

Abstract
Parkinson’s disease (PD) is characterized by a prominent degeneration of nigrostriatal dopamine (DA) neurons with an accompanying neuroinflammation. Despite clinical and preclinical studies of neuroprotective strategies for PD, there is no effective treatment for preventing or slowing the progression of neurodegeneration. The inverse correlation between caffeine consumption and risk of PD suggests that caffeine may exert neuroprotection. Whether caffeine is neuroprotective in a chronic progressive model of PD has not been evaluated nor is it known if delayed caffeine treatment can stop DA neuronal loss. We show that a chronic unilateral intra-cerebroventricular infusion of 1-methyl-4-phenylpyridinium in the rat brain for 28 days produces a progressive loss of DA and tyrosine hydroxylase in the ipsilateral striatum and a loss of DA cell bodies and microglial activation in the ipsilateral substantia nigra. Chronic caffeine consumption prevented the degeneration of DA cell bodies in the substantia nigra. Importantly, neuroprotection was still apparent when caffeine was introduced after the onset of the neurodegenerative process. These results add to the clinical relevance for adenosine receptors as a disease-modifying drug target for PD.
Keywords: animal model, PD, caffeine, progressive neurodegeneration, MPP+, dopamine neurons, microglia, miniosmotic pump
INTRODUCTION
Parkinson’s disease (PD) is a devastating neurodegenerative disorder characterized by extensive loss of the nigrostriatal dopamine (DA) neurons and neuroinflammation (Appel et al., 2009; Hirsch and Hunot, 2009). At present, the only therapy for PD is symptomatic and such treatments eventually fail. Disease-modifying approaches that can slow or stop the progression of neurodegeneration are desperately needed.
Neuropathology in PD occurs long before any substantive clinical symptoms appear. It is estimated that at the time of symptom presentation there may be 60-80% loss of striatal DA (Hornykiewicz, 1979). Experimental PD animal models have provided a plethora of information on mechanisms of DA neurodegeneration. However, most of these models are based on acute neurotoxicant exposure and may not accurately portray the chronic pathology that is seen in the human PD brain. Moreover, the introduction of disease-modifying drugs to PD patients will occur long after neurodegeneration has been initiated and under conditions of an on-going pathological process. To better predict the efficacy of potential disease-modifying agents in PD, it is important that a progressive PD model be used and that the drug is introduced during the pathological processes of neurodegeneration and neuroinflammation. We have developed a chronic progressive rat model of PD in which MPP+ is infused into the left cerebral ventricle for 28 days. In this model, there is a selective loss of nigrostriatal DA neurons accompanied by neuroinflammation in the nigrostriatal DA brain regions ipsilateral to the side of infusion (Yazdani et al., 2006; Zeevalk et al., 2007). In the present studies, we have used this model to examine whether DA neurons can be rescued from neurodegeneration by concurrent or delayed caffeine treatment
In the past decade, interest in caffeine has emerged as a possible neuroprotective compound. Epidemiological studies show an inverse relationship between caffeine consumption and the risk of developing PD which suggests caffeine may exert neuroprotection in humans (Morelli et al., 2011). In acute animal models of PD, caffeine has been found to be neuroprotective. Treatment of mice with caffeine protects DA neurons from the acute neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Chen et al., 2001; Kalda et al., 2006; Xu et al., 2010). In the acute 6-hydroxydopamine rat model of PD, chronic caffeine treatment protects against the loss of striatal DA neurochemistry and nigral DA cell bodies produced by a single intrastriatal infusion of 6-hydroxydopamine (Aguiar et al., 2006; Joghataie et al., 2004). In the one chronic study, caffeine was shown to protect mouse nigral DA cell bodies in the chronic pesticide (paraquat/maneb) exposure model (Kachroo et al., 2010). However, in all of these studies, caffeine was introduced prior to or concurrent with the neurotoxicant.
In the present study, we have further characterized the progressive nature of damage in the chronic PD rat model and have investigated the ability of caffeine to prevent neurodegeneration as well as to rescue DA neurons. Our findings demonstrate that caffeine protects the nigral DA cell bodies even when treatment is initiated later in the neurodegenerative process.
METHODS
Animals
Male Sprague–Dawley rats (weighing ~300 g at the beginning of the study) (Taconic Farms, Germantown, NY) were maintained on a 12-h light–dark cycle with food and water available ad libitum. Experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the animal care committee of UMDNJ.
Surgeries and MPP+ Infusions
Vehicle (sodium-iodide) or MPP+-iodide (Sigma-Aldrich) dissolved in saline was infused into the left cerebral ventricle via tubing linked to an Alzet osmotic minipump (model 2ML4) implanted subcutaneously as previously described (Yazdani et al., 2006; Zeevalk et al., 2007). Stereotaxic cannula placement was at coordinates relative to bregma: anterior -0.5 mm, lateral left +1.4 mm, depth –3.9 mm (Paxinos and Watson, 1986). Stereotaxic surgery and minipump placement were performed under anesthesia. MPP+-iodide or vehicle was administered at a dose of 75 μg/day for 28 days with a drug delivery rate of 2.5 μl/h. With the exception of the time course studies, animals were killed 27-28 days after cannula placement.
