

Abstract
In HeLa cells the combinatorial knockdown of Bcl-xL and Mcl-1 is sufficient to induce spontaneous apoptosis. Quinoxaline derivatives were screened for the induction of Mcl-1 dependent apoptosis using a cell line without functional Bcl-xL. Quinoxaline urea analog 1h was able to specifically induce apoptosis in an Mcl-1 dependent manner. We demonstrate that even small changes to 1h results in dramatic los of activity. In addition, 1h and ABT-737 synergistically inhibit cell growth and induce apoptosis. Our results also suggest that 1h could have therapeutic potential against ABT-737 refractory cancer.
Keywords: Quinoxaline, Structure activity relationship, Mcl-1, Bcl-xL, ABT-737
1. Introduction
Benzopyrazine or commonly known as quinoxaline is a naphthalene isostere with carbon atoms 1 and 4 replaced with nitrogen atoms. Quinoxalines are an important class of heterocycles found in natural products. Examples include the cyclic peptide triostin A and the recently isolated Izumiphenazines A–C (Figure 1).1 The quinoxaline core is also found in several drugs currently on the market. They include brimonidine used to treat glaucoma, quinacillin that has antibacterial properties and the smoking cessation agent varenicline (Figure 1).2 Several quinoxaline analogs are in preclinical and clinical development against a variety of diseases. Kinase inhibitors BMS345541 and NVP-BSK805 are being explored as an antitumor agent and to treat polycythemia respectively.3 R(+)XK469 is an example of a quinoxaline analog in clinics to treat patients with advanced refractory solid tumors (Figure 1).4 Additionally compounds containing the quinoxaline core were identified as hits from recent high throughput screening campaigns.5 We identified a quinoxaline urea analog (NCGC55879-01) as a BRCA1 inhibitor from a quantitative high throughput screen.6 These studies show that the quinoxaline analogs act as ligands for a variety of biological targets and have activities against several diseases, which fit definition of a privileged scaffold.1a This led to the synthesis of focused libraries of quinoxaline analogs to further explore the chemical space and structure activity relationship studies.7
Figure 1.

The BH3 mimetic ABT-737 and quinoxaline core containing natural products, drugs on the market, preclinical agents, a clinical compound and a hit from a high throughput screen.
An intricate network of protein-protein interactions between pro-apoptotic and pro-survival family proteins maintains a balance between cell survival and cell death.8 There are three types of apoptotic proteins, the anti-apoptotic (Bcl-xL, Bcl-2, Bcl-2 and Mcl-1), the initial pro-apoptotic proteins (Bad, Bim, Puma and Noxa) and cell death proteins (Bak and Bax). The proapoptotic proteins Bax and Bak undergo oligomerization upon activation by apoptotic stimuli.9 This triggers permeabilization of the mitochondrial membrane, release of apoptotic factors such as cytochrome c,10 activation of effector caspases and cell death.11 The sequestration of pro-apoptotic proteins by the pro-survival Bcl-2 family proteins suppresses mitochondrial damage and ensures cell survival.12 Increased levels of pro-survival proteins (Bcl-2, Bcl-xL or Mcl-1) are associated with maintenance of malignant diseases, resistance to chemotherapy and poor clinical outcome.13 These observations triggered a concerted effort to identify small molecule inhibitors and develop strategies to perturb the levels of pro-survival Bcl-2 family of proteins.
The most successful Bcl-2 inhibitors discovered to date are the ABT series of compounds from the Abbott laboratories. The crystal structure of Bcl-xL-BH3 peptide complex revealed the presence of a hydrophobic core formed by the BH1–3 domains of Bcl-xL that binds a α-helix peptide found in BH3 only proteins.14 A structure guided fragment based design led to the development of ABT-737 a small molecule BH3 mimetic.15 ABT-737 had nanomolar binding affinities for Bcl-xL and Bcl-2 but did not bind Mcl-1.16In vitro preclinical studies showed that ABT-737 induced apoptosis in multiple cancer cell lines at micromolar concentrations.17 Interestingly, down regulation of Mcl-1 resulted in sensitization to ABT-737 both in vitro and in vivo.18
A combination of knockdown and biochemical studies on the entire collection of prosurvival protein and BH3 only proteins were conducted to understand their roles in apoptosis.19 These studies revealed that knock down of Mcl-1 or Bcl-xL did not induce apoptosis however the combinatorial knockdown of Mcl-1 and Bcl-xL resulted in spontaneous apoptosis (without an external stimulus).20 These are consistent with the sensitization of cells to ABT-737 upon Mcl-1 knock down. They also highlight the emerging functional importance of Mcl-1 and the need to develop inhibitors that perturb Mcl-1 levels.21
A major challenge in directly targeting Mcl-1 is the complexity associated with its regulation. Mcl-1 levels and its functions are regulated rapidly through changes in transcription, a number of posttranslational modifications that affect its localization, its stability and its ability to form a variety of protein-protein interactions (Figure 2).22 On the other hand the various proteins that regulate Mcl-1 levels are attractive targets for inhibitor development.21 In the present study, we screened a focused quinoxaline library using a cell line without functional Bcl-xL, Bcl-2 and Bcl-w (accomplished by the inducible expression of the BH3 only protein Bad3SA). We identified a quinoxaline urea analog 1h as a Mcl-1 pathway inhibitor. Mechanism specific inhibition by 1h was established using cell lines in which levels of functional Mcl-1 and Bcl-xL was regulated by the inducible expression of BH3 only proteins Noxa and Bad3SA respectively. Structure activity relationship revealed the functional groups on 1h required for activity. Follow-up studies showed that 1h reduced Mcl-1 levels in a dose- and time-dependent manner. Compound 1h also sensitized cells to ABT-737 and the combination of 1h and ABT-737 resulted in rapid cleavage of Poly (ADP-Ribose) Polymerase (PARP) a biochemical marker of apoptosis. We are currently working on establishing the molecular target of 1h, which will be reported in due course.
Figure 2.
