

Background
Celecoxib is a nonsteroidal anti-inflammatory drug (NSAID) with anti-inflammatory, analgestic, and antipyretic properties. It is approved for the treatment of osteoarthritis, rheumatoid arthritis, ankylosing spondylitis, and acute pain [1-3]. Celecoxib has also shown promise in prevention of cancer, and has been used as an adjunct to surgery to reduce the number of adenomatous colorectal polyps in patients with the hereditary colon cancer susceptibility syndrome, familial adenomatous polyposis (FAP) [4-6]. The anti-inflammatory and pain-relieving properties of celecoxib result from inhibition of prostaglandin (PG) synthesis by selective inhibition of PG G/H synthase-2 (encoded by gene PTGS2). The two PTGS isoforms, PTGS1 and PTGS2, are bisfunctional enzymes with both cyclooxygenase (COX) and hydroperoxidase activities, but they are commonly referred to as COX; see ‘Pharmacodynamics’ section) [1,7,8]. Celecoxib is a member of the subclass of NSAIDs, which were purposefully designed as COX-2-selective inhibitors (pdCOX-2 inhibitors) and that are frequently called coxibs [9,10]. Most traditional NSAIDs (tNSAIDs) inhibit both COX isoforms; however, some of them show a degree of COX-2 selectivity that is similar to that of celecoxib, although they were developed before COX-2 was discovered [11]. pdCOX-2 inhibitors provide anti-inflammatory effects that are comparable with tNSAIDs that inhibit both COX isoforms while reducing the risk of serious gastrointestinal toxicity.
Following its introduction to US market in December 1998, celecoxib quickly became one of the most frequently prescribed drugs for the relief of pain and inflammation [12], although the data supporting a favorable gastrointestinal toxicity profile were much weaker than those of other compounds within the class [13]. Celecoxib, as well as other selective and nonselective NSAIDs, have been under intense scrutiny since 2004, when two pdCOX-2-selective inhibitors, rofecoxib (VIOXx) and valdecoxib, were withdrawn from the market due to an increased risk of cardiovascular events including myocardial infarction [14-16]. Etoricoxib and lumiracoxib were never approved in the US due to cardiovascular safety concerns. Celecoxib is the only pdCOX-2 inhibitor currently available in the US. For many patients with both severe arthritis and intolerance to nonselective NSAIDs due to gastrointestinal side effects, pdCOX-2 inhibitors provide significant clinical benefit. The clinical care of patients requiring anti-inflammatory pain therapy, as well as those at high risk of colorectal adenomas, would be greatly aided by measurements that identify the patients who will benefit from celecoxib, yet not suffer adverse events. This summary briefly reviews the pharmacokinetics of celecoxib (Fig. 1) and discusses the candidate genes mediating the diverse pharmacological profile of celecoxib (Fig. 2). Knowledge of the pharmacogenomics of these pathways may help to achieve personalization and optimization of celecoxib therapy.
Fig. 1.
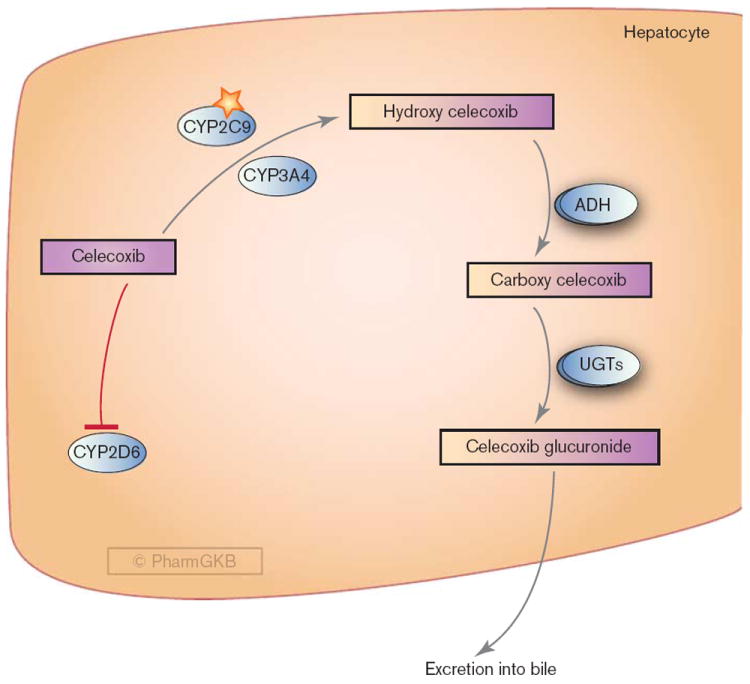
Hepatic metabolism of celecoxib. ADH, alcohol dehydrogenases; UGTs, UDP glucuronosyltransferases. A fully interactive version is available online at http://www.pharmgkb.org/pathway/PA165816736.
Fig. 2.
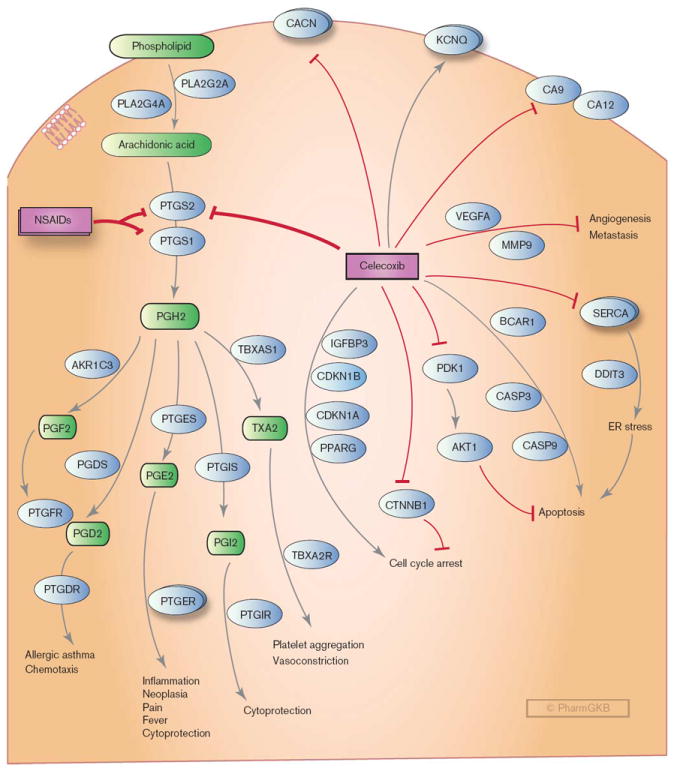
Stylized cell depicting the mechanism of action of celecoxib and candidate genes interacting with celecoxib and involved in the proposed anticancer mechanisms of celecoxib, including induction of apoptosis, cell cycle arrest, regulation of angiogenesis, and induction of endoplasmic reticulum (ER) stress. CACN: L-type calcium channels; KCNQ: voltage-gated potassium channels; MMP9, metalloproteinase; NSAIDs, nonsteroidal anti-inflammatory drugs; PGH2, prostaglandin H2; PGE2, prostaglandin E2; PGI2, prostacyclin; PGD2, prostaglandin D2; PGF2, prostaglandin F2; PTGER, prostaglandin E receptors; SERCA, sarcoplasmic/ER calcium ATPases; TXA2, thromboxane A2; VEGFA, vascular endothelial cell growth factor. A fully interactive version is available online at http://www.pharmgkb.org/pathway/PA152241951.