Caffeine Treatment
Caffeine (Sigma-Aldrich), administered in the drinking water (1 g/l), was prepared fresh every 3-4 days. The dose chosen was to approximate caffeine intake in humans and equates to 60-80 mg/kg/day. In humans with high caffeine intake, doses approximate 10-20 mg/kg/day (Stavric et al., 1988). While the dose in our rats is higher than that consumed by humans, the half-life of caffeine in the rat is ~1 h whereas in humans it is ~5 h (Xu et al., 2010). Thus, a higher dose in rats is needed to achieve blood or brain concentrations (~22 μM) similar to those achieved in the serum of coffee drinkers (Bienvenu et al., 1990; Costenla et al., 2010; Gandhi et al., 2010).
Neurochemistry
In rats used for neurochemistry and midbrain DA cell counts, brains were rapidly removed and sectioned at mid-hypothalamus. Left and right striata were dissected from the forebrain, weighed and frozen at −80°C until analyzed. The hind brain containing the substantia nigra (SN) was immersion fixed for immunohistochemistry. Striatal tyrosine hydroxylase (TH) was determined by ELISA and monoamines and metabolites determined by HPLC with electrochemical detection, as previously described (Alfinito et al., 2003).
Immunohistochemistry for nigral DA cells and activated microglia
The hind brain was used for performing TH+ cell counts using immunohistochemical methods similar to those described previously (Yazdani et al., 2006). Briefly, 30-μm-thick coronal sections were cut through the entire SN. Every fourth section through the rostral–caudal extent of the SN was stained with an antibody against TH (1:4000; Protos Biotech Corp.) to identify the DA neurons.
Some rats were transcardially perfused for evaluating microglial response and TH staining in both the striatum and the SN. Animals were deeply anesthetized and were transcardially perfused with 100 ml of 0.9% NaCl in 0.1M sodium phosphate buffer, pH 7.3, containing 50 units/ml heparin. This was followed by 500 ml of 4% paraformaldehyde in 0.1M sodium phosphate buffer, pH 7.3. The brains were dissected out and taken through a series of 10, 20, and 30% sucrose in 0.1M sodium phosphate buffer, pH 7.3, for cryoprotection. Cryostat sections were cut in the coronal plane at a thickness of 20 μm. Sections from the striatum and the SN were immunostained with an antibody against ED1 (1:100; AbD Serotec) to identify activated microglia. Some sections were double labeled by immunofluorescence for TH and ED1. Digital photographs were taken using a Zeiss Axioplan microscope (Carl Zeiss, Inc.) equipped with an epi-fluorescence illuminator and Axiovision software.
Stereology
StereoInvestigator software (version 9.0. MicroBrightfield Inc., Williston, VT) was used to count TH-immunoreactive (TH-IR) SN cells. Cells were counted with a 40x objective using a Leica DMRE microscope. The cell counting frame was 50 × 50 × 5 μm with a 2 μm upper and lower guard zone. A cell was defined as a TH-IR somata with a clearly visible unstained nucleus. The TH cell counts were taken from 6 sections, spaced 4 apart (120 μm) and 200–250 cells were counted in the SN on the left side of the brain. The SN region was defined according to previous anatomical demarcation in the rat (German and Manaye, 1993).
Statistical analysis
Differences among means were analyzed using one-way analysis of variance (ANOVA) or two-way ANOVA. Two-way ANOVA revealed significant differences between treatment groups and sides (left lesioned and right non-lesioned sides). However, one-way ANOVA on data from the right sides revealed no significant differences across treatment groups allowing use of one-way ANOVA for comparisons in the left lesioned side across treatment groups. When ANOVA showed significant differences, comparisons between means were tested by the Tukey–Kramer or Bonferoni multiple comparisons post hoc test. In all analyses, the null hypothesis was rejected at the 0.05 level. All values are expressed as the mean ± SEM.
RESULTS
Loss of striatal DA and TH is linear over time with continuous icv MPP+ administration
Reductions in the content of DA and TH in the striatum ipsilateral to the infusion progressed over time in rats infused with MPP+ (75 μg/day) into the left cerebral ventricle (Fig 1). Because no significant reductions in these measures were seen in the contralateral striata in any of the groups, the data are plotted as left/right ratios to simplify data presentation. um. Reductions in DA in the left striatum (plotted as left/right ratios) were significant by 2 weeks with progressively greater reductions seen in subsequent weeks (Fig. 1). TH loss occurred more slowly, but the loss of both TH and DA exhibited significant correlation coefficients over time; r2 values of 0.98 and 0.99, respectively (p<.0001). Thus, MPP+ infusion produces a progressive loss of striatal DA and TH making this model ideal for evaluating neuroprotective agents when administered before or during the period of MPP+ insult.
Fig. 1. Continuous icv MPP+ administration produces a progressive and linear reduction in striatal DA and TH.