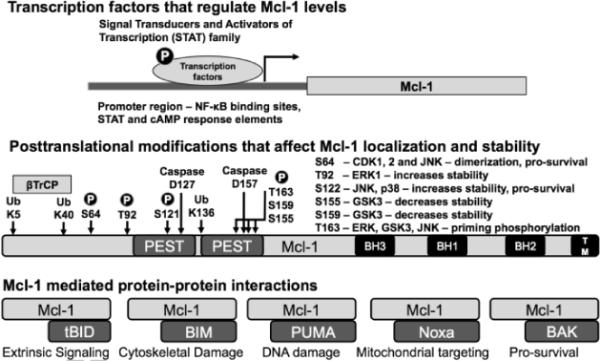
An overview of the modes of regulation of Mcl-1 function.
2. Results and Discussion
2.1 Quinoxaline analogs screened for Mcl-1 depended apoptosis
Recent reports have shown that quinoxaline analogs have growth inhibitory and apoptotic activity against several cancer cell lines.23, 7b The quinoxaline analogs 1a – 1o screened here for anti-Mcl-1 activity were previously reported by us to have antiproliferative activity.7b The emergence of Mcl-1 as a potential target led us to screen quinoxaline analogs for Mcl-1 specific inhibitors. We used a cell line (Dox-Bad3SA) that expressed Bad3SA (a BH3 only protein that binds Bcl-xL) under doxycycline control. In the presence of doxycycline the Dox-Bad3SA cell line will express Bad3SA, which will bind and inactivate Bcl-xL leaving functional Mcl-1 to control cell survival.19, 20 Therefore compounds that are Mcl-1 pathway specific inhibitors will show enhanced apoptosis in Dox-Bad3SA cells only in the presence of doxycycline. For this screen Dox-Bad3SA cells were incubated for 6 hours with the inhibitors (10 μM) in the presence and absence of doxycycline. Induction of apoptosis was measured using caspase 3/7 activity assay (normalized for cell number using alamarBlue).24 Camptothecin (50 μM) and a Bayer IKKβ inhibitor IKK2VII (10 μM)25 a compound with a well characterized target were used as controls in this screen. Camptothecin increases the stability of Noxa protein which, competes with the Mcl-1-Bak complex to release free bak and thereby induce apoptosis.25c
The quinoxaline library is described in Table 1. The results from the screen are summarized as a bar chart in Figure 3. The black bars represent caspase activity in Dox-Bad3SA HeLa cells (−Dox) with functional Bcl-xL and Mcl-1 while the grey bars represent the same with only functional Mcl-1 (+Dox). As expected, camptothecin shows increased caspase 3/7 activity in the cells with only functional Mcl-1 when compared to cells with function Bcl-xL and Mcl-1. On the other hand IKK2VII shows the opposite effect. Quinoxaline analogs 1d, 1f, 1j, 1l and 1n show little to no effect on Dox-Bad3SA both in the presence or absence of doxycycline and are therefore classified as inactive. Compounds 1a, 1c, 1g, and 1m show increased caspase activity in Dox-Bad3SA cells both in the presence and absence of doxycycline and are therefore classified as non-specific inhibitors. Compounds 1b, 1e and 1i are trending to be IKK2VII like suggesting that they have Mcl-1 agonist like function. Compounds 1h, 1k and 1o show the desired Mcl-1 dependent caspase activation. It is interesting to note that going from −H to −F to −Br in 1e to 1g to 1h respectively resulted in a systematic trending towards Mcl-1 dependent caspase activation. Also, a comparison of caspase activation by compounds 1k and 1o suggests the need to explore the R1 position. Therefore, decided to generate a second set of quinoxaline analogs explore analog 1h further.
Table 1.
Focused library of quinoxaline analogs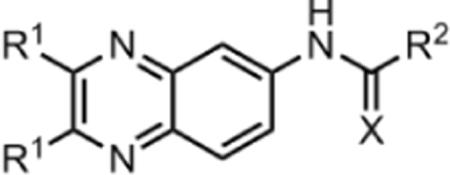
| Entry | R1 | X | R1 |
|---|---|---|---|
| 1a | Methyl | O | -CH3 |
| 1b | Methyl | O | -NH-phenyl |
| 1c | Methyl | O | -NH-(4-benzyl)-piperidine |
| 1d | 2-Furanyl | O | -CH3 |
| 1e | 2-Furanyl | O | -NH-phenyl |
| 1f | 2-Furanyl | S | -NH-phenyl |
| 1g | 2-Furanyl | O | -NH-(4-fluoro)-Ph |
| 1h | 2-Furanyl | O | -NH-(4-bromo)-Ph |
| 1i | 2-Furanyl | O | -NH-4-biphenyl |
| 1j | 2-Furanyl | S | -NH-(4-nitro)-Ph |
| 1k | 2-Furanyl | O | -N-Pyrrolidine |
| 1l | 2-Furanyl | O | -N-Morpholine |
| 1m | 2-Furanyl | O | -NH-(4-benzyl)-piperidine |
| 1n | Phenyl | S | -NH-(4-nitro)-Ph |
| 1o | Phenyl | O | -N-Pyrrolidine |
Figure 3.
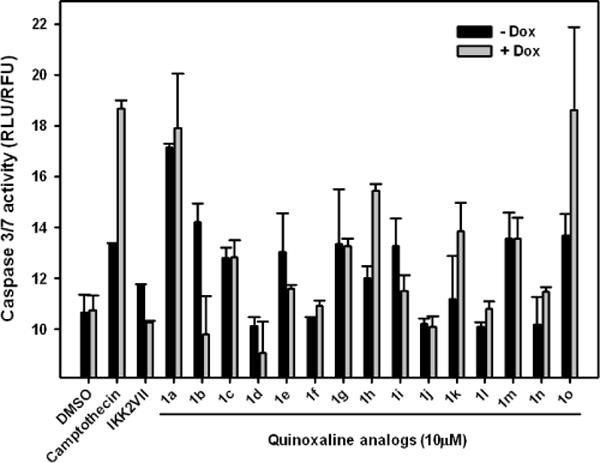
Induction of Mcl-1 dependent apoptosis by quinoxaline analogs. Dox-Bad3SA cells were treated with indicated compounds in the absence (−Dox) and presence (+Dox) of doxycycline. Caspase activity was measured 6 h after treatment.