Pharmacokinetics
After oral administration, celecoxib is rapidly absorbed and achieves peak serum concentration in approximately 3 h. It is extensively metabolized in the liver, with very little drug (< 3%) being eliminated unchanged [17]. The major routes of excretion for celecoxib are feces and urine [18]. Celecoxib is metabolized primarily through methyl hydroxylation to form hydroxycelecoxib. This reaction is largely catalyzed by CYP2C9, although CYP3A4 also plays a minor (< 25%) role [7,17,19] (Fig. 1). Hydroxycelecoxib is further oxidized to form carboxycelecoxib by cytosolic alcohol dehydrogenases ADH1 and ADH2 [19], then conjugated with glucuronic acid by UDP glucuronosyltransferases to form the 1-O-glucuronide. None of the metabolites are pharmacologically active [7].
As celecoxib metabolism is predominantly mediated by CYP2C9, polymorphisms in CYP2C9 are likely to have a direct impact on celecoxib pharmacokinetics and variability in drug responses. Individuals who are poor metabolizers of CYP2C9 substrates (e.g. CYP2C9*3 allele carriers) have an increased exposure to celecoxib when compared with those with normal CYP2C9 activity [19-21] (see ‘Pharmacogenomics’ section). Drugs that inhibit CYP2C9 should therefore be used with caution in patients taking celecoxib.
Although not a substrate of CYP2D6, celecoxib inhibits this metabolic enzyme [22]. Drugs that are metabolized by CYP2D6 (e.g. metoprolol [22]) should also be used with caution in patients receiving celecoxib due to a potential risk of drug interaction.
Pharmacodynamics
Celecoxib exerts its anti-inflammatory and analgesic activities through blocking the synthesis of various inflammatory prostanoids (PG) [7,8,23]. The prostanoids, which include PGs and thromboxane, are the end products of fatty acid metabolism produced by tissue-specific COX enzymatic activity. These products are important physiological and pathological mediators that are involved in a wide range of biological processes including inflammation, pain, cancer, glaucoma, osteoporosis, cardiovascular diseases, and asthma [24]. The production of the prostanoids (PG) is dependent on the availability of arachidonic acid (AA). Following stimulation of the cell membrane by inflammatory or mitogenic signals, the first step in PG synthesis is the release of AA from the cellular phospholipids through the action of either secretory (sPLA2, encoded by gene PLA2G2A) or cytoplasmic (cPLA2, encoded by gene PLA2G4A) phospholipases. Once AA is released, the two isoenzymes, COX-1 (encoded by PTGS1) and COX-2 (encoded by PTGS2), catalyze the production of the prostanoids (Fig. 2) [25]. As indicated above, this involves two sequential reactions. The initial COX reaction converts AA into PGG2. The second reaction reduces PGG2 to PGH2. PGH2 is then converted into active metabolites PGE2, prostacyclin (PGI2), thromboxane (T×A2), PGD2, and PGF2α by the action of tissue-specific PG synthases [26,27]. These active metabolites interact with specific prostanoid G-protein-coupled receptors to mediate diverse physiological responses, such as inflammation, fever, blood pressure regulation, clotting, and gastrointestinal protection.
The COX-1 and COX-2 enzymes exhibit distinct expression profiles and roles in physiological processes. COX-1 is constitutively expressed in many cell types and is the major COX isoform in gastric tissue. It is responsible for the protection of the gastric mucosa, which led to the development of the ‘COX-2 hypothesis’ that drugs targeted against COX-2 only would have reduced upper gastrointestinal toxicity [8]. Although COX-2 is highly inducible by inflammatory stimuli such as cytokines, growth factors, and tumor promoters [28-31], it is also constitutively expressed in some tissues, such as the vessel wall, the kidney, or the heart. Indeed, the depression of the physiological formation of COX-2-dependent prostanoids in these tissues has been identified as the molecular mechanism underlying the thrombotic cardiovascular complications of COX-2 inhibition [16]. Seven placebocontrolled, randomized trials with three chemically distinct pdCOX-2 inhibitors, including celecoxib, have documented the cardiovascular risk. Of note, celecoxib is now used at lower doses than in the trials that showed its cardiovascular hazard. Celecoxib has 30-fold greater inhibitory activity against COX-2 compared with COX-1, and inhibits COX-1 only minimally at therapeutic concentrations [32,33]. Although the selectivity for COX-2 measured in vitro is lower for celecoxib compared with other drugs in the coxib class (e.g. rofecoxib, valdecoxib, lumiracoxib, and etoricoxib), it is very similar at therapeutic concentrations in vivo. Celecoxib also retains more ability to inhibit COX-1 compared with other coxibs; however, the consequences of this with regard to its therapeutic efficacy and toxicity are not well understood [34-36].
Antineoplastic actions
Selective COX-2 inhibitors, especially celecoxib, have been evaluated as a potential cancer chemopreventive and therapeutic agent in clinical trials for various malignancies. Nonselective NSAIDs such as sulindac have been used since the 1980s as adjuncts to surgery for prevention of intestinal tumors in patients with FAP, a genetic condition that often leads to colorectal cancer [6]. Celecoxib has been shown to significantly reduce the number of colorectal polyps in patients with FAP as well as those with sporadic colorectal adenomas [4-6]. Celecoxib has also demonstrated anticancer effects in established invasive tumors, including colon carcinoma, lung carcinoma, and prostate cancer, both in vitro and in vivo [8,37-42]. The exact mechanisms for its anticancer activity are not clear and may involve both COX-dependent and COX-independent mechanisms [41,43,44]. A wide range of tumor-associated molecular events are modulated by celecoxib in in-vitro assays, but these have yet to be placed within a coherent context that clearly describes clinical responses and most COX-independent effects were only observed at supratherapeutic concentrations in vitro (reviewed by Grosch et al. [44]). Figure 2 shows that the candidate genes involved in the proposed anticarcinogenic mechanisms of celecoxib include induction of apoptosis, cell cycle arrest, regulation of angiogenesis, and induction of endoplasmic reticulum (ER) stress. Celecoxib-mediated inhibition of cell cycle progression has been observed in cell culture experiments along with an increased expression of cell cycle inhibitors, p21 (encoded by gene CDKN1A) and p27 (encoded by gene CDKN1B), and/or decreased expression of cyclins (encoded by gene CCNA1, CCNB1, and CCND1) [40,45-47]. Increased degradation of the oncoprotein, β-catenin (encoded by gene CTNNB1) is also observed in celecoxib-treated human colon cancer cells, and this is associated with marked reductions in tumor cell proliferation [48]. Again, a major caveat is that these studies were conducted at concentrations in vitro that were 10–100 times higher than plasma concentrations measured in humans. Induction of apoptosis by celecoxib is associated with activation of proapoptosis molecules such as caspases and CHOP (encoded by gene DDIT3) [49], as well as inhibition of antiapoptotic molecules, such as PDK1 (encoded by gene PDPK1) and its downstream target AKT1 [38,50-52]. Finally, inhibition of angiogenesis and tumor cell invasiveness may also contribute to the antitumor activity of celecoxib. Celecoxib treatment decreased the expression of vascular endothelial cell growth factor [53-55] and inhibition of matrix metalloproteinase 9 [56] in cancer tissues and cell lines.