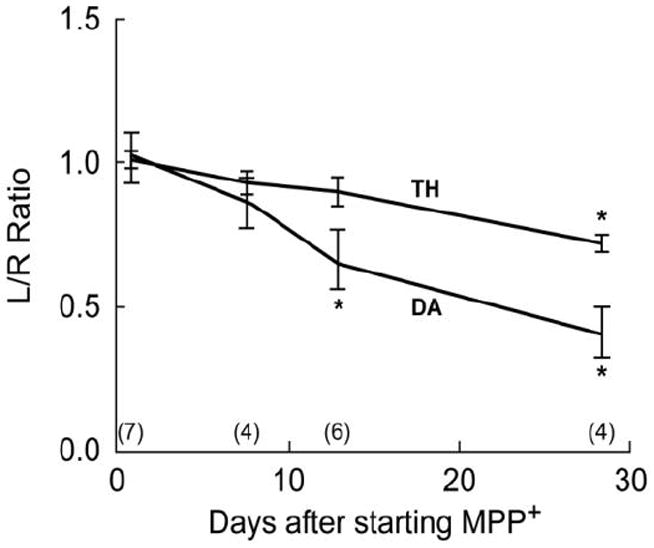
Rats received continuous MPP+ infusions (75 μg/day) into the left cerebral ventricle and were killed at 7, 14, or 28 days after starting the MPP+ infusion. DA and TH protein were measured in both the left and right striatum. Results are plotted as the ratio of DA or TH content in the left striatum to the right striatum (L/R Ratio) as a function of days of MPP+ exposure from the number of rats indicated in parenthesis. Data at time 0 are L/R ratios from naïve rats. DA and TH content in the right striata of the treatment groups did not differ significantly from right striata of naïve rats nor did they differ across the treatment groups. Linear regression analysis showed correlation coefficient r2 values of 0.98 (p<0.01) for DA and of 0.99 (p<0.01) for TH. *P<0.01 vs L/R ratio in naive rats.
Neither MPP+ nor caffeine treatment alters rat body weights
As shown in Table 1, there were no significant differences in the end-of-study body weights in the different groups of rats (naïve, vehicle-treated, MPP+ or caffeine plus MPP+). These data indicate that neither MPP+ nor caffeine treatment had an adverse effect on weight gain in the rats.
Table 1.
Treatment effects on body weight. Rats were weighed at the end of the study (28 days after surgery). Results are the mean ± SD of the number of rats shown in parenthesis. Treatment did not significantly alter weight gain over the 28-day course of treatment.
| Treatment Group | Weight (gms) |
|---|---|
| Naives | 418 ± 28 (10) |
| Vehicle-infused | 403 ± 18 (8) |
| MPP+ | 408 ± 19 (6) |
| Caffeine 1 wk/MPP+ | 395 ± 48 (7) |
| Caffeine 3 wk/MPP+ | 397 ± 46 (5) |
Caffeine treatment initiated simultaneously or during the course of ongoing neurodegeneration reduces loss of nigral DA neurons
MPP+ produced a significant reduction in the number of TH-immunostained cells (49 ± 9%) in the left SN of animals killed 27-28 days after starting infusion (Fig. 2). Oral caffeine (1 g/l in the drinking water) from the onset of MPP+ infusions prevented the loss of the nigral TH-immunostained cell bodies. More importantly, supplying caffeine at 1 or 3 weeks after initiating MPP+ infusions also reduced the loss of nigral TH-immunostained cells. These time points (1 and 3 wks) were selected for starting caffeine treatment as they represent early and later stages of loss of striatal DA based upon results shown in Fig. 1. These data demonstrate that degeneration of nigral DA neurons can be halted or slowed even after the neurodegenerative process has begun.
Fig. 2. Caffeine protects nigral DA neurons when administered prior to or during the course of neurodegeneration initiated by MPP+.
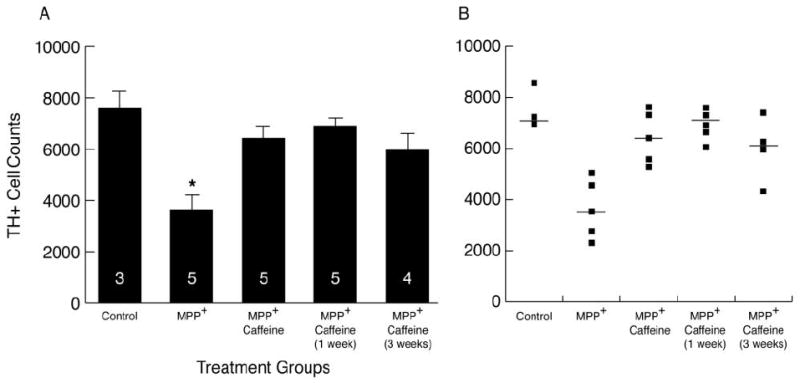
Rats were infused with MPP+ (75 μg/day) for 4 wks. Caffeine in the drinking water (1 g/l) was provided from the start of the infusion, or beginning 1 or 3 weeks later. (A) Results are the mean ± SEM of TH+ cell counts in the left SN, with the number of rats indicated in the columns. Controls are naïve untreated rats. When caffeine was given along with MPP+, or delayed by 1 or 3 weeks after beginning MPP+ infusions, there was less neurodegeneration of nigral DA neurons compared to MPP+ given alone. *P<0.01 control vs. group treated with only MPP+. (B) Data are plotted from individual animals, and also show the median cell count (horizontal line) for each of the 5 treatment groups.