2.2 Synthesis and evaluation of quinoxaline urea 1h analogs
To explore the functional groups at the R1 position and the positional effect of the bromine atom in 1h we generated a second set of quinoxaline analogs. The quinoxaline core was generated by condensation of symmetrical diones 2a–e with 4-nitrobenzene-1,2-diamine (3). Reduction of the resulting nitro compounds 4a–e to the amine 5a–e followed by condensation with substituted isocyanates yielded the quinoxaline urea analogs 1p–v (Scheme 1).7b
Scheme 1.
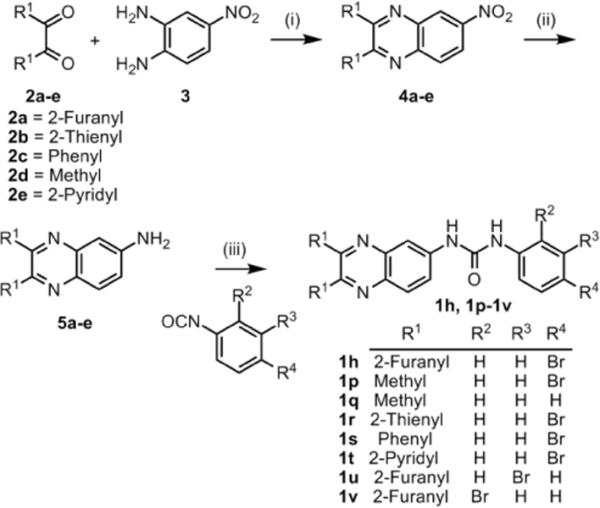
Synthesis of 1h analogs
Reagents and conditions: (i) ethanol, reflux (85 – 99 %); (ii) H2/Pd-C, ethanol, 25 – 28°C (85 – 96 %); (iii) CH2Cl2, 25 – 28°C (62 – 83 %)
The compounds were screened as described in the previous section. Except cell death was quantified after a 12 hour incubation by Hoechst staining. The results from this screen are summarized in Figure 4. Replacing the furanyl rings at the R1 position with methyl groups in analog 1p resulted in a complete loss of activity. Not surprisingly removal of both the bromine atom at R4 and furanyl rings at R1 in analog 1q also resulted in a complete loss of activity. Replacing the oxygen atoms in the furanyl rings with sulfur atoms in analog 1r resulted in a significant (> 90%) loss of activity. On the other hand, replacing the furanly rings with phenyl rings in analog 1s resulted in retention of activity. Interestingly replacing the phenyl rings with pyridyl rings in analog 1t also resulted in a complete loss of activity. A comparison of the activities of 1h, 1r, 1s and 1t suggests that a chelation driven conformational change could be responsible for the loss of activity in 1r and 1t. We next probed the positional effects of the bromine atom in compounds 1u (meta) and 1v (ortho). Moving the bromine atom to the meta position in analog 1u did not alter the activity however moving it to the ortho position in analog 1v resulted in > 90% loss of activity. This suggests that the –NH group in the urea could be involved in possible hydrogen bonding interactions with the molecular target of 1h.
Figure 4.
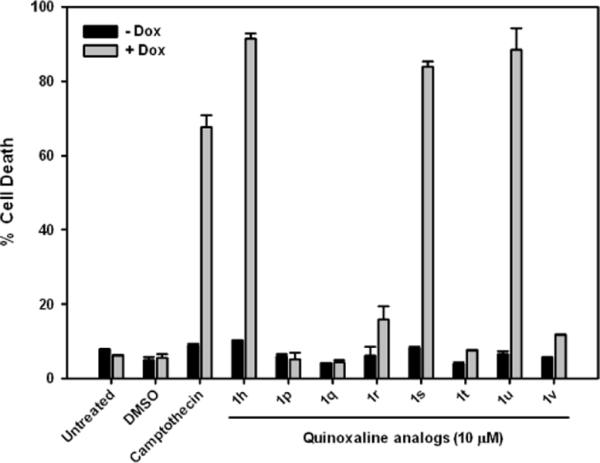
Structure-activity relationship with quinoxaline analogs (1p–1v). Dox-Bad3SA cells were treated with indicated compounds in the absence (−Dox) and presence (+Dox) of doxycycline. Cell death was measured 12 h after treatment by Hoechst staining. (See Figure S1 for images)
2.3 Follow-up studies to establish Mcl-1 dependent induction of apoptosis by analog 1h
To demonstrate Mcl-1 dependent induction of apoptosis by 1h we used two additional cell lines (Dox-GFP and Dox-Noxa).19, 20 Figure 5 summarizes the functional prosurvival proteins present in these inducible cell lines. The Dox-GFP control cell line expresses GFP when treated with doxycycline and has functional Bcl-xL and Mcl-1. The Dox-Noxa cell line expresses Noxa when treated with doxycycline, which sequesters Mcl-1 and therefore has only functional Bcl-xL. The Dox-Bad3SA cell line expresses Bad3SA and has only functional Mcl-1.
Figure 5.
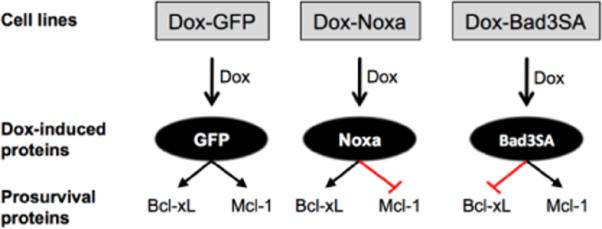
Doxycycline inducible cell lines used to identify Mcl-1 specific modulators. Doxycycline (Dox) treatment of cells leads to the induction of GFP, Noxa and Bad3SA expression.
The three cell lines, Dox-GFP, Dox-Noxa and Dox-Bad3SA were treated with doxycycline (1μg/mL) for 3h to induce the expression of GFP, Noxa and Bad3SA respectively. The cells were then treated with DMSO, ABT-737 (1μM) or 1h (10μM) for an additional 12h. Under the assay conditions neither ABT-737 nor 1h induced cell death in Dox-GFP cells. This is because ABT-737 inhibits only Bcl-xL allowing functional Mcl-1 to prevent cell death and 1h inhibits only Mcl-1 allowing functional Bcl-xL to prevent cell death. As expected, ABT-737 induced cell death in Dox-Noxa cells, but not in Dox-Bad3SA cells. This is consistent with results reported in the literature about the specificity of ABT-737 for Bcl-2/Bcl-xL and not Mcl-1.15 Compound 1h induced cell death only in Dox-Bad3SA cells, but not in Dox-Noxa cells demonstrating its specificity for Mcl-1 dependent apoptosis (Figure 6).