Besides COX-2, celecoxib can directly bind to and inhibit a few other targets that may play important roles in the antitumor response mechanism. PDK1 is a direct target of celecoxib and inhibition of PDK1/Akt signaling correlated with celecoxib-induced apoptosis in both colon and prostate tumor cell lines [38,50]. However, significantly higher concentrations of celecoxib (IC50 in micromolar range) are required for inhibition of PDK1 compared with that required to inhibit COX-2 (IC50 in nanomolar range). Celecoxib also binds to and inhibits sarcoplasmic/ER calcium ATPase [57]. This binding can lead to rapid leakage of calcium into the cytosol, triggers ER stress, and ultimately leads to apoptosis [58]. This activity is highly specific for celecoxib and has not been associated with other COX inhibitors. Carbonic anhydrases (CA), enzymes that catalyze the reversible hydration of carbon dioxide, are also inhibited by celecoxib (IC50 in the low nanomolar range) [59,60]. Some of the CAs (e.g. CA9 and CA12) are associated with tumor development [61,62]. However, no study has clearly demonstrated the relationship between inhibition of CAs and anticancer activity of celecoxib, and celecoxib used at therapeutic concentrations also did not appear to have a clinically significant inhibitory action on renal CAs [63].
Cardiovascular toxicity
Data from clinical trials and case–control studies have associated the use of selective COX-2 inhibitors, rofecoxib, valdecoxib, and celecoxib with an increased incidence of myocardial infarction, stroke, and death due to cardiovascular causes [5,64-67]. These toxicities were uncovered as secondary endpoints during trials testing coxibs for colorectal adenoma prevention and arthritis treatment. The US Food and Drug Administration currently mandates black-box warnings of increased cardiovascular hazards for the entire NSAIDs class. Available data suggest that this risk may increase with the duration of use and may also vary by a patient’s individual baseline cardiovascular risk [16].
For celecoxib, the increased cardiovascular risk seems to be exposure dependent; both the dose and the dosing interval may be important factors in cardiovascular risk. In the Adenoma Prevention with Celecoxib (APC) trial, celecoxib 400 mg twice a day exhibited a greater than three-fold risk for combined endpoints of cardiovascular death, myocardial infarction, stroke, or heart failure compared with placebo, and 200 mg twice a day with a greater than two-fold risk [5,67]. Patients with higher baseline cardiovascular risk factors also tended to exhibit an increased risk. An evaluation of 5-year outcome data from the APC trial found a significant association between baseline cardiovascular risk factors and celecoxib-associated cardiovascular events [67]. The prevention of colorectal sporadic adenomatous polyps trial showed that the risk for cardiac disorders was higher in those taking celecoxib 400 mg once daily than in those on placebo [4,68]. In contrast, a number of clinical studies and a meta-analysis failed to demonstrate clear evidence of an increased thrombotic cardiovascular risk with celecoxib doses of less than or equal to 400 mg daily compared with placebo [42,69,70]. These analyses included data comparing coxibs with other nonselective NSAIDs. It is unclear whether nonselective NSAIDs also increase cardiovascular risk; therefore, these data cannot assess the relative safety of coxibs. In a more conclusive study, a pooled analysis of six randomized trials comparing celecoxib with placebo concluded that cardiovascular risk for celecoxib-treated patients increases with dose, and that a once-daily dose is associated with lower cardiovascular risk than the twice-daily dose [18,71]. Patients in the high baseline cardiovascular risk group exhibited a disproportionately higher risk of an adverse event, whereas celecoxib did not cause a significant increase in cardiovascular events in the low-risk group, suggesting the importance of considering baseline cardiovascular risk for appropriate patient selection. In response to these data, the American Heart Association recommends that patients treated with celecoxib use the lowest effective dose for the shortest duration to minimize the potential risk for an adverse cardiovascular event. Patients with existing cardiovascular disease or risk factors for cardiovascular disease may be at greater risk and alternative therapy should be considered.
The mechanism underlying the increased cardiovascular risk of COX-2 inhibition has been studied extensively. The clinical events associated with coxib cardiovascular toxicity are primarily thrombotic in nature. The depression of COX-2-derived cardioprotective PGs, particularly PGI2, and perhaps PGE2, by the coxibs removes a physiological restraint on mediators that induce thrombosis, increase blood pressure, and promote atherogenesis [8,16,24,72-74]. One of these mediators is TxA2, which is synthesized by COX-1 action in platelets. Long-term treatment with COX-2-specific inhibitors may create a prothrombotic environment and predispose patients to elevated cardiovascular risk. This may be particularly harmful for patients already predisposed to thrombosis due to the presence of atherosclerotic plaques in coronary or cerebral arteries. It is likely that the clinical safety of celecoxib may depend on a fine balance of multiple factors, especially given the complexity of the molecular system regulating atherothrombotic processes [75]. Inter-individual variability in drug metabolism, differences in the half-life of the drug, the effect on blood pressure [76], or endothelial function [77] may all contribute to the toxicity profile of coxibs or other NSAIDs. Simultaneous COX-1 inhibition may ameliorate the cardiovascular hazard of COX-2 inhibition [16]. For example, compared with other more selective COX-2 inhibitors (e.g. rofecoxib and valdecoxib), celecoxib has some activity against COX-1 and also seems to exhibit a relatively safer cardiovascular profile in randomized-controlled trials, especially when used at lower than 400 mg daily [69,78]. Observational studies suggest that the cardiovascular risk of the tNSAIDs is heterogeneous. Indeed, some tNSAIDs may have cardiovascular toxicity similar to that of the coxibs [79]. In addition to a simple dose–effect relationship, COX-independent ‘protective’ mechanisms have been discussed. For example, experiments showed that celecoxib, but not rofecoxib, caused a reduction in the excitability and contractility of cultured vascular smooth muscle cells and decreased vascular tone in vitro, by opening the voltage-gated KCNQ5 K + channels and blocking the L-type calcium channels [80,81]. However, research is needed to assess whether effects on ion channel can be observed in vivo and indeed result in vasodilation and reduced systemic blood pressure.