Caffeine does not modify MPP+-induced decreases in striatal DA or TH
In contrast to the protection seen in the SN, caffeine did not significantly modify the MPP+-induced changes in the striatum. MPP+ produced significant reductions in TH (by 35%), DA (by 50%), DOPAC (by 56%) and HVA (by 61%) in the left striatum as compared to the left striatum in naive rats (see Fig.3A). The administration of chronic caffeine did not modify TH, DA or DOPAC in the right non-lesioned striatum indicating that the caffeine treatment did not modify synthesis or turnover of DA (Fig. 3B). In rats treated with caffeine and MPP+, the reductions in DA and TH in the left striatum was generally less than in the MPP+ group but did not, however, differ significantly from the MPP+ group. In vehicle-treated rats, there were no significant neurochemical differences between left and right striata (L/R ratios for TH: 1.12 ± 0.19 and for DA: 1.05 ± 0.18. mean ± SD; 5 rats) indicating that cannula placement did not significantly damage DA nerve terminals. Fig. 3 also illustrates that the striatal serotonin system was not affected by MPP+ administration (no significant loss of 5HT or 5HIAA in the MPP+-treated rats), indicating selectivity of this dose of MPP+ towards DA neurons as has been previously reported (Yazdani et al., 2006).
Fig. 3. Caffeine does not alter the MPP+-induced reductions in striatal TH, DA or DA metabolites.
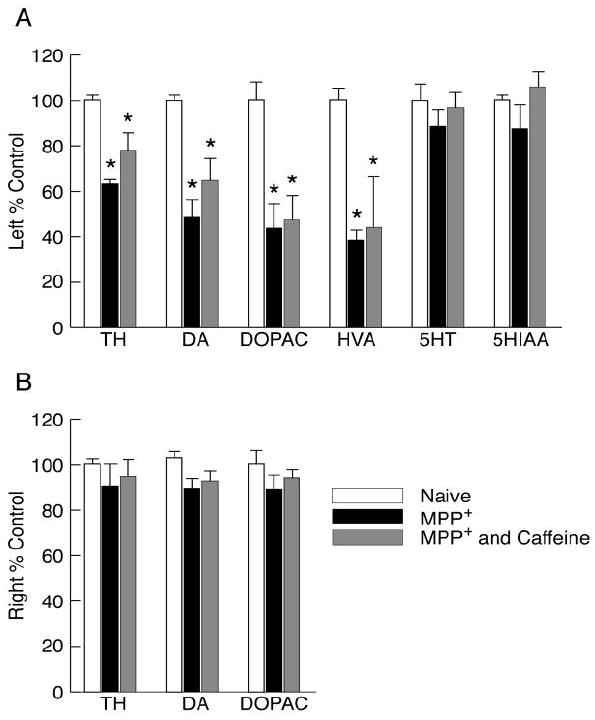
Rats were treated as described in Fig.2. (A) Results are from the left striatum and are presented as the % of control ± SEM (naives, n=3; MPP+, n=7; simultaneous MPP+ and caffeine, n=6). Control values are (mean ± SEM in ng/mg tissue): TH, 174 ± 6; DA, 16.7 ± 0.3; DOPAC, 1.7 ± 0.1; HVA, 1.6 ± 0.1; 5HT, 0.8 ± 0.1; 5HIAA, 1.6 ± 0.1. * p<0.05 vs. respective side of controls. (B) Results are presented as % control from right sides of naïve rats or rats treated with MPP+ or MPP+ and caffeine.
Caffeine attenuates microglia activation in the SN but not in the striatum of MPP+-treated rats
To evaluate the microglial response, brain sections containing the SN or striatum were immunostained with an antibody to ED1 which detects activated microglia and were counterstained with a TH antibody. Immunohistochemical staining revealed increased numbers of activated microglia in the left SN and left striatum of the MPP+-treated and the caffeine/MPP+-treated rats. Figure 4 illustrates data from representative animals in the two groups (n=2 rats/group). In the photomicrograph in the upper panel, ED1 immunostained cells were prevalent in the left SN whereas only very few were seen in the right SN, as would be expected in non-lesioned or non-damaged tissue. It can also be seen that there is a loss of TH-immunostained cells, especially in the medial region of the SN. In rats treated with MPP+ and caffeine, there were fewer cells that immunostained for ED1. Additionally, there is preservation of TH-immunostained cells in the rats treated with caffeine and MPP+.
Fig. 4. Caffeine appears to attenuate the microglia response in the SN but not in the striatum.