Figure 6.
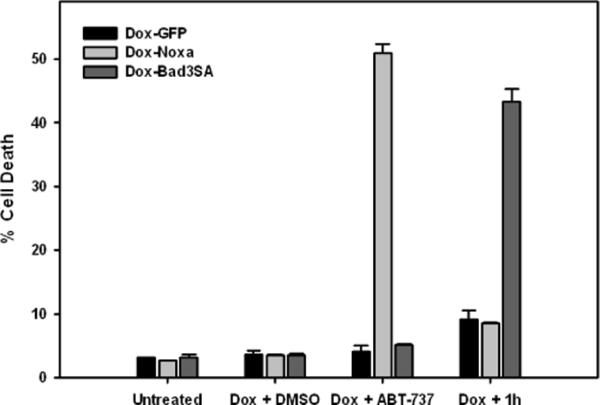
Compound 1h induces cell death in a Mcl-1 specific manner. Doxycycline inducible cell lines were treated as indicated for 12 h and stained with Hoechst dye to quantify number of dead cells. (See Figure S2 for images)
To determine if analog 1h perturbs Mcl-1 levels we conducted dose response (1, 5 and 10 μM of 1h with a 12 hour incubation) and time course (3, 6 and 12 hour incubation at 10 μM of 1h) studies. The lysates were subjected to SDS-PAGE and probed for prosurvival proteins (Mcl-1, Bcl-xL and Bcl-2) by Western blot analyses. We observed a dose- and time-dependent decrease of Mcl-1 levels in cells treated with 1h (Figure 7). For the time depdendent studies we observed varying amounts of cell death in HeLa cells therefore we used HeLa cells that over express Bcl-xL.
Figure 7.
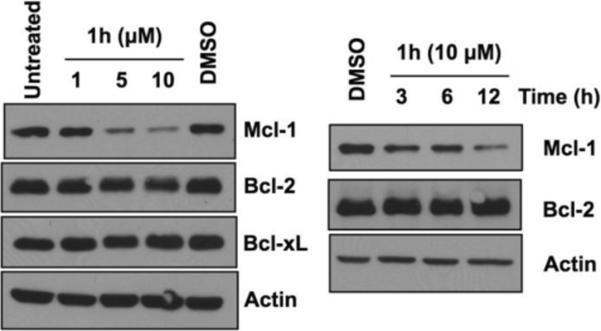
Down regulation of Mcl-1 by 1h. (left) HeLa cells were incubated with the indicated concentrations of compound 1h for 12 h. (right) HeLa cells that over-express Bcl-xL were treated with compound 1h (10 μM) for the indicated time points. Cells were then harvested and lysates were resolved on SDS-PAGE gels. DMSO was used as a vehicle control. Actin was used as loading control.
2.4 Synergism studies with 1h and ABT-737
Since 1h selectively perturbs Mcl-1 levels in cells we hypothesized that 1h and ABT-737 will synergistically inhibit growth and induce apoptosis. To test this hypothesis, we treated HeLa cells with various concentration combinations of 1h and ABT-737 and monitored cell growth (Figure 8A). Data from this study was used to determine combination indices (CI) for 1h and ABT-737 (Figure 8B).26 CI < 1 indicates synergy; CI ~ 1 indicates additive effects while CI > 1 indicates antagonism.26 CI values ranging from 0.24–0.84 were obtained when micromolar concentrations of both drugs were used, however at lower concentrations CI values > 1 were observed. At the present time we do not fully understand the observed CI > 1. The growth inhibitory effects of the compounds ABT-737 (12.5 μM) and 1h (10 μM) were monitored individually and in combination (CI = 0.84) over a five-day period (Figure 8C). An inactive compound 1p was used as a control in this experiment. The results showed a synergistic inhibition of cell growth only with the combination of 1h and ABT-737.
Figure 8.

Synergism studies with 1h and ABT-737. (A) Matrix of combination treatment with 1h and ABT-737 in a cell growth assay. (B) Combination index (CI) determination using matrix data. (C) Time dependent HeLa cell growth at CI = 0.84 concentrations.
We next explored if the synergistic growth inhibition extends to the induction of apoptosis by 1h and ABT-737. Activation of caspases is considered one of the hallmarks of apoptosis. The executioner caspases cleave multiple structural and repair proteins resulting in programmed cell death.27 HeLa cells were treated with either DMSO, 1h, 1p, ABT-737, 1h + ABT-737 or 1p +ABT-737 and caspase 3/7 activity was measured after a six hour incubation (Figure 9 see inset). The compounds individually had very little effect on the induction of caspases, however the combination of 1h and ABT-737 resulted in ~4-fold increase in caspase activity. No such effect was observed with the 1p and ABT-737 combination. We also measured cell death by Hoechst staining and found a dose-dependent increase of cell death with the combination of 1h and ABT-737 (Figure 9). In summary we observed a synergistic induction of apoptosis with 1h and ABT-737.
Figure 9.
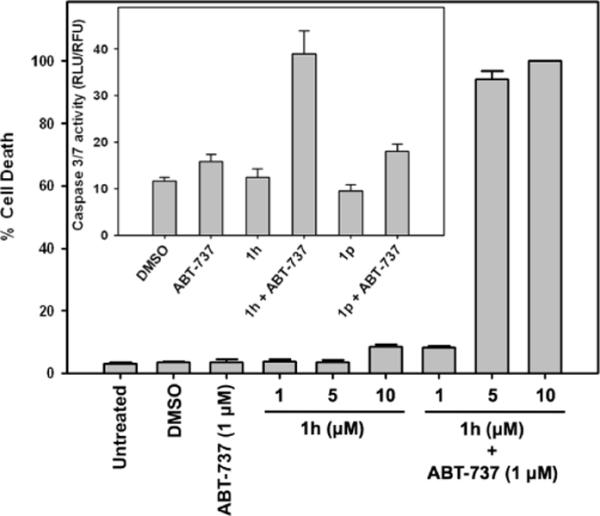
Apoptosis studies with 1h and ABT-737. HeLa cells were treated as indicated and cell death was measured by Hoechst staining or caspase activity assay (inset). For the caspase assay 1 μM of ABT-737 and 10 μM of 1h and 1p were used.