Pharmacogenomics
Multiple reports have shown that there is considerable variability both within and between individuals in the pharmacokinetic and pharmacodynamic responses toward celecoxib [34,35]. A large portion of the variability may be attributable to stable host factors including genetics. For example, polymorphisms in celecoxib-metabolizing enzymes or targets may affect its efficacy or toxicity. Several studies have reported associations between genomic variations of CYP2C9 and plasma drug levels of celecoxib, and a few have explored the role of other metabolizing enzymes and pharmacodynamic candidate genes/variants in drug efficacy, resistance, and adverse drug reactions. On the basis of data described below, celecoxib was one of the first drugs for which the manufacturers’ drug information recommended caution on the basis of pharmacogenetic information. The caution is raised for patients who are known or suspected to be poor CYP2C9 metabolizers and specifically states that clinicians should ‘consider starting treatment at half the lowest recommended dose in poor metabolizers (i.e. CYP2C9*3/*3), and consider using alternative management in patients with juvenile rheumatoid arthritis who are poor metabolizers’ (CELEBREXR Package Insert, http://pfizer.com/files/products/uspi_celebrex.pdf).
CYP2C9 and pharmacokinetics
Celecoxib is metabolized primarily by CYP2C9, a phase I metabolizing enzyme responsible for the clearance of many drugs. The gene encoding the CYP2C9 enzyme is highly polymorphic, including several functional variants of significant pharmacogenetic importance [82,83]. Among individuals of European ancestry, the two most common variants associated with reduced enzyme activity are CYP2C9*2 (rs1799853) and CYP2C9*3 (rs1057910) [82-84]. Both in-vitro and in-vivo studies found that celecoxib pharmacokinetics are altered in individuals carrying the CYP2C9 variant alleles [19-21,85,86]. In-vitro study showed that CYP2C9-dependent methyl hydroxylation of celecoxib decreased by 34 and 90% in the presence of cDNA-expressed CYP2C9*2 and CYP2C9*3, respectively [21]. In human liver microsomes, the rate of celecoxib hydroxylation decreased in cells with heterozygous CYP2C9*1/*3 (59% decrease) and CYP2C9*1/*2 (47% decrease) genotypes compared with those with CYP2C9*1/*1 [21]. There was also a marked reduction (up to 5.3 times) in hydroxycelecoxib formation in a liver sample with a homozygous CYP2C9*3/*3 genotype [20,21,86]. From two single-dose clinical studies (100 and 200 mg, respectively), the area under the curve (AUC) of celecoxib in individuals with CYP2C9*1/*3 and CYP2C9*3/*3 genotypes was more than or equal to two-fold higher than in those with the CYP2C9*1/*1 genotype [20,21]. Similar results were observed in a steady-state pharmacokinetic study with twice-daily doses of celecoxib (200 mg) [85]. Yet another study showed that the AUC in a pediatric patient with the CYP2C9*3/*3 genotype was more than 10-fold higher than that from two CYP2C9*1/*1 patients following a single celecoxib dose (250 mg/m2) [86]. The effect of CYP2C9*2 on celecoxib pharmacokinetics seems to be much weaker than that of CYP2C9*3 in most systems examined so far. Three studies showed that the CYP2C9*1/*2 genotype had minimal impact on celecoxib hydroxylation and clearance in vivo [20,21,85] and the two studies found that the AUC of celecoxib in CYP2C9*2/*2 patients did not differ statistically from the ones with the CYP2C9*1/*1 genotype [20,85]. Surprisingly, a placebo-controlled crossover study showed that the CYP2C9*2 variant was associated with elevated plasma concentrations of celecoxib (200 mg) 4 h after administration, whereas the CYP2C9*3 variant had no effect on plasma concentrations [34]. The impact of other CYP2C9 polymorphisms, such as *5, *6, *9, and *11, on the pharmacokinetics of celecoxib has not been evaluated.
CYP2C9 and clinical outcomes
Although the data presented above include some inconsistencies, it appears that the enzyme coded by the *2 allele, CYP2C9*2, shows a moderate decrease in activity, whereas the enzyme coded by the *3 allele, especially in the homozygous state, leads to a more marked decrease in activity when compared with the wild-type CYP2C9 (also called CYP2C9*1) [87]. Patients expressing one or more reduced function alleles of the enzyme may have reduced celecoxib clearance, resulting in increased drug exposure and a greater risk for side effects [84]. The strongest data for a clinically relevant association between the CYP2C9 genotype and celecoxib activity come from the APC trial. This study randomized 2035 patients with a high risk of colorectal adenoma formation to either low-dose celecoxib (200 mg, twice daily), high-dose celecoxib (400 mg, twice daily), or placebo. The primary endpoint of the trial was the occurrence of one or more colorectal adenomas during a 3-year on-treatment interval. This trial also evaluated the influence of CYP2C9*2 and *3 variants on celecoxib response [88]. Among all genotypes, celecoxib was associated with a dose-dependent reduction in adenoma, with high-dose celecoxib yielding a 5.6% greater reduction in the 3-year cumulative incidence of adenoma compared with low-dose celecoxib. However, the additional benefit of the higher dose was restricted to those with the CYP2C9*3 genotype (relative risk = 0.51; confidence interval = 0.30–0.87). Patients with the CYP2C9*3 genotype treated with high-dose celecoxib showed a 19.7% greater reduction in adenoma incidence than those treated with low-dose celecoxib. In contrast, the high dose was not associated with significant risk reduction among patients with CYP2C9*2 or wild-type genotypes. These data, therefore, showed that the beneficial effect of higher drug dose was realized only in the slow metabolizers.
There is limited evidence linking CYP2C9 genotypes to variability in celecoxib-induced toxicity, such as gastrointestinal bleeding and cardiovascular toxicity. In patients using celecoxib for acute arthritis management, the presence of CYP2C9*3 and CYP2C9*2 alleles was associated with a significantly higher risk of developing gastrointestinal bleeding [89-92]. However, these data were not conclusive, because the sample size of celecoxib users among all NSAIDs users included in these studies was small. There was no relationship found between the CYP2C9 variant genotype and the risk of gastrointestinal bleeding in patients from the large APC trial, which had the advantage of a placebo comparison arm [88]. The APC trial also examined the relationship between the CYP2C9 genotype and celecoxib-associated cardiovascular toxicity. Compared with placebo, a higher incidence of cardiovascular and thrombotic events was observed for those with the CYP2C9 variant genotype using high-dose celecoxib. Despite the considerable size of the trial, however, the overall cardiovascular toxicity event rate was low, making it impossible to determine whether or not the CYP2C9 genotype affected either the event rate or the dose response.
