

Abstract
20-hydroxyeicosatetraenoic acid (20-HETE) is a cytochrome P450 (CYP) metabolite of arachidonic acid that that contributes to infarct size following focal cerebral ischemia. However, little is known about the role of 20-HETE in global cerebral ischemia or neonatal hypoxia-ischemia (H-I). The present study examined the effects of blockade of the synthesis of 20-HETE with HET0016 in neonatal piglets after H-I to determine if it protects highly vulnerable striatal neurons. Administration of HET0016 after H-I improved early neurological recovery and protected neurons in putamen after 4 days of recovery. HET0016 had no significant effect on cerebral blood flow. CYP 4A immunoreactivity was detected in putamen neurons, and direct infusion of 20-HETE in the putamen increased phosphorylation of Na+,K+-ATPase and NMDA receptor NR1 subunit selectively at PKC-sensitive sites but not at PKA-sensitive sites. HET0016 selectively inhibited the H-I–induced phosphorylation at these same sites at 3 h of recovery and improved Na+,K+-ATPase activity.At 3 h, HET0016 also suppressed H-I–induced Erk1/2 activation and protein markers of nitrosative and oxidative stress. Thus, 20-HETE can exert direct effects on key proteins involved in neuronal excitotoxicity in vivo and contributes to neurodegeneration after global cerebral ischemia in immature brain.
Keywords: cerebral blood flow; cytochrome P450 4A; HET0016; Na+, K+-ATPase; NMDA receptor; piglet
Neonatal hypoxic-ischemic encephalopathy causes significant infant mortality and morbidity (Dixon et al. 2002). Ca2+ entry secondary to activation of N-methyl-D-aspartate (NMDA) receptor channels and other channels contributes to neuronal cell death following H-I (Szatkowski and Attwell 1994; Yang et al. 2011). A consequence of increased intracellular Ca2+ is stimulation of phopholipase A2 and release of arachidonic acid. Indeed, the levels of fatty acids markedly increase following ischemia and reperfusion. In addition to metabolism by cyclooxygenases and lipoxygenases, arachidonic acid can undergo ω-hydroxylation by cytochrome P450 (CYP) enzymes of the 4A family to produce 20-hydroxyeicosatetraenoic acid (20-HETE).
Several lines of evidence provide a rationale for the possibility that 20-HETE production could be involved in neurodegeneration following cerebral ischemia. CYP 4A isoforms are expressed in cerebral vascular smooth muscle (Dunn et al. 2008), and 20-HETE has been well documented to be a potent vasoconstrictor of cerebral microvessels (Harder et al. 1994) through mechanisms partly dependent on protein kinase C (PKC) (Lange et al. 1997). Thus, 20-HETE has been postulated to augment focal ischemic injury by reducing cerebral blood flow (CBF) (Poloyac et al. 2006; Marumo et al. 2010). 20-HETE has also been shown to serve as an intracellular signaling molecule in some types of non-neuronal cells. For example, it exerts a profound depressive effect on Na+,K+-ATPase activity in kidney through PKC-mediated phosphorylation of Ser23 on the Na+,K+-ATPase α subunit (Nowicki et al. 1997). In addition, 20-HETE can induce mitogen-activated protein kinase (MAPK) activation in vascular smooth muscle cells and extracellular signal-regulated kinase 1/2 (Erk1/2) activation in renal epithelial cells (Muthalif et al. 1998; Akbulut et al. 2009). Considering that CYP 4A is expressed in neurons after brain ischemia (Omura et al. 2006), the possibility exists that 20-HETE may exert effects directly on neurons independent of its effect on CBF. This possibility is supported by work demonstrating that 20-HETE synthesis inhibition reduces injury from oxygen-glocose deprivation in hippocampal slice cultures (Renic et al. 2011) and reduces infarct volume in experimental models of focal cerebral ischemia (Miyata et al. 2005; Omura et al. 2006; Poloyac et al. 2006; Tanaka et al. 2007) without producing intraischemic vasodilation (Cao et al. 2009; Renic et al. 2009). Whether 20-HETE contributes to neuronal injury after global cerebral ischemia or in a neonatal model of hypoxia-ischemia (H-I), however, is unknown.