To further explore the role of 1h in the induction of Mcl-1 dependent apoptosis and the observed synergism with ABT-737, we determined expression levels of apoptotic proteins. Activation of caspases leads to the cleavage of specific proteins such as PARP, a nuclear protein involved in DNA repair. PARP is one of the earliest proteins targeted for cleavage by caspases.28 X-linked inhibitor of apoptosis protein (XIAP) binds directly to caspase-3 and blocks access to the substrates.29 Peptides derived from XIAP also bind to the catalytic site of caspase-7, thereby inactivating its enzymatic function.30 Therefore, we probed PARP and XIAP levels in cells treated with DMSO, ABT-737, 1h, 1h + ABT-737 1p, 1p + ABT-737 by Western blot analysis (Figure 10). PARP cleavage was seen only in the combination of 1h and ABT-737 and we observed reduced XIAP levels only in 1h treated cells. This suggests that inhibiting Bcl-xL does not have any effect on XIAP levels and 1h could be perturbing an upstream target that regulates both Mcl-1 and XIAP. Together these studies show synergistic effects of 1h and ABT-737 in inducing apoptosis in these cells.
Figure 10.
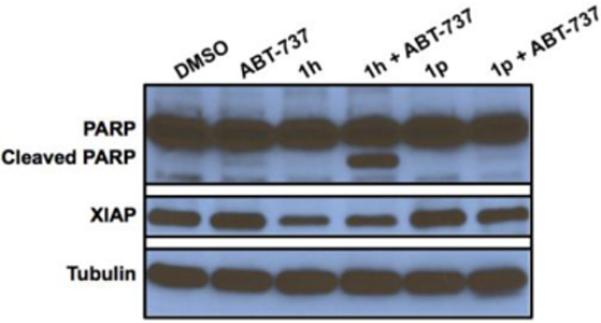
PARP cleavage by 1h and ABT-737 and down regulation of XIAP in HeLa cells by 1h evaluated by Western blotting. Tubulin was used as a loading control.
2.5 Conclusion
In this report, we screened a small quinoxaline library for Mcl-1 dependent apoptosis. We identified compound 1h that had activities comparable to the positive control camptothecin which was used at 5-fold higher concentrations. We generated a second set of compounds and identified the key functional groups on 1h required for its activity. We found that small changes to 1h resulted in significant loss of activity. Using doxycycline responsive cell lines that express GFP, Noxa and Bad3SA, we established mechanism specificity of compound 1h. HeLa cells treated with 1h showed a dose- and time-dependent decrease in Mcl-1 levels. We also showed that 1h and ABT-737 synergistically inhibited cell growth and induced apoptosis. Reduction of XIAP levels in cells treated with 1h is consistent with activation of caspases and rapid cleavage of PARP. In summary, we report the identification and characterization of quinoxaline urea analog 1h that could have therapeutic value against ABT-737 refractory cancers
3. Experimental Methods
Chemistry General Methods
All reagents were purchased from commercial sources and were used without further purification. Flash chromatography was carried out on silica gel (200–400 mesh). Thin layer chromatography (TLC) were run on pre-coated EMD silica gel 60F254 plates and observed under UV light. 1H-NMR (500MHz) and 13C-NMR (125 MHz) spectra were recorded in chloroform-d or DMSO-d6 on an Varian-500 spectrometer. LC-MS for the compounds were generated on an Agilent 1200 series system with UV detector (214 nm and 254 nm) and an Agilent 6130 quadrupole mass detector. All the compounds tested were > 97 % pure as determined by LC. Columns and conditions used to determine purity of the compounds reported are summarized below.
Solvent A: Acetonitrile with 0.01% TFA; Solvent B: Water with 0.01% TFA. Flow rate: 1 mL/min
Column-condition A: Agilent Zorbax 300SB_C18, narrow bore, 2.1 × 150 mm, 5 micron. 15 min gradient: 2% to 95% of A.
Column-condition B: Agilent Zorbax Eclipse PAH rapid resolution, 4.6 × 150 mm, 3.5 micron. 10 min gradient: 5% to 95% of A.
The spectral characterization data for compounds 1a – 1o and 1q can be found in the supplementary material of reference 7b.
General Procedure of quinoxalinylurea derivatives 6a–i
To a stirring solution of 5a–e (1.0 mmol) in dichloromethane (20–30 ml) under nitrogen the corresponding phenylisocyanate (1.2–1.3 mmol) was added. The mixture was maintained at room temperature for 12–48 h and the reaction monitored for completion by TLC. The precipitated solid was filtered, dried and purified by flash column chromatography to yield the desired compounds.
1-(4-bromophenyl)-3-(2,3-dimethylquinoxalin-6-yl)urea 1p
1H-NMR (DMSO-d6, 500 MHz) δ: 2.61(s, 3H), 2.63 (s, 3H), 7.46 (s, 4H), 7.67 (dd, J1 = 2.44 Hz, J2 = 8.78 Hz, 1H), 7.85 (d, J = 8.78 Hz, 1H), 8.13 (d, J = 2.44 Hz, 1H), 8.98 (s, 1H), 9.15 (s, 1H). 13C-NMR (DMSO-d6, 125 MHz) δ: 23.1, 23.5, 114.1, 114.2, 120.9 121.0, 122.5, 129.1, 132.2, 132.3, 137.3, 139.6, 140.4, 142.0, 152.2, 153.0, 154.7 MS calcd C17H15BrN4O, 370.0; found ESI-MS m/z: 371.0 (M+1). Retention times, A: 6.9 min and B: 9.3 min.