Pharmacodynamic candidate genes
There are limited data with regard to the impact of pharmacodynamic candidate genes on celecoxib response and toxicity. A study with 50 healthy volunteers showed that the Pro17Leu variant in the signal peptide of the COX-1 enzyme was associated with a failure by either celecoxib or rofecoxib to inhibit thromboxane formation [34]. Variants in COX-2 (PTGS2), the primary target of celecoxib, were also examined for their impact on celecoxib response in this study; however, no association was found between variant rs5273 in PTGS2 and any phenotype [34]. Another variant in PTGS2, − 765G > C (rs20417), did not modulate celecoxib effects on PG production in an ex-vivo whole blood assay [93]. Finally, 15-hydroxyprostaglandin dehydrogenase (15-PGDH, encoded by gene HPGD), an enzyme responsible for the metabolism of PGs and an inhibitor of the colonic COX-2 pathway, has been associated with resistance to celecoxib [94]. In mice lacking 15-PGDH, celecoxib adenoma preventive activity is abrogated. 15-PGDH levels are variable among adenoma patients, and low 15-PGDH levels correlated with celecoxib resistance in a small subset of patients with recurrent colon adenomas from the APC trial. Further work is needed to validate this result and uncover the genomic determinants of 15-PGDH expression.
Summary
The selective COX-2 inhibitor, celecoxib, shows a complex but important pharmacogenomics profile. The use of this highly effective anti-inflammatory and antitumor drug is limited by concerns about its potential for increased cardiovascular risk. Although the mechanism of action of celecoxib is well studied and many large clinical trials have examined its efficacy, we still lack a complete understanding of the sources of variability in the occurrence of adverse effects as well as the differential risk profile for all drugs in the NSAID class. Preliminary work showed that genetic factors influence response to celecoxib and may be useful in selecting individuals at risk for adverse events. However, the existing pharmacogenetic studies of celecoxib toxicity are still exploratory, with limited sample size and limited number of events. NSAIDs are widely used, and the lower gastrointestinal toxicity of the coxibs represents an important potential advantage over nonselective NSAIDs, as long as we can identify those at an increased risk for cardiovascular toxicity. Larger outcome trials and comprehensive analysis of both pharmacokinetic and pharmacodynamic candidate genes are needed to identify genetic markers to predict variations in response and celecoxib-induced adverse events.
Acknowledgments
The authors thank Fen Liu for assistance with the graphics. PharmGKB is supported by the National Institutes of Health/National Institute of General Medical Sciences (U01GM61374).
Footnotes
Conflicts of interest
There are no conflicts of interest.
References
- 1.Fort J. Celecoxib, a COX-2-specific inhibitor: the clinical data. Am J Orthop (Belle Mead NJ) 1999;28:13–18. [PubMed] [Google Scholar]
- 2. [1 June 2011];CELEBREXR Package Insert. Available at http://pfizer.com/files/products/uspi_celebrex.pdf.
- 3.Clemett D, Goa KL. Celecoxib: a review of its use in osteoarthritis, rheumatoid arthritis and acute pain. Drugs. 2000;59:957–980. doi: 10.2165/00003495-200059040-00017. [DOI] [PubMed] [Google Scholar]
- 4.Arber N, Eagle CJ, Spicak J, Racz I, Dite P, Hajer J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355:885–895. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 5.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–884. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 6.Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 7.Davies NM, McLachlan AJ, Day RO, Williams KM. Clinical pharmacokinetics and pharmacodynamics of celecoxib: a selective cyclooxygenase-2 inhibitor. Clin Pharmacokinet. 2000;38:225–242. doi: 10.2165/00003088-200038030-00003. [DOI] [PubMed] [Google Scholar]
- 8.Grosser T. The pharmacology of selective inhibition of COX-2. Thromb Haemost. 2006;96:393–400. [PubMed] [Google Scholar]
- 9.Lanas A. Clinical experience with cyclooxygenase-2 inhibitors. Rheumatology (Oxford) 2002;41(Suppl 1):16–22. discussion 35–42. [PubMed] [Google Scholar]
- 10.Lanas A, Sopena F. Nonsteroidal anti-inflammatory drugs and lower gastrointestinal complications. Gastroenterol Clin North Am. 2009;38:333–352. doi: 10.1016/j.gtc.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 11.FitzGerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345:433–442. doi: 10.1056/NEJM200108093450607. [DOI] [PubMed] [Google Scholar]
- 12.Crofford LJ. Specific cyclooxygenase-2 inhibitors: what have we learned since they came into widespread clinical use? Curr Opin Rheumatol. 2002;14:225–230. doi: 10.1097/00002281-200205000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Juni P, Rutjes AW, Dieppe PA. Are selective COX 2 inhibitors superior to traditional non steroidal anti-inflammatory drugs? BMJ. 2002;324:1287–1288. doi: 10.1136/bmj.324.7349.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krotz F, Schiele TM, Klauss V, Sohn HY. Selective COX-2 inhibitors and risk of myocardial infarction. J Vasc Res. 2005;42:312–324. doi: 10.1159/000086459. [DOI] [PubMed] [Google Scholar]
- 15.Juni P, Nartey L, Reichenbach S, Sterchi R, Dieppe PA, Egger M. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet. 2004;364:2021–2029. doi: 10.1016/S0140-6736(04)17514-4. [DOI] [PubMed] [Google Scholar]
- 16.Grosser T, Fries S, FitzGerald GA. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paulson SK, Hribar JD, Liu NW, Hajdu E, Bible RH, Jr, Piergies A, Karim A. Metabolism and excretion of [(14)C]celecoxib in healthy male volunteers. Drug Metab Dispos. 2000;28:308–314. [PubMed] [Google Scholar]