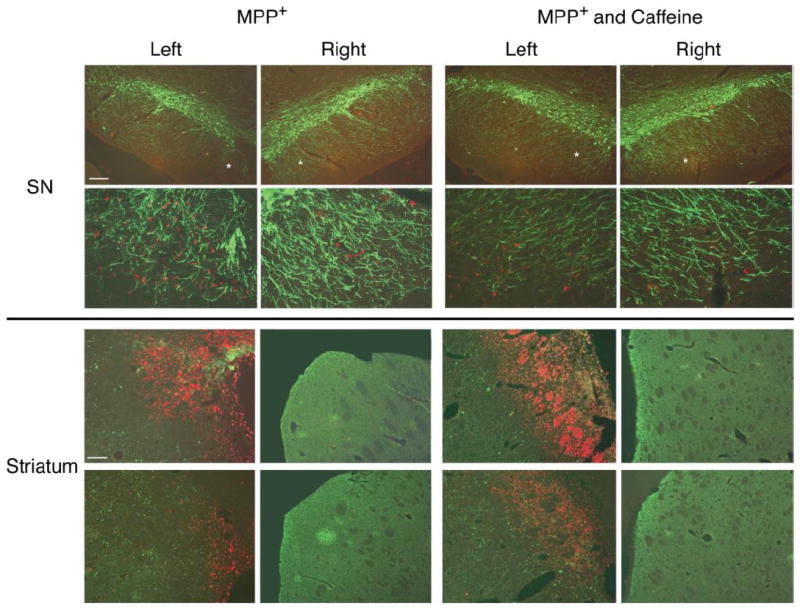
Rats were treated as described in Fig. 2 except that caffeine treatment was started 1 week after onset of MPP+ infusion. ED1 immunostaining for microglia is red. TH+ immunostaining for DA neurons is green. (Upper panels) Representative photomicrograph from one of two rats treated with only MPP+. Left side is ipsilateral to the icv infusion. Top photmicrographs are at lower magnification. ED1 immunostained cells are prominently noted in the left side whereas few are seen in the right side. Also, note the reduction of TH+ cells in the medial region of the SN on the left side as compared to the right SN. The findings in the second rat were similar. Representative photomicrographs from one of two rats that received both caffeine and MPP+. Note that there are fewer ED1 immunostained cells in the left SN region in the MPP+ and caffeine treated rat than in the MPP+ rat. Also note the preservation of TH+ cells in the medial region of the SN as compared to MPP+ rats. Similar findings were observed in the second rat. (Lower panels) Caffeine does not attenuate the microglia response in the striatum. Areas shown are from sections at two different rostral-caudal levels from one rat; the upper photomicrographs are from near the icv cannula placement region whereas the lower photomicrographs are from sections rostral to cannula placement. Note intense ED1 staining in the left striatum of the MPP+-treated rat near the cannula placement site with much less intense staining at the more rostral site. Note absence of ED1 staining in the right striatum. Also note loss of TH staining in the left striatum vs. the right striatum. The rat treated with MPP+ and caffeine shows similar ED1 staining as in the rat treated with only MPP+, but the TH staining is somewhat more intense in this animal vs. MPP+ alone. As in the MPP+ treated rat, there is a paucity of ED1 staining in the right striatum.
In the striatum, there was intense ED1 immunostaining, particularly in regions near the ventricle, in the MPP+-treated rats. Very few ED1 immunostained cells are seen in the right striatum, consistent with the lack of damage in the non-lesioned striatum. Also, in the right striatum, there is intense TH immunostaining whereas in the left striatum, TH immunostaining is markedly reduced. In rats that were treated with MPP+ and caffeine, the ED1 immunostaining in cells in the left striatum appear similar to that seen in rats treated with only MPP+. Likewise, the reduced striatal TH staining in rats treated with MPP+ and caffeine is similar to that seen in rats treated with only MPP+. We also found that the expression of glial markers (GFAP for astrocytes and MAC-1 for activated microglia) were elevated by approximately 3-fold in the left striata of MPP+ treated rats and that caffeine treatment did not significantly modify these effects (data not shown). We note that these are preliminary findings obtained from a small number of animals and that the effect of caffeine on the microglia response requires further characterization and quantification.
Discussion
Arresting the progression of neurodegeneration in PD remains a critical, unmet goal and is a genuine challenge to research in PD. We have found that caffeine treatment protects against the loss of nigral DA neurons in a chronic progressive rat model of PD. Most importantly, caffeine treatment was protective even when introduced late into the neurodegenerative process suggesting that it may be capable of arresting or slowing neurodegeneration. To our knowledge, this is the first evidence that delayed pharmacological therapy can retard degeneration of DA neurons.
The chronic MPP+ rat model provides an excellent progressive model of neurodegeneration. We have previously shown a progressive loss over time of TH-immunostained neurons in the SN ipsilateral to the infusion and that the reduction in the number of neurons is due to loss of TH-containing cell bodies and not just to loss of TH immunostaining as confirmed by counting Nissl-stained non-TH-immunostained neurons (Yazdani et al., 2006). We now show that the decline of DA content and TH protein in the ipsilateral striatum is linear over the 4-week time period studied, with the loss of TH being less than DA. There was a significant loss of striatal DA after 2 weeks of MPP+ treatment, but striatal TH was not significantly reduced until 4 weeks of treatment. Interestingly, when caffeine was given after 1 and 3 weeks of MPP+ treatment, when striatal TH was still minimally affected compared to DA, caffeine reduced the degeneration of nigral DA neurons seen after 4 weeks of MPP+ treatment. These data suggest that: (a) loss of striatal DA nerve terminal function occurs before loss of the nigral DA neurons; (b) nigral neurodegeneration becomes apparent at about 3 weeks after MPP+ treatment; and (c) caffeine given at or before this time blocks the nigral neurodegenerative process without restoring the striatal nerve terminal neurochemistry. Thus, after the neurodegenerative process has begun, as indicated by the striatal DA neurochemical reductions, initiation of caffeine treatment can still block the loss of nigral DA neurons.