In the present study, we tested the hypothesis that treatment with a 20-HETE synthesis inhibitor after H-I in neonatal piglets attenuates neuronal damage. The neonatal H-I model involves induction of systemic hypoxia followed by asphyxic cardiac arrest and resuscitation. We focused on putamen because this is the most vulnerable region to H-I (Martin et al. 1997) and because increases in PKC-dependent phosphorylation on and decreases in Na+,K+-ATPase activity persist in this region 3 hours after resuscitation (Yang et al. 2007), thereby allowing evaluation of whether 20-HETE modulates Na+,K+-ATPase activity in a manner analogous to that found in kidney (Nowicki et al. 1997). In addition, increases in phosphorylation at PKC-dependent sites on NMDA receptors persist at 3 hours (Yang et al. 2007) and permit evaluation of the possible role of 20-HETE in mediating phosphorylation in non-vascular cells. The 20-HETE synthesis inhibitor N-hydroxy-N′-(4-n-butyl-2-methylphenyl)formamidine (HET0016) (Miyata et al. 2001) was selected because of its ability to reduce infarct volume after focal cerebral ischemia in adult rats when administered systemically (Poloyac et al. 2006; Renic et al. 2009) and to protect neurons from oxygen-glucose deprivation (Renic et al. 2011). Thus, we tested the additional hypotheses that HET0016 administration 1) attenuates H-I–induced increases in phosphorylation at PKC-sensitive sites on Na+,K+-ATPase and NR1 of the NMDA receptor while sparing protein kinase A- (PKA) sensitive sites, 2) improves recovery of Na+,K+-ATPase activity, 3) reduces markers of oxidative stress, and 4) results in less phosphorylation of Erk1/2. Lastly, we tested whether administration of 20-HETE directly into non-ischemic putamen via microdialysis could replicate the HET0016-inhibitable phosphorylation changes seen after H-I.
Materials and Methods
Experimental protocol
All experimental protocols were approved by the Animal Care and Use Committee of the Johns Hopkins University and performed in accordance with National Institutes of Health guidelines. A total of 93 male piglets (2–2.5 kg) were studied at 4–7 days of age. As described previously (Yang et al. 2007; Yang et al. 2010), the piglets were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneal), orally intubated, and underwent catheterization of a femoral artery and vein under aseptic conditions. To induce H-I, inspired O2 was decreased to 10.0 ± 0.2% for 40 min, followed by ventilation with 21% O2 for 5 min (required for cardiac resuscitation) and airway occlusion (asphyxia) for 7 min (to produce cardiac arrest). Piglets were then resuscitated by mechanical ventilation with 50% O2, manual chest compressions, and, if necessary, intravenous injection of epinephrine until return of spontaneous circulation. After resuscitation, inspired O2 was gradually reduced to 30% to maintain arterial O2 saturation. Arterial blood pressure, blood gases, glucose and rectal temperature were monitored until piglets regaining consciousness. Those surviving for 3 hours for biochemical measurements and 6 hours for CBF measurements were maintained sedated with a continuous infusion of fentanyl (10 μg/kg/h, intravenously). Sham-operated animals were subjected to catheterization but not hypoxia or asphyxia.
HET0016 (Cayman, Ann Arbor, MI) was injected intravenously at a dose of 1 mg/kg (low dose) or 10 mg/kg (high dose) at 5 min after resuscitation or an equivalent time in sham-operated piglets. Vehicle-treated piglets received the equivalent volume of 10% (2-hydroxypropyl)-β-cyclodextrin in 0.9% saline. The dose of 1 mg/kg of HET0016 was based on previous work showing efficacy in focal ischemia models (Renic et al. 2009). The higher dose of 10 mg/kg was also selected to determine if protection at the lower dose was maximal. Since no dose-dependent difference was found in the degree of neuroprotection, only the low dose was used in studies of biochemical changes at 3 h of recovery. For the CBF study, the higher dose was used to enhance the likelihood of detecting an effect.