- 18.Solomon SD, Wittes J, Finn PV, Fowler R, Viner J, Bertagnolli MM, et al. Cardiovascular risk of celecoxib in 6 randomized placebocontrolled trials: the cross trial safety analysis. Circulation. 2008;117:2104–2113. doi: 10.1161/CIRCULATIONAHA.108.764530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandberg M, Yasar U, Stromberg P, Hoog JO, Eliasson E. Oxidation of celecoxib by polymorphic cytochrome P450 2C9 and alcohol dehydrogenase. Br J Clin Pharmacol. 2002;54:423–429. doi: 10.1046/j.1365-2125.2002.01660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kirchheiner J, Stormer E, Meisel C, Steinbach N, Roots I, Brockmoller J. Influence of CYP2C9 genetic polymorphisms on pharmacokinetics of celecoxib and its metabolites. Pharmacogenetics. 2003;13:473–480. doi: 10.1097/00008571-200308000-00005. [DOI] [PubMed] [Google Scholar]
- 21.Tang C, Shou M, Rushmore TH, Mei Q, Sandhu P, Woolf EJ, et al. In-vitro metabolism of celecoxib, a cyclooxygenase-2 inhibitor, by allelic variant forms of human liver microsomal cytochrome P450 2C9: correlation with CYP2C9 genotype and in-vivo pharmacokinetics. Pharmacogenetics. 2001;11:223–235. doi: 10.1097/00008571-200104000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Werner U, Werner D, Rau T, Fromm MF, Hinz B, Brune K. Celecoxib inhibits metabolism of cytochrome P450 2D6 substrate metoprolol in humans. Clin Pharmacol Ther. 2003;74:130–137. doi: 10.1016/S0009-9236(03)00120-6. [DOI] [PubMed] [Google Scholar]
- 23.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzgerald GA. Prostaglandins: modulators of inflammation and cardiovascular risk. J Clin Rheumatol. 2004;10:S12–S17. doi: 10.1097/01.rhu.0000130685.73681.8b. [DOI] [PubMed] [Google Scholar]
- 25.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 26.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 27.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 28.Liu J, Seibold SA, Rieke CJ, Song I, Cukier RI, Smith WL. Prostaglandin endoperoxide H synthases: peroxidase hydroperoxide specificity and cyclooxygenase activation. J Biol Chem. 2007;282:18233–18244. doi: 10.1074/jbc.M701235200. [DOI] [PubMed] [Google Scholar]
- 29.Smith WL, Dewitt DL. Prostaglandin endoperoxide H synthases-1 and -2. Adv Immunol. 1996;62:167–215. doi: 10.1016/s0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- 30.Smith WL, Song I. The enzymology of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat. 2002;68-69:115–128. doi: 10.1016/s0090-6980(02)00025-4. [DOI] [PubMed] [Google Scholar]
- 31.Thorn CF, Grosser T, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for PTGS2. Pharmacogenet Genomics. 2011;21:607–613. doi: 10.1097/FPC.0b013e3283415515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capone ML, Tacconelli S, Di Francesco L, Sacchetti A, Sciulli MG, Patrignani P. Pharmacodynamic of cyclooxygenase inhibitors in humans. Prostaglandins Other Lipid Mediat. 2007;82:85–94. doi: 10.1016/j.prostaglandins.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 33.Tacconelli S, Capone ML, Sciulli MG, Ricciotti E, Patrignani P. The biochemical selectivity of novel COX-2 inhibitors in whole blood assays of COX-isozyme activity. Curr Med Res Opin. 2002;18:503–511. doi: 10.1185/030079902125001335. [DOI] [PubMed] [Google Scholar]
- 34.Fries S, Grosser T, Price TS, Lawson JA, Kapoor S, DeMarco S, et al. Marked interindividual variability in the response to selective inhibitors of cyclooxygenase-2. Gastroenterology. 2006;130:55–64. doi: 10.1053/j.gastro.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Grosser T. Variability in the response to cyclooxygenase inhibitors: toward the individualization of nonsteroidal anti-inflammatory drug therapy. J Investig Med. 2009;57:709–716. doi: 10.2310/JIM.0b013e3181b04d1f. [DOI] [PubMed] [Google Scholar]
- 36.Rouzer CA, Marnett LJ. Cyclooxygenases: structural and functional insights. J Lipid Res. 2009;50(Suppl):S29–S34. doi: 10.1194/jlr.R800042-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu GS, Zou SQ, Liu ZR, Tang ZH, Wang JH. Celecoxib inhibits proliferation and induces apoptosis via prostaglandin E2 pathway in human cholangiocarcinoma cell lines. World J Gastroenterol. 2003;9:1302–1306. doi: 10.3748/wjg.v9.i6.1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kulp SK, Yang YT, Hung CC, Chen KF, Lai JP, Tseng PH, et al. 3-phosphoinositide-dependent protein kinase-1/Akt signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer Res. 2004;64:1444–1451. doi: 10.1158/0008-5472.can-03-2396. [DOI] [PubMed] [Google Scholar]
- 39.Levitt RJ, Buckley J, Blouin MJ, Schaub B, Triche TJ, Pollak M. Growth inhibition of breast epithelial cells by celecoxib is associated with upregulation of insulin-like growth factor binding protein-3 expression. Biochem Biophys Res Commun. 2004;316:421–428. doi: 10.1016/j.bbrc.2004.02.062. [DOI] [PubMed] [Google Scholar]
- 40.Patel MI, Subbaramaiah K, Du B, Chang M, Yang P, Newman RA, et al. Celecoxib inhibits prostate cancer growth: evidence of a cyclooxygenase-2-independent mechanism. Clin Cancer Res. 2005;11:1999–2007. doi: 10.1158/1078-0432.CCR-04-1877. [DOI] [PubMed] [Google Scholar]
- 41.Schonthal AH. Direct non-cyclooxygenase-2 targets of celecoxib and their potential relevance for cancer therapy. Br J Cancer. 2007;97:1465–1468. doi: 10.1038/sj.bjc.6604049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harris RE. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology. 2009;17:55–67. doi: 10.1007/s10787-009-8049-8. [DOI] [PubMed] [Google Scholar]
- 43.Schonthal AH. Antitumor properties of dimethyl-celecoxib, a derivative of celecoxib that does not inhibit cyclooxygenase-2: implications for glioma therapy. Neurosurg Focus. 2006;20:E21. doi: 10.3171/foc.2006.20.4.14. [DOI] [PubMed] [Google Scholar]
- 44.Grosch S, Maier TJ, Schiffmann S, Geisslinger G. Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. J Natl Cancer Inst. 2006;98:736–747. doi: 10.1093/jnci/djj206. [DOI] [PubMed] [Google Scholar]
- 45.Dvory-Sobol H, Cohen-Noyman E, Kazanov D, Figer A, Birkenfeld S, Madar-Shapiro L, et al. Celecoxib leads to G2/M arrest by induction of p21 and down-regulation of cyclin B1 expression in a p53-independent manner. Eur J Cancer. 2006;42:422–426. doi: 10.1016/j.ejca.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 46.Grosch S, Tegeder I, Niederberger E, Brautigam L, Geisslinger G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. Faseb J. 2001;15:2742–2744. doi: 10.1096/fj.01-0299fje. [DOI] [PubMed] [Google Scholar]