1-(4-bromophenyl)-3-(2,3-di-(thiophen-2-yl)quinoxalin-6-yl)urea 1r
1H-NMR (DMSO d6, 500 MHz) δ: 7.10 (dd, J1 = 4.88Hz, J2 = 8.78Hz, 2H), 7.16 (d, J = 2.93 Hz, 1H), 7.19 (d, J = 3.41 Hz, 1H), 7.48 (bs, 4H), 7.75 (m, 3H), 7.96 (d, J = 8.78 Hz, 1H), 8.28 (d, J = 2.44 Hz, 1H), 9.08 (s, 1H), 9.34 (s, 1H). 13C-NMR (DMSO-d6, 125 MHz) δ: 113.4, 114.4, 120.9, 121.1, 124.6, 128.4, 128.5, 129.5, 129.6, 129.8, 130.5, 132.2, 132.3, 136.8, 139.4, 139.6, 141.6, 141.7, 141.9, 142.2, 144.4, 147.0, 152.9. MS calcd C23H15BrN4OS2, 506.0; found ESI-MS m/z: 507.0 (M+1). Retention times, A: 10.7 min and B: 11.4 min.
1-(4-bromophenyl)-3-(2,3-diphenylquinoxalin-6-yl)urea 1s
1H-NMR (DMSO d6, 500 MHz) δ: 7.32–7.39 (m, 6H), 7.42–7.46 (m, 4H), 7.48 (bs, 4H), 7.82 (d, J = 8.78 Hz, 1H), 8.06 (d, J =8.78 Hz, 1H), 8.35 (s, 1H), 9.07 (s, 1H), 9.35 (s, 1H). 13C-NMR (DMSO-d6, 125 MHz) δ: 113.9, 114.4, 120.9, 121.1, 124.4, 128.7, 128.7, 129.2, 129.4, 130.0, 130.3, 130.4, 132.3, 137.5, 139.5, 139.6, 142.0, 142.2, 151.5, 153.0, 153.8. MS calcd C27H19BrN4O, 494.1; found ESI-MS m/z: 495.1 (M+1). Retention times, A: 10.4 min and B: 11.1 min.
1-(4-bromophenyl)-3-(2,3-di-(pyridin-2-yl)quinoxalin-6-yl)urea 1t
1H-NMR (DMSO-d6, 500 MHz) δ: 7.32 (m, 2H), 7.49 (bs, 4H), 7.88 (dd, J = 8.78 Hz, 1H), 7.91–7.99 (m, 4H), 8.12 (d, J = 8.78 Hz, 1H), 8.25 (dd, J = 4.88 Hz, J2 = 5.37Hz, 2H), 8.41 (s, 1H), 9.10 (s, 1H), 9.41 (s, 1H). 13C-NMR (DMSO-d6, 125 MHz) δ: 113.9, 114.4, 121.1, 123.7, 123.9, 124.4, 124.6, 125.0, 130.1, 132.3, 137.2, 137.4, 137.5, 139.4, 142.0, 142.4, 148.7, 150.8, 153.0, 153.3, 157.8, 157.9. MS calcd C25H17BrN6O, 496.1; found ESI-MS m/z: 497.1 (M+1). Retention times, A: 7.1 min and B: 8.5 min.
1-(3-bromophenyl)-3-(2,3-di-(furan-2-yl)quinoxalin-6-yl)urea 1u
1H-NMR (DMSO d6, 500 MHz) δ: 6.66–6.70 (m, 4H), 7.19 (d, J = 7.81 Hz, 1H), 7.27 (t, J = 7.81 Hz, 1H), 7.38 (d, J = 8.78 Hz, 1H), 7.82 (dd, J = 2.44 Hz, J2 = 8.78Hz, 1H), 7.83–7.88 (m, 3H), 8.01 (d, J = 8.78 Hz, 1H), 8.30 (d, J = 2.44 Hz, 1H) 9.16 (s, 1H), 9.43 (s, 1H). 13C-NMR (DMSO-d6, 125 MHz) δ: 112.6, 112.7, 112.8, 113.4, 113.8, 118.0, 121.4, 122.4, 124.9, 125.5, 129.5, 129.9, 131.5, 136.9, 140.06, 141.7, 141.8, 142.3, 143.0, 145.1, 145.5, 151.2, 151.3, 152.9. MS calcd C23H15BrN4O3, 474.0; found ESI-MS m/z: 475.0 (M+1). Retention times, A: 9.2 min and B: 10.1 min.
1-(2-bromophenyl)-3-(2,3-di-(furan-2-yl)quinoxalin-6-yl)urea 1v
1H-NMR (DMSO d6, 500 MHz) δ: 6.66–6.70 (m, 4H), 7.02 (t, J = 7.81 Hz, 1H), 7.39 (t, J = 8.3Hz, 1H), 7.65 (d, J = 7.81 Hz, 1H), 7.77 (dd, J1 = 2.44 Hz, J2 = 9.28 Hz, 1H), 7.88 (d, J = 9.28Hz, 2H), 8.03 (d, J = 8.78 Hz, 1H), 8.09 (d, J = 8.3 Hz, 1H), 8.36 (d, J = 2.44 Hz, 1H), 8.38 (s, 1H), 10.05 (s, 1H). 13C-NMR (DMSO-d6, 125 MHz) δ: 112.7, 112.8, 112.9, 113.5, 113.7, 114.3, 123.4, 124.8, 125.4, 128.9, 130.1, 133.3, 137.0, 137.1, 140.7, 141.8, 142.3, 143.1, 145.2, 145.6, 151.2, 151.3, 152.8. MS calcd C23H15BrN4O3, 474.0; found ESI-MS m/z: 475.1 (M+1). Retention times, A: 9.0 min and B: 10.2 min.
Biological activity
Cell death assays
Doxycyline inducible HeLa cell lines (Dox-GFP, Dox-Noxa, Dox-BadS3A)19, 20 were induced with doxycycline (Dox = 1μg/mL) for 3 hours. Cells were then treated as indicated for 12 hours. Cells were fixed and stained with Hoechst dye and the number of condensed nuclei was counted for each treatment to determine percent cell death.
Cell growth inhibition assay
Human cervical tumor cells (HeLa) were cultured in RPMI-1640 medium containing 10% FBS and maintained in a 37°C incubator with 5% CO2. Cells were plated at 2000 cells/well in 96 well plates and incubated overnight. The next day, cells were treated as indicated. The treated cells were assayed for viability using the alamar blue assay. Briefly, 10μL reagent was added to each well and the plate was returned to the incubator for 3hrs after which fluorescence at 544ex/590em was measured using a SpectraMax M5e (Molecular Devices) plate reader. The alamar Blue assay was repeated each day for 5 days. Growth is expressed as raw fluorescent units (RFU).