Neuronal cell death in putamen progresses between 6 and 24 h after reoxygenation (Martin et al. 2000). Hence, biochemical measurements were made at 3 h before neuronal loss would affect the measurements. Neuronal damage was evaluated after 4 days of recovery to allow for a possible delay in the maturation of injury after HET0016 treatment.
Blood flow measurement
In separate cohorts of piglets, regional CBF was measured by laser-Doppler flowmetry (LDF) (Moor Instruments, England) and by FluoSpheres Blood Flow Determination kit (Molecular Probes, Eugene, OR). The laser-Doppler probe was placed on the cortical surface through a small craniotomy and an incision in the dura 8 mm anterior and 4 mm lateral from bregma. LDF was monitored continuously throughout the experiment. Fluorescent microspheres permitted regional CBF to be measured at six discrete times during the experiment with a procedure modified from previous work (Berkowitz et al. 1991; Glenny et al. 1993). In brief, a catheter was inserted from a femoral artery into the left ventricle for microsphere injection. Approximately 1 × 106 microspheres (15 μm diameter) were injected into the left ventricle at 20 min before hypoxia, and at 30, 60, 120, 240, and 360 min of recovery. The microspheres labeled with different fluorochromes (Crimson, Blue-Green, Red, Orange, Blue, Scarlet, and Yellow-Green) were randomly injected at the six different times. Reference blood samples were withdrawn at a rate of 2 ml/min from the femoral artery for 2 min during and after the injection. After harvesting the brain at 6 h of recovery, the putamen was dissected, digested in 2 M ethanolic KOH with 0.5% Tween 80, and centrifuged to obtain a suspension of microspheres. Fluorescent dyes were released from the microspheres in the tissue and arterial blood samples with the addition of 2-ethoxyethyl acetate and quantified by spectrofluorometry with excitation of each fluorochrome at the appropriate wavelength (Van Oosterhout et al. 1995).
20-HETE infusion
Holes were drilled in the skull 8 mm anterior and 7.5 mm lateral from bregma in separate sets of anesthetized piglets without H-I. The tip of the microdialysis probes (CMA 12; CMA/Microdialysis, Solna, Sweden) with a 4-mm membrane length was inserted 16.5 mm below the dura into putamen. The probes were perfused with artificial cerebrospinal fluid without recirculation at a rate of 2.5 μl/min starting at 1 h after placement for 1 h, followed by a freshly made solution of 30 μM 20-HETE (Cayman) or 10% ethanol vehicle for another hour. The brains were then perfused transcardially with ice-cold phosphate-buffered saline (PBS), and the putamen was rapidly dissected and processed for Western blot analysis. For each brain, optical densities on blots were normalized by the optical densities in the contralateral putamen without microdialysis perfusion.
Neurobehavioral assessment
A neurologic deficit score (0 = best outcome, 154 = worst outcome) was used to quantify overall neurologic function based on seven different components: 1) level of consciousness; 2) brain stem function; 3) sensory responses; 4) motor function; 5) behavioral activities; 6) spatial orientation; and 7) excitability (Agnew et al. 2003; Yang et al. 2007; Yang et al. 2010).
Immunohistochemical and double immunofluorescence staining
Anesthetized piglets were perfused transcardially with ice-cold PBS and 4% paraformaldehyde at 3 h or 4 days of recovery. Brains were removed, bisected mid-sagittally, and cut into 1-cm slabs. The left forebrain was embedded in paraffin for histology with hematoxylin and eosin (H&E) staining and neuropathological assessment of damaged neurons. The right forebrain was cryoprotected, frozen, and cut into serial 40-μm coronal sections through the putamen.
Double immunofluorescence staining was used to assess if CYP4A, p38 MAP kinase (p38), c-Jun N-terminal kinase (JNK), or p44/42 MAPK (ERK1/2) localized in striatal neurons. Sections were blocked in 10% normal horse serum and incubated with primary antibodies overnight at 4°C. Primary antibodies included rabbit anti-CYP4A (1:500, Thermo, Rockford, IL), mouse anti-NeuN (1:500, Chemicon, Temecula, CA), rabbit anti-p38, rabbit anti-JNK, and rabbit anti-Erk1/2 (1:300, Cell Signaling, Danvers, MA). Antibody binding was visualized by incubating sections with Cy3-conjugated (1:500, Jackson ImmunoResearch, West Grove, PA) or Alexa Fluor 488-conjugated (1:500, Invitrogen, Eugene, OR) secondary antibodies.