- 47.Han C, Leng J, Demetris AJ, Wu T. Cyclooxygenase-2 promotes human cholangiocarcinoma growth: evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 2004;64:1369–1376. doi: 10.1158/0008-5472.can-03-1086. [DOI] [PubMed] [Google Scholar]
- 48.Maier TJ, Janssen A, Schmidt R, Geisslinger G, Grosch S. Targeting the beta-catenin/APC pathway: a novel mechanism to explain the cyclooxygenase-2-independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. FASEB J. 2005;19:1353–1355. doi: 10.1096/fj.04-3274fje. [DOI] [PubMed] [Google Scholar]
- 49.Jendrossek V, Handrick R, Belka C. Celecoxib activates a novel mitochondrial apoptosis signaling pathway. Faseb J. 2003;17:1547–1549. doi: 10.1096/fj.02-0947fje. [DOI] [PubMed] [Google Scholar]
- 50.Arico S, Pattingre S, Bauvy C, Gane P, Barbat A, Codogno P, Ogier-Denis E. Celecoxib induces apoptosis by inhibiting 3-phosphoinositide-dependent protein kinase-1 activity in the human colon cancer HT-29 cell line. J Biol Chem. 2002;277:27613–27621. doi: 10.1074/jbc.M201119200. [DOI] [PubMed] [Google Scholar]
- 51.Hsu AL, Ching TT, Wang DS, Song X, Rangnekar VM, Chen CS. The cyclooxygenase-2 inhibitor celecoxib induces apoptosis by blocking Akt activation in human prostate cancer cells independently of Bcl-2. J Biol Chem. 2000;275:11397–11403. doi: 10.1074/jbc.275.15.11397. [DOI] [PubMed] [Google Scholar]
- 52.Zhu J, Song X, Lin HP, Young DC, Yan S, Marquez VE, Chen CS. Using cyclooxygenase-2 inhibitors as molecular platforms to develop a new class of apoptosis-inducing agents. J Natl Cancer Inst. 2002;94:1745–1757. doi: 10.1093/jnci/94.23.1745. [DOI] [PubMed] [Google Scholar]
- 53.Wang L, Chen W, Xie X, He Y, Bai X. Celecoxib inhibits tumor growth and angiogenesis in an orthotopic implantation tumor model of human colon cancer. Exp Oncol. 2008;30:42–51. [PubMed] [Google Scholar]
- 54.Wei D, Wang L, He Y, Xiong HQ, Abbruzzese JL, Xie K. Celecoxib inhibits vascular endothelial growth factor expression in and reduces angiogenesis and metastasis of human pancreatic cancer via suppression of Sp1 transcription factor activity. Cancer Res. 2004;64:2030–2038. doi: 10.1158/0008-5472.can-03-1945. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Y, Ran J, Tang C, Wu J, Honghua L, Xingwen L, et al. Effect of celecoxib on E-cadherin, VEGF, microvessel density and apoptosis in gastric cancer. Cancer Biol Ther. 2007;6:269–275. doi: 10.4161/cbt.6.2.3629. [DOI] [PubMed] [Google Scholar]
- 56.Peluffo GD, Stillitani I, Rodriguez VA, Diament MJ, Klein SM. Reduction of tumor progression and paraneoplastic syndrome development in murine lung adenocarcinoma by nonsteroidal antiinflammatory drugs. Int J Cancer. 2004;110:825–830. doi: 10.1002/ijc.20226. [DOI] [PubMed] [Google Scholar]
- 57.Johnson AJ, Hsu AL, Lin HP, Song X, Chen CS. The cyclo-oxygenase-2 inhibitor celecoxib perturbs intracellular calcium by inhibiting endoplasmic reticulum Ca2 + -ATPases: a plausible link with its anti-tumour effect and cardiovascular risks. Biochem J. 2002;366:831–837. doi: 10.1042/BJ20020279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanaka K, Tomisato W, Hoshino T, Ishihara T, Namba T, Aburaya M, et al. Involvement of intracellular Ca2 + levels in nonsteroidal anti-inflammatory drug-induced apoptosis. J Biol Chem. 2005;280:31059–31067. doi: 10.1074/jbc.M502956200. [DOI] [PubMed] [Google Scholar]
- 59.Knudsen JF, Carlsson U, Hammarstrom P, Sokol GH, Cantilena LR. The cyclooxygenase-2 inhibitor celecoxib is a potent inhibitor of human carbonic anhydrase II. Inflammation. 2004;28:285–290. doi: 10.1007/s10753-004-6052-1. [DOI] [PubMed] [Google Scholar]
- 60.Weber A, Casini A, Heine A, Kuhn D, Supuran CT, Scozzafava A, Klebe G. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: new pharmacological opportunities due to related binding site recognition. J Med Chem. 2004;47:550–557. doi: 10.1021/jm030912m. [DOI] [PubMed] [Google Scholar]
- 61.Pastorekova S, Zatovicova M, Pastorek J. Cancer-associated carbonic anhydrases and their inhibition. Curr Pharm Des. 2008;14:685–698. doi: 10.2174/138161208783877893. [DOI] [PubMed] [Google Scholar]
- 62.Pastorekova S, Kopacek J, Pastorek J. Carbonic anhydrase inhibitors and the management of cancer. Curr Top Med Chem. 2007;7:865–878. doi: 10.2174/156802607780636708. [DOI] [PubMed] [Google Scholar]
- 63.Alper AB, Jr, Tomlin H, Sadhwani U, Whelton A, Puschett J. Effects of the selective cyclooxygenase-2 inhibitor analgesic celecoxib on renal carbonic anhydrase enzyme activity: a randomized, controlled trial. Am J Ther. 2006;13:229–235. doi: 10.1097/01.mjt.0000182359.63457.01. [DOI] [PubMed] [Google Scholar]
- 64.Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. 1522 p following 1528. [DOI] [PubMed] [Google Scholar]
- 65.Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 66.Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 67.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Breazna A, Kim K, et al. Five-year efficacy and safety analysis of the Adenoma Prevention with Celecoxib Trial. Cancer Prev Res (Phila) 2009;2:310–321. doi: 10.1158/1940-6207.CAPR-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arber N, Spicak J, Racz I, Zavoral M, Breazna A, Gerletti P, et al. Five-year analysis of the prevention of colorectal sporadic adenomatous polyps trial. Am J Gastroenterol. 2011;106:1135–1146. doi: 10.1038/ajg.2011.116. [DOI] [PubMed] [Google Scholar]
- 69.McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA. 2006;296:1633–1644. doi: 10.1001/jama.296.13.jrv60011. [DOI] [PubMed] [Google Scholar]
- 70.White WB, West CR, Borer JS, Gorelick PB, Lavange L, Pan SX, et al. Risk of cardiovascular events in patients receiving celecoxib: a meta-analysis of randomized clinical trials. Am J Cardiol. 2007;99:91–98. doi: 10.1016/j.amjcard.2006.07.069. [DOI] [PubMed] [Google Scholar]