Caspase 3/7 activation
HeLa cells (2000 cells/well) were treated in 96 well plates as indicated for 6 hours. Caspase Glo reagent (Promega, Inc.) was added and luminescence was measured using a Spectramax M5 (Molecular Devices) plate reader after 1 hr. Raw luminescence values (RLU) were normalized to alamarBlue (RFU).
PARP cleavage
HeLa cells were treated as indicated for 6 hours. Cells were harvested by collecting media, trypsinizing cells, and centrifuging to obtain a combined cell pellet from all steps. Cells were lysed in radio immuno precipitation assay (RIPA) buffer (150 mM NaCl, 1.0 % Triton X-100, 0.5 % sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH 8.0) and protein content was subjected to SDS-PAGE. PARP cleavage was determined via Western blotting using anti-PARP antibody (Calbiochem #AM30).
Apoptosis protein expression analysis
HeLa cells were treated as indicated for 24 hours. Cells were harvested by collecting media, trypsinizing cells, and centrifuging to obtain a combined cell pellet from all steps. Cells were lysed in RIPA buffer and protein content was subjected to SDS-PAGE. Mcl-1 and XIAP expression levels were determined via Western blotting using anti-Mcl-1 antibody (Santa Cruz Biotechnology, Inc. sc-819) and anti-XIAP antibody (Santa Cruz Biotechnology, Inc. sc-58537). Anti-α-tubulin antibody (Cell Signaling Technology, Inc. #3873) was used as a control.
Supplementary Material
Acknowledgments
This project was supported in part by NIH R01CA127239 and the Eppley Cancer Center pilot grant. We would like to thank the Eppley NMR facility, Dr. Srikumar Raja for his help with determining CI, Smitha Kizhake for carrying out LC-MS analyses and the Natarajan lab members for helpful discussions.
ABBREVIATIONS
- Mcl-1
Myeloid cell leukemia 1
- Bcl-xL
B-cell lymphoma extra large
- Bcl-2
B-cell lymphoma 2
- BH
Bcl-2 homology
- XIAP
X-linked Inhibitor of Apoptosis Protein
- PARP
Poly (ADP-Ribose) Polymerase
- Dox
Doxycycline
- GFP
Green Fluorescent Protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Welsch ME, Snyder SA, Stockwell BR. Curr. Opin. Chem. Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zolova OE, Mady AS, Garneau-Tsodikova S. Biopolymers. 2010;93:777–790. doi: 10.1002/bip.21489. [DOI] [PubMed] [Google Scholar]; (c) Dawson S, Malkinson JP, Paumier D, Searcet M. Nat. Prod. Rep. 2007;24:109–126. doi: 10.1039/b516347c. [DOI] [PubMed] [Google Scholar]; (d) Abdelfattah MS, Kazufumi T, Ishibashi M. J. Nat. Prod. 2010;73:1999–2002. doi: 10.1021/np100400t. [DOI] [PubMed] [Google Scholar]
- 2.(a) Greenfield DS, Liebmann JM, Ritch R. J. Glaucoma. 1997;6:250–258. [PubMed] [Google Scholar]; (b) Smith JT, Hamilton-Miller JM, Knox R. Nature. 1964;203:1148–1150. doi: 10.1038/2031148a0. [DOI] [PubMed] [Google Scholar]; (c) Flore MC, Baker TB. N. Engl. J. Med. 2011;365:1222–1231. doi: 10.1056/NEJMcp1101512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, Qiu Y, Zusi FC. J. Biol. Chem. 2003;278:1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]; (b) Baffert F, Régnier CH, De Pover A, Pissot-Soldermann C, Tavares GA, Blasco F, Brueggen J, Chéne P, Drueckes P, Erdmann D, Furet P, Gerspacher M, Lang M, Ledieu D, Nolan L, Ruetz S, Trappe J, Vangrevelinghe E, Wartmann M, Wyder L, Hofmann F, Radimerski T. Mol. Can. Ther. 2010;9:1945–1955. doi: 10.1158/1535-7163.MCT-10-0053. [DOI] [PubMed] [Google Scholar]
- 4.Undevia SD, Innocenti F, Ramirez J, House L, Desai AA, Skoog LA, Singh DA, Karrison T, Kindler HL, Ratain MJ. Eur. J. Biol. Cancer. 2008;44:1684–1692. doi: 10.1016/j.ejca.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Johnston PA, Foster CA, Tierno MB, Shun TY, Shinde SN, Paquette WD, Brummond KM, Wipf P, Lazo JS. Assay Drug Dev. Technol. 2009;7:250–265. doi: 10.1089/adt.2008.186. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Johnston PA, Soares KM, Shinde SN, Foster CA, Shun TY, Takyi HK, Wipf P, Lazo JS. Assay Drug Dev. Technol. 2008;6:505–518. doi: 10.1089/adt.2008.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simeonov A, Yasgar A, Jadhav A, Lokesh GL, Klumpp C, Michael S, Austin CP, Natarajan A, Inglese J. Anal. Biochem. 2008;375:60–70. doi: 10.1016/j.ab.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Cavazzuti A, Paglietti G, hunter WN, Gamarro F, Piras S, Loriga M, Allecca S, Corona P, Mcluskey K, Tulloch L, Gibellini F, Ferrari S, Costi MP. Proc. Natl. Acad. Sci. U. S. A. 2008;105:1448–1453. doi: 10.1073/pnas.0704384105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen Q, Bryant VC, Lopez H, Kelly DL, Luo X, Natarajan A. Bioorg. Med. Chem. Lett. 2011;21:1929–1932. doi: 10.1016/j.bmcl.2011.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) You L, Cho EJ, Leavitt J, Ma LC, Montelione GT, Anslyn EV, Krug RM, Ellington A, Robertus JD. Bioorg. Med. Chem. Lett. 2011;21:3007–3011. doi: 10.1016/j.bmcl.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen LH, Chang CM, Salunke DB, Sun CM. ACS Comb. Sci. 2011;13:391–398. doi: 10.1021/co200022u. [DOI] [PubMed] [Google Scholar]