Western blotting
Putamen was rapidly dissected from H-I piglets at 3 h of recovery and time-matched sham-operated piglets after transcardial cold PBS perfusion. Tissues were homogenized and fractionated into membrane-enriched and cytosol-enriched fractions, as described previously (Yang et al. 2007; Yang et al. 2010). Twenty-microgram protein samples were separated by 4–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The membranes were probed with the following primary antibodies: mouse anti-NR1 (1:2000, BD Pharmingen, San Jose, CA), rabbit anti-phospho-NR1 Ser896, anti-phospho-NR1 Ser897 (1:2000, Upstate, Lake Placid, NY), mouse anti-Na+,K+ - ATPase α1 (1:50,000, Sigma, St Louis, MO) or Na+,K+- ATPase α3 (1:50,000, Affinity Bioreagents, Golden, CO), rabbit anti-phospho Na+,K+- ATPase α Ser23 or Na+,K+- ATPase α Ser943 (1:2000, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-p38, anti-phospho-p38 Thr180/Tyr182, anti-JNK, anti-phospho-JNK Thr183/Tyr185, anti-Erk1/2, anti-phospho-Erk1/2 Thr202/Tyr204 (1:2000, Cell signaling), mouse anti-synaptophysin (1:20,000, Chemicon), mouse anti-nitrotyrosine (1:40,000, Upstate), or anti-β-actin (1:5000, Santa Cruz). Synaptophysin and βactin were used as a protein loading standard for membrane- and cytosol- enriched fractions, respectively. Optical density values were normalized to the value of a sham-operated, vehicle-treated piglet on each gel. Each experiment was performed with tissue from four to six piglets per group.
Protein oxidation assay
Oxidative modification of proteins in putamen after H-I was determined with an OxyBlot protein oxidation detection kit (Chemicon) for carbonyl groups as described previously (Mueller-Burke et al. 2008).
Na+, K+-ATPase biochemical assay
The biochemical activity of Na+,K+-ATPase in samples of putamen was modified from the method described previously (Golden et al. 2001). In brief, cell membrane-enriched putamen samples was reacted in 1.5 ml buffer containing 20 mM Tris-HCl (pH7.4), 0.57 mM EDTA, 5 mM MgCl2, 133 mM NaCl, with/without 1.0 mM ouabain. The reaction was carried out in the presence of 3 mM ATP at 37°C for 15 min. The enzymatic hydrolysis of ATP was terminated by adding 1.5 ml of 20% trichloroacetic acid followed by centrifugation. The activity of ATPase was measured as a function of liberated inorganic phosphate by the colorimetric assay.
Data and statistical analysis
Profile counting on H&E-stained paraffin sections was used to estimate viable neurons in putamen at 4 days of recovery. Sections were coded and labeled in a manner that protects the blinding. In level-matched sections, the number of ischemic and non-ischemic neuronal profiles was counted randomly in an observer-blinded fashion in seven non-overlapping microscopic fields at 1000× power in anterior, median, and posterior putamen sections (bregma level: +15 mm, +9 mm, and +5 mm) (Salinas-Zeballos et al. 1986). The values were averaged from 21 fields to obtain a single value of viable neurons per square millimeter for each piglet to be used in the statistical analysis.
All values are expressed as means ± SD. The neurologic deficit score was analyzed by two-way analysis of variance (ANOVA) for repeated measures. Other measurements were analyzed with Student t-test or one-way ANOVA followed by the Student-Newman-Keuls multiple range test. P < 0.05 was considered statistically different.