- 71.Trelle S, Reichenbach S, Wandel S, Hildebrand P, Tschannen B, Villiger PM, et al. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. 2011;342:c7086. doi: 10.1136/bmj.c7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci USA. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mukherjee D. Selective cyclooxygenase-2 (COX-2) inhibitors and potential risk of cardiovascular events. Biochem Pharmacol. 2002;63:817–821. doi: 10.1016/s0006-2952(02)00842-0. [DOI] [PubMed] [Google Scholar]
- 74.Vardeny O, Solomon SD. Cyclooxygenase-2 inhibitors, nonsteroidal anti-inflammatory drugs, and cardiovascular risk. Cardiol Clin. 2008;26:589–601. doi: 10.1016/j.ccl.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 75.Grosser T, Yu Y, Fitzgerald GA. Emotion recollected in tranquility: lessons learned from the COX-2 saga. Annu Rev Med. 2010;61:17–33. doi: 10.1146/annurev-med-011209-153129. [DOI] [PubMed] [Google Scholar]
- 76.Sowers JR, White WB, Pitt B, Whelton A, Simon LS, Winer N, et al. The effects of cyclooxygenase-2 inhibitors and nonsteroidal anti-inflammatory therapy on 24-hour blood pressure in patients with hypertension, osteoarthritis, and type 2 diabetes mellitus. Arch Intern Med. 2005;165:161–168. doi: 10.1001/archinte.165.2.161. [DOI] [PubMed] [Google Scholar]
- 77.Hermann M, Camici G, Fratton A, Hurlimann D, Tanner FC, Hellermann JP, et al. Differential effects of selective cyclooxygenase-2 inhibitors on endothelial function in salt-induced hypertension. Circulation. 2003;108:2308–2311. doi: 10.1161/01.CIR.0000101683.30157.0B. [DOI] [PubMed] [Google Scholar]
- 78.Kimmel SE, Berlin JA, Reilly M, Jaskowiak J, Kishel L, Chittams J, Strom BL. Patients exposed to rofecoxib and celecoxib have different odds of nonfatal myocardial infarction. Ann Intern Med. 2005;142:157–164. doi: 10.7326/0003-4819-142-3-200502010-00005. [DOI] [PubMed] [Google Scholar]
- 79.Graham DJ, Campen D, Hui R, Spence M, Cheetham C, Levy G, et al. Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: nested case-control study. Lancet. 2005;365:475–481. doi: 10.1016/S0140-6736(05)17864-7. [DOI] [PubMed] [Google Scholar]
- 80.Brueggemann LI, Mackie AR, Mani BK, Cribbs LL, Byron KL. Differential effects of selective cyclooxygenase-2 inhibitors on vascular smooth muscle ion channels may account for differences in cardiovascular risk profiles. Mol Pharmacol. 2009;76:1053–1061. doi: 10.1124/mol.109.057844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brueggemann LI, Mani BK, Mackie AR, Cribbs LL, Byron KL. Novel actions of nonsteroidal anti-inflammatory drugs on vascular ion channels: accounting for cardiovascular side effects and identifying new therapeutic applications. Mol Cell Pharmacol. 2010;2:15–19. [PMC free article] [PubMed] [Google Scholar]
- 82.Lee CR, Goldstein JA, Pieper JA. Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics. 2002;12:251–263. doi: 10.1097/00008571-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 83.Kirchheiner J, Brockmoller J. Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther. 2005;77:1–16. doi: 10.1016/j.clpt.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 84.Ali ZK, Kim RJ, Ysla FM. CYP2C9 polymorphisms: considerations in NSAID therapy. Curr Opin Drug Discov Devel. 2009;12:108–114. [PubMed] [Google Scholar]
- 85.Brenner SS, Herrlinger C, Dilger K, Murdter TE, Hofmann U, Marx C, Klotz U. Influence of age and cytochrome P450 2C9 genotype on the steady-state disposition of diclofenac and celecoxib. Clin Pharmacokinet. 2003;42:283–292. doi: 10.2165/00003088-200342030-00003. [DOI] [PubMed] [Google Scholar]
- 86.Stempak D, Bukaveckas BL, Linder M, Koren G, Baruchel S. Cytochrome P450 2C9 genotype: impact on celecoxib safety and pharmacokinetics in a pediatric patient. Clin Pharmacol Ther. 2005;78:309–310. doi: 10.1016/j.clpt.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 87.Xie HG, Prasad HC, Kim RB, Stein CM. CYP2C9 allelic variants: ethnic distribution and functional significance. Adv Drug Deliv Rev. 2002;54:1257–1270. doi: 10.1016/s0169-409x(02)00076-5. [DOI] [PubMed] [Google Scholar]
- 88.Chan AT, Zauber AG, Hsu M, Breazna A, Hunter DJ, Rosenstein RB, et al. Cytochrome P450 2C9 variants influence response to celecoxib for prevention of colorectal adenoma. Gastroenterology. 2009;136:e2121. doi: 10.1053/j.gastro.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Agundez JA, Garcia-Martin E, Martinez C. Genetically based impairment in CYP2C8- and CYP2C9-dependent NSAID metabolism as a risk factor for gastrointestinal bleeding: is a combination of pharmacogenomics and metabolomics required to improve personalized medicine? Expert Opin Drug Metab Toxicol. 2009;5:607–620. doi: 10.1517/17425250902970998. [DOI] [PubMed] [Google Scholar]
- 90.Agundez JA, Martinez C, Garcia-Martin E, Ladero JM. Cytochrome P450 CYP2C9 polymorphism and NSAID-related acute gastrointestinal bleeding. Gastroenterology. 2007;133:2071–2072. doi: 10.1053/j.gastro.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 91.Martinez C, Blanco G, Ladero JM, Garcia-Martin E, Taxonera C, Gamito FG, et al. Genetic predisposition to acute gastrointestinal bleeding after NSAIDs use. Br J Pharmacol. 2004;141:205–208. doi: 10.1038/sj.bjp.0705623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pilotto A, Seripa D, Franceschi M, Scarcelli C, Colaizzo D, Grandone E, et al. Genetic susceptibility to nonsteroidal anti-inflammatory drug-related gastroduodenal bleeding: role of cytochrome P450 2C9 polymorphisms. Gastroenterology. 2007;133:465–471. doi: 10.1053/j.gastro.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 93.Skarke C, Reus M, Schmidt R, Grundei I, Schuss P, Geisslinger G, Lotsch J. The cyclooxygenase 2 genetic variant −765G > C does not modulate the effects of celecoxib on prostaglandin E2 production. Clin Pharmacol Ther. 2006;80:621–632. doi: 10.1016/j.clpt.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 94.Yan M, Myung SJ, Fink SP, Lawrence E, Lutterbaugh J, Yang P, et al. 15-Hydroxyprostaglandin dehydrogenase inactivation as a mechanism of resistance to celecoxib chemoprevention of colon tumors. Proc Natl Acad Sci USA. 2009;106:9409–9413. doi: 10.1073/pnas.0902367106. [DOI] [PMC free article] [PubMed] [Google Scholar]