaspari TA, Widdop RE. Angiotensin-(1–7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension. 2005;45(5):960–966. doi: 10.1161/01.HYP.0000160325.59323.b8. [DOI] [PubMed] [Google Scholar]
- 51.Bosnyak S, Jones ES, Christopolous A, Aguilar MI, Thomas WG, Widdop RE. Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clinical Science. 2011;121:297–303. doi: 10.1042/CS20110036. [DOI] [PubMed] [Google Scholar]