- 8.Danial NN, Korsmeyer SJ. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 9.Roucou X, Montessuit S, Antonsson B, Martinou JC. Biochem. J. 2002;368:915–921. doi: 10.1042/BJ20020972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesing PH. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 11.Nuñez G, Bendict MA, Hu Y, Inohara N. Oncogene. 1998;17:3237–3245. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- 12.Youle RJ, Strasser A. Nat. Rev. Mol. Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 13.(a) Awan FT, Kay NE, Davis ME, Wu W, Geyer SM, Leung N, Jelinek DF, Tschumper RC, Secreto CR, Lin TS, Grever MR, Shanafel TD, Zent CS, Call TG, Heerema NA, Lozanski G, Byrd JC, Lucas DM. Blood. 2009;113:535–537. doi: 10.1182/blood-2008-08-173450. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fennell DA. Clin. Lung Cancer. 2003;4:307–313. doi: 10.3816/clc.2003.n.012. [DOI] [PubMed] [Google Scholar]; (c) Kausch I, Jiang H, Thode B, Doehn C, Krüger S, Jocham D. Eur. Urol. 2005;47:703–709. doi: 10.1016/j.eururo.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 14.Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon SH, Nettesheim D, Chang BS, Thompson CB, Wong SL, Ng SL, Fesik SW. Nature. 1996;381:335–341. doi: 10.1038/381335a0. [DOI] [PubMed] [Google Scholar]
- 15.(a) Petros AM, Dinges J, Augeri d. J., Baumeister SA, Betebenner DA, Bures MG, Elmore SW, Hajduk PJ, Joseph MK, Landis SK, Nettesheim DG, Rosenberg SH, Shen W, Thomas S, Wang X, Zanze I, Zhang H, Fesik SW. J. Med. Chem. 2006;49:656–663. doi: 10.1021/jm0507532. [DOI] [PubMed] [Google Scholar]; (b) Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joesph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 16.Van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, Warner RB, Ng SC, Fesik SW, Elmore SW, Rosenberg SH, Tse C. Cancer Res. 2007;67:1176–1183. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 18.(a) Zheng L, yang W, Zhang C, Ding WJ, Zhu H, Lin NM, Wu HH, He QJ, Yang B. Cancer Lett. 2011;309:27–36. doi: 10.1016/j.canlet.2011.05.011. [DOI] [PubMed] [Google Scholar]; (b) Tromp JM, Geest CR, Breij EC, Elias JA, van Laar J, Luijks DM, Kater AP, Beaumont T, Van Oers MH, Eldering E. Clin. Cancer Res. 2011;18:487–489. doi: 10.1158/1078-0432.CCR-11-1440. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Lopez H, George NM, liu X, Pang X, Luo X. Cell Death Differ. 2011;18:864–873. doi: 10.1038/cdd.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez H, Zhang L, George NM, Liu X, Pang X, Evans JJ, Targy NM, Luo X. J. Biol. Chem. 2010;285:15016–15026. doi: 10.1074/jbc.M109.086231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Azmi AS, Wang Z, Philip PA, Mohammad RM, Sarkar FH. Expert Opin. Emerg. Drugs. 2011;16:59–70. doi: 10.1517/14728214.2010.515210. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dai Y, Grant S. Cancer Res. 2007;67:2908–2911. doi: 10.1158/0008-5472.CAN-07-0082. [DOI] [PubMed] [Google Scholar]
- 22.Thomas LW, Lam C, Edwards SW. FEBS Lett. 2010;584:2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 23.(a) Grande F, Aiello F, De Grazia OD, Brizzi A, Garofalo A, Neamati N. Bioorg. Med. Chem. 2007;15:288–294. doi: 10.1016/j.bmc.2006.09.073. [DOI] [PubMed] [Google Scholar]; (b) Gavara L, Saugues E, Alves G, Debiton E, Anizon F, Moreau P. Eur. J. Med. Chem. 2010:5520–5526. doi: 10.1016/j.ejmech.2010.08.067. [DOI] [PubMed] [Google Scholar]
- 24.Bryant VC, Kishore Kumar GD, Nyong AM, Natarajan A. Bioorg. Med. Chem. Lett. 2012;22:245–248. doi: 10.1016/j.bmcl.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Murata T, Shimada M, Sakakibara S, Yoshino T, Masuda T, Shintani T, Sato H, Koriyama Y, Fukushima K, Nunami N, Yamauchi M, Fuchikami K, Komura H, Watanabe A, Ziegelbauer KB, Bacon KB, Lowinger TB. Bioorg. Med. Chem. Lett. 2004;14:4019–4022. doi: 10.1016/j.bmcl.2004.05.041. [DOI] [PubMed] [Google Scholar]; (b) Sanda T, Lida S, Ogura H, Asamitsu K, Murata T, Bacon KB, Ueda R, Okamoto T. Clin. Cancer Res. 2005;11:1974–1982. doi: 10.1158/1078-0432.CCR-04-1936. [DOI] [PubMed] [Google Scholar]; (c) Okumura K, Huang S, Sinicrope FA. Clin. Cancer Res. 2008;14:8132–8142. doi: 10.1158/1078-0432.CCR-08-1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Chou TC. Pharmacol. Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]; (b) Chou TC. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]; (c) Raja SM, Clubb RJ, Ortega-Cava C, Williams SH, Bailey T. a., Duan L, Zhao x., Reddi a. L., Nyong a. M., Natarajan A, Band v., Band H. Cancer Biol. Ther. 2011;11:263–276. doi: 10.4161/cbt.11.2.13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi Y. Mol. Cell. 2002;9:459–470. doi: 10.1016/s1097-2765(02)00482-3. [DOI] [PubMed] [Google Scholar]
- 28.Durlez PJ, Shah GM. Biochem. Cell Biol. 1997;75:337–349. [PubMed] [Google Scholar]
- 29.(a) Dubrez-Daloz L, Dupoux A, Cartier J. Cell Cycle. 2008;7:1036–1046. doi: 10.4161/cc.7.8.5783. [DOI] [PubMed] [Google Scholar]; (b) Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 30.Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, Shi Y. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.