Neuroprotection by caffeine in the MPP+ rat model is seen primarily at the level of the DA cell bodies in the SN. The reasons for the regional differences in protection by caffeine are not known. One possibility is that the icv route exposes the striatum to higher MPP+ concentrations than the SN and thus to a greater toxic insult. However, pharmacological protection by several diverse compounds is greater in the SN than in the striatum in mice treated systemically with acute or subacute doses of MPTP. For example, pharmacological intervention with adenosine A2A receptor antagonists, rosglitazone (an agonist at peroxisome proliferators-activated receptor-gamma), or an inhibitor of monoacylglycerol lipase (which reduces brain prostaglandin synthesis) completely protect against loss of nigral TH immunostained neurons but only minimally protect the striatal DA nerve terminals from MPTP (Dehmer et al., 2000; Nomura et al., 2011; Pierri et al., 2005; Schintu et al., 2009; Yu et al., 2008). These findings may indicate a region-selective effect of the drugs or alternatively that the striatal DA nerve terminals are much more sensitive to MPTP/MPP+. In mice with targeted mitochondrial damage to DA neurons, loss of striatal DA markers occurs long before any loss of nigral DA cell bodies, suggesting that the DA nerve terminals are particularly sensitive to mitochondrial dysfunction (Pickrell et al., 2011). Inhibition of complex I of the mitochondrial electron transport chain and hence of mitochondrial function by MPP+ is a principal mechanism underlying neurodegeneration in DA neurons (Vyas et al., 1986). Recent data indicate that MPP+ also damages mitochondrial transport in DA axons (Kim-Han et al., 2011), providing another possible explanation for why the DA nerve terminals are more vulnerable to MPP+-induced damage than are the cell bodies in the SN.
Caffeine treatment provides neuroprotection to DA neurons in the SN. Based on our preliminary data, we think that this neuroprotection is due to a diminished immune response in the SN although additional studies are required to further characterize and quantify the glial responses. We propose this because adenosine, which is prominently released in injured brain regions, mediates many of its neuroinflammatory actions through activation of A2A receptors on microglia (Fiebich et al., 1996; Hasko et al., 2008; Orr et al., 2009; Saura et al., 2005; Trincavelli et al., 2008). Caffeine or selective A2A receptor antagonists prevent or attenuate microglia responses such as proliferation, retard their recruitment to sites of injury and reduce the production of pro-inflammatory cytokines (Brothers et al., 2010; Rebola et al., 2011). Moreover, chronic caffeine partially reverses the microglia activation seen in the brains of older rats (Brothers et al., 2010). That caffeine’s protection may be mediated by its blockade of A2A receptors is supported by findings that A2A receptor antagonists or genetic ablation of A2A receptors protect nigral DA neurons and reduce neuroinflammation in MPTP-treated mice (Carta et al., 2009; Chen et al., 2001; Pierri et al., 2005; Yu et al., 2008). While the mechanism(s) by which caffeine treatment modifies the microglia response is not fully established, blockade of microglia A2A receptors is a candidate target site. Alternatively, it may be that the microglial response is not as pronounced in the caffeine-MPP+ treated rats because damage in the SN is considerably less than in the rats treated only with MPP+, thus leading to a much lower recruitment of microglial cells to the SN. Additional studies are needed to sort out this “chicken and egg” question.
In addition to A2A receptors located on microglia, neuronal A2A receptors may also participate in the neuroprotection afforded by caffeine. Stimulation of pre- and/or post-synaptic A2A receptors in the striatum drives activity in the striato-pallidal-subthalamic-nigral pathway (the indirect pathway) causing nigral glutamate release (Morelli et al., 2011). Nigral glutamate is elevated in animal PD models due to the loss of the inhibitory actions of DA on D2 receptors which then leave unopposed the excitatory drive by A2A receptors on the indirect pathway (ibid). Excessive stimulation of the nigral glutamatergic N-methyl-D-aspartate (NMDA) receptors located on DA neurons can be excitotoxic, especially to metabolically compromised DA neurons as would occur in the MPP+-treated rats. Selective blockade of nigral NMDA receptors protects DA neurons from metabolic stress, indicating the importance of nigral glutamate and excitotoxicity to DA neurons (Zeevalk et al., 2000). In the striatum, blockade of striatal post-synaptic A2A receptors reduces nigral glutamate release (Morelli et al., 2011). Indeed, in mice lacking neuronal A2A receptors in the forebrain, neurotoxicity by sub-acute MPTP is attenuated (Carta et al., 2009). Selective blockade of nigral A2A receptors also protects DA neurons against metabolic stress, an action thought to impact on nigral glutamate release (Alfinito et al., 2003). Thus, blocking striatal and/or nigral A2A receptors located on neurons may reduce nigral glutamate release and excitotoxic damage.
Caffeine metabolites may also contribute to neuroprotection. Paraxanthine (1,7-dimethylxanthine), which is the major metabolite, protects DA neurons in vitro and in vivo by non-adenosine receptor dependent mechanisms (Geraets et al., 2006; Guerreiro et al., 2008; Xu et al., 2010). In vitro studies demonstrate paraxanthine is a potent inhibitor of poly (ADP-ribose) polymerase-1 (PARP-1; at an IC50 of 15μM) and a less potent activator of ryanodine receptors (Geraets et al., 2006; Guerreiro et al., 2008).
Based on the data presented here, we propose that caffeine’s neuroprotection in the MPP+ rat model of PD is mediated, at least in part, by blockade of A2A receptors and an attenuation of neuroinflammation in the SN. If the effects of caffeine in our model are mediated by A2A receptors, then it is possible that the administration of A2A antagonists to PD patients would modify the neuropathology and slow disease progression. Selective A2A antagonists may be superior to caffeine as they would not block the neuroprotective effects of adenosine as mediated through A1 receptors (Alfinito et al., 2003). Clinical studies are currently underway to evaluate several A2A receptor antagonists for their effects both on symptom relief and in arresting disease progression (reviewed in Morelli et al., 2011). Based on our findings with caffeine, we would predict that the A2A antagonists should slow disease progression.
Highlights.
Chronic icv MPP+ infusion produces progressive loss of striatal dopamine markers
Caffeine treatment protects against MPP+-induced loss of nigral dopamine neurons
Delayed caffeine treatment rescues dopamine neurons from MPP+
Acknowledgments
This work was supported by NIH grants NS052733, NS058329 and ES005022 and in part by the James Webb Fund of the Dallas Foundation, the Dallas Area Parkinsonism Society. The authors thank Evelyn Ho for validation of the nigral DA cell count data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguiar LM, Nobre HV, Jr, Macedo DS, Oliveira AA, Freitas RM, Vasconcelos SM, Cunha GM, Sousa FC, Viana GS. Neuroprotective effects of caffeine in the model of 6-hydroxydopamine lesion in rats. Pharmacol Biochem Behav. 2006;84:415–419. doi: 10.1016/j.pbb.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Alfinito PD, Wang SP, Manzino L, Rijhsinghani S, Zeevalk GD, Sonsalla PK. Adenosinergic protection of dopaminergic and GABAergic neurons against mitochondrial inhibition through receptors located in the substantia nigra and striatum, respectively. J Neurosci. 2003;23:10982–10987. doi: 10.1523/JNEUROSCI.23-34-10982.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel SH, Beers DR, Henkel JS. T cell-microglial dialogue in Parkinson’s disease and amyotrophic lateral sclerosis: are we listening? Trends in Immunology. 2009;31:7–17. doi: 10.1016/j.it.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu T, Pons G, Rey E, Richard MO, d’Athis P, Olive G. Effect of hypophysectomy on caffeine elimination in rats. Fundam Clin Pharmacol. 1990;4:393–399. doi: 10.1111/j.1472-8206.1990.tb00693.x. [DOI] [PubMed] [Google Scholar]
- Brothers HM, Marchalant Y, Wenk GL. Caffeine attenuates lipopolysaccharide-induced neuroinflammation. Neurosci Lett. 2010;480:97–100. doi: 10.1016/j.neulet.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta AR, Kachroo A, Schintu N, Xu K, Schwarzschild MA, Wardas J, Morelli M. Inactivation of neuronal forebrain A receptors protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neurochem. 2009;111:1478–1489. doi: 10.1111/j.1471-4159.2009.06425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Xu K, Petzer JP, Staal R, Xu YH, Beilstein M, Sonsalla PK, Castagnoli K, Castagnoli N, Jr, Schwarzschild MA. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J Neurosci. 2001;21:RC143. doi: 10.1523/JNEUROSCI.21-10-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costenla AR, Cunha RA, de Mendonca A. Caffeine, adenosine receptors, and synaptic plasticity. Journal of Alzheimer’s Disease. 2010;20(Suppl 1):S25–34. doi: 10.3233/JAD-2010-091384. [DOI] [PubMed] [Google Scholar]
- Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB. Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J Neurochem. 2000;74:2213–2216. doi: 10.1046/j.1471-4159.2000.0742213.x. [DOI] [PubMed] [Google Scholar]
- Fiebich BL, Biber K, Lieb K, van Calker D, Berger M, Bauer J, Gebicke-Haerter PJ. Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2a-receptors. Glia. 1996;18:152–160. doi: 10.1002/(SICI)1098-1136(199610)18:2<152::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Gandhi KK, Williams JM, Menza M, Galazyn M, Benowitz NL. Higher serum caffeine in smokers with schizophrenia compared to smoking controls. Drug Alcohol Depend. 2010;110:151–155. doi: 10.1016/j.drugalcdep.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraets L, Moonen HJJ, Wouters EFM, Bast A, Hageman GJ. Caffeine metabolites are inhibitors of the nuclear enzyme poly(ADP-ribose)polymerase-1 at physiological concentrations. Biochem Pharmacol. 2006;72:902–910. doi: 10.1016/j.bcp.2006.06.023. [DOI] [PubMed] [Google Scholar]
- German DC, Manaye KF. Midbrain dopaminergic neurons (nuclei A8, A9, and A10): three-dimensional reconstruction in the rat. J Comp Neurol. 1993;331:297–309. doi: 10.1002/cne.903310302. [DOI] [PubMed] [Google Scholar]
- Guerreiro S, Toulorge D, Hirsch E, Marien M, Sokoloff P, Michel PP. Paraxanthine, the primary metabolite of caffeine, provides protection against dopaminergic cell death via stimulation of ryanodine receptor channels. Mol Pharmacol. 2008;74:980–989. doi: 10.1124/mol.108.048207. [DOI] [PubMed] [Google Scholar]
- Hasko G, Linden J, Cronstein B, Pacher P, Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nature Reviews Drug Discovery. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurology. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. The Neurobiology of Dopamine. In: Horn AS, Korf J, Westerink BHC, editors. Brain dopamine in Parkinson’s disease and other neurological disturbances. Academic Press; 1979. pp. 633–654. [Google Scholar]
- Joghataie MT, Roghani M, Negahdar F, Hashemi L. Protective effect of caffeine against neurodegeneration in a model of Parkinson’s disease in rat: behavioral and histochemical evidence. Parkinsonism & Related Disorders. 2004;10:465–468. doi: 10.1016/j.parkreldis.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Kachroo A, Irizarry MC, Schwarzschild MA. Caffeine protects against combined paraquat and maneb-induced dopaminergic neuron degeneration. Exp Neurol. 2010;223:657–661. doi: 10.1016/j.expneurol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalda A, Yu L, Oztas E, Chen J-F. Novel neuroprotection by caffeine and adenosine A(2A) receptor antagonists in animal models of Parkinson’s disease. J Neurol Sci. 2006;248:9–15. doi: 10.1016/j.jns.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Kim-Han JS, Antenor-Dorsey JA, O’Malley KL. The Parkinsonian mimetic, MPP+, specifically impairs mitochondrial transport in dopamine axons. J Neurosci. 2011;31(19):7212–7221. doi: 10.1523/JNEUROSCI.0711-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli M, Carta AR, Kachroo A, Schwarzschild MA. Pathophysiological roles for purines: adenosine, caffeine and urate. Prog Brain Res. 2011;183:183–208. doi: 10.1016/S0079-6123(10)83010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MCG, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr AG, Orr AL, Li X-J, Gross RE, Traynelis SF. Adenosine A(2A) receptor mediates microglial process retraction. Nat Neurosci. 2009;12:872–878. doi: 10.1038/nn.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates-Second Edition. Academinc Press, Inc; Orlando, FL: 1986. [Google Scholar]
- Pickrell AM, Pinto M, Hida A, Moraes CT. Striatal Dysfunctions Associated with Mitochondrial DNA Damage in Dopaminergic Neurons in a Mouse Model of Parkinson’s Disease. The Journal of Neuroscience. 2011;31:17649–17658. doi: 10.1523/JNEUROSCI.4871-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierri M, Vaudano E, Sager T, Englund U. KW-6002 protects from MPTP induced dopaminergic toxicity in the mouse. Neuropharmacology. 2005;48:517–524. doi: 10.1016/j.neuropharm.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Rebola N, Simões AP, Canas PM, Tomé AR, Andrade GM, Barry CE, Agostinho PM, Lynch MA, Cunha RA. Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. J Neurochem. 2011;117:100–111. doi: 10.1111/j.1471-4159.2011.07178.x. [DOI] [PubMed] [Google Scholar]
- Saura J, Angulo E, Ejarque A, Casado V, Tusell JM, Moratalla R, Chen JF, Schwarzschild MA, Lluis C, Franco R, Serratosa J. Adenosine A2A receptor stimulation potentiates nitric oxide release by activated microglia. J Neurochem. 2005;95:919–929. doi: 10.1111/j.1471-4159.2005.03395.x. [DOI] [PubMed] [Google Scholar]
- Schintu N, Frau L, Ibba M, Caboni P, Garau A, Carboni E, Carta AR, Schintu N, Frau L, Ibba M, Caboni P, Garau A, Carboni E, Carta AR. PPAR-gamma-mediated neuroprotection in a chronic mouse model of Parkinson’s disease. Eur J Neurosci. 2009;29:954–963. doi: 10.1111/j.1460-9568.2009.06657.x. [DOI] [PubMed] [Google Scholar]
- Stavric B, Klassen R, Watkinson B, Karpinski K, Stapley R, Fried P. Variability in caffeine consumption from coffee and tea: possible significance for epidemiological studies. Food Chem Toxicol. 1988;26:111–118. doi: 10.1016/0278-6915(88)90107-x. [DOI] [PubMed] [Google Scholar]
- Trincavelli ML, Melani A, Guidi S, Cuboni S, Cipriani S, Pedata F, Martini C. Regulation of A(2A) adenosine receptor expression and functioning following permanent focal ischemia in rat brain. J Neurochem. 2008;104:479–490. doi: 10.1111/j.1471-4159.2007.04990.x. [DOI] [PubMed] [Google Scholar]
- Vyas I, Heikkila RE, Nicklas WJ. Studies on the neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: inhibition of NAD-linked substrate oxidation by its metabolite, 1-methyl-4-phenylpyridinium. J Neurochem. 1986;46:1501–1507. doi: 10.1111/j.1471-4159.1986.tb01768.x. [DOI] [PubMed] [Google Scholar]
- Xu K, Xu YH, Chen JF, Schwarzschild MA. Neuroprotection by caffeine: time course and role of its metabolites in the MPTP model of Parkinson’s disease. Neuroscience. 2010;167:475–481. doi: 10.1016/j.neuroscience.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani U, German DC, Liang CL, Manzino L, Sonsalla PK, Zeevalk GD. Rat model of Parkinson’s disease: chronic central delivery of 1-methyl-4-phenylpyridinium (MPP+) Exp Neurol. 2006;200:172–183. doi: 10.1016/j.expneurol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Yu L, Shen H-Y, Coelho JE, Araujo IM, Huang Q-Y, Day Y-J, Rebola N, Canas PM, Rapp EK, Ferrara J, Taylor D, Muller CE, Linden J, Cunha RA, Chen J-F. Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann Neurol. 2008;63:338–346. doi: 10.1002/ana.21313. [DOI] [PubMed] [Google Scholar]
- Zeevalk GD, Manzino L, Sonsalla PK. NMDA receptors modulate dopamine loss due to energy impairment in the substantia nigra but not striatum. Exp Neurol. 2000;161:638–646. doi: 10.1006/exnr.1999.7283. [DOI] [PubMed] [Google Scholar]
- Zeevalk GD, Manzino L, Sonsalla PK, Bernard LP. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: relevance to Parkinson’s disease. Exp Neurol. 2007;203:512–520. doi: 10.1016/j.expneurol.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]