Results
Successful resuscitation was achieved in 49 of 60 piglets (82%) subjected to H-I. The amount of epinephrine used during resuscitation did not differ among groups subsequently treated with vehicle (111 ± 150 μg/kg), the 1 mg/kg (low dose) of HET0016 (130 ± 95 μg/kg), or the 10 mg/kg (high dose) of HET0016 (80 ± 123 μg/kg).
Neuronal damage in putamen after H-I
In the cohorts used for neurobehavior and histology, mortality during the 4-day recovery period was 2 of 9 piglets in the vehicle group, 2 of 10 piglets in the low-dose HET0016 treatment group, 0 of 8 piglets in the high-dose HET0016 treatment group. Mortality rates include piglets that were euthanized because of inability to extubate within 10 h of recovery or severe generalized seizures [2/0 (no extubation/seizures), 1/1, and 0/0 in the vehicle, low-, and high-dose HET0016 treatment groups, respectively]. In the survivors, no significant difference was observed in arterial PO2, PCO2, pH, or mean arterial pressure during H-I or early recovery, and values at 1–3 h of recovery were in the normal physiological range (Table 1).
Table 1.
Physiologic parameters during and after hypoxia-ischemia
| Baseline | Hypoxia 37 min |
21% O2 5 min |
Asphyxia | Post-resuscitation
|
|||
|---|---|---|---|---|---|---|---|
| 1 h | 2 h | 3 h | |||||
| MABP (mmHg) | |||||||
| Vehicle | 75 ± 9 | 77 ± 7 | 77 ± 9 | 21 ± 9 | 64 ± 11 | 61 ± 6 | 61 ± 7 |
| HET0016-Low | 71 ± 6 | 75 ± 20 | 79 ± 12 | 23 ± 7 | 62 ± 5 | 62 ± 7 | 61 ± 7 |
| HET0016-High | 69 ± 9 | 70± 11 | 70 ± 17 | 25 ± 8 | 65 ± 8 | 66 ± 10 | 66 ± 7 |
| Arterial PO2 (mmHg) | |||||||
| Vehicle | 136 ± 14 | 23 ± 2 | 77 ± 9 | 9 ± 4 | 116 ± 15 | 123 ± 14 | 136 ± 9 |
| HET0016-Low | 140 ± 15 | 23 ± 4 | 77 ± 15 | 6 ± 4 | 123 ± 16 | 139 ± 13 | 139 ± 21 |
| HET0016-High | 137 ± 18 | 23 ± 4 | 74 ± 14 | 7 ± 2 | 125 ± 16 | 131 ± 15 | 140 ± 20 |
| Arterial PCO2 (mmHg) | |||||||
| Vehicle | 38 ± 3 | 44 ± 4 | 42 ± 6 | 95 ± 7 | 36 ± 4 | 37 ± 6 | 37 ± 4 |
| HET0016-Low | 40 ± 3 | 40 ± 6 | 42 ± 8 | 96 ± 9 | 38 ± 6 | 37 ± 4 | 36 ± 5 |
| HET0016-High | 40 ± 5 | 42 ± 4 | 44 ± 3 | 95 ± 13 | 39 ± 7 | 38 ± 4 | 37 ± 3 |
| Arterial pH | |||||||
| Vehicle | 7.45 ± 0.03 | 7.30 ± 0.09 | 7.29 ± 0.10 | 6.92 ± 0.07 | 7.42 ± 0.09 | 7.46 ± 0.07 | 7.47 ± 0.08 |
| HET0016-Low | 7.43 ± 0.03 | 7.22 ± 0.07 | 7.21 ± 0.10 | 6.86 ± 0.06 | 7.37 ± 0.06 | 7.43 ± 0.04 | 7.45 ± 0.07 |
| HET0016-High | 7.44 ± 0.06 | 7.30 ± 0.07 | 7.25 ± 0.22 | 6.93 ± 0.15 | 7.40 ± 0.07 | 7.46 ± 0.06 | 7.46 ± 0.05 |
Values are mean ± SD; n = 7 in Vehicle group; n = 8 in HET0016 low- and high-dose groups.
After 4 days of recovery, sham-operated piglets treated with vehicle and HET0016 exhibited normal cytoarchitecture and cellular morphology in the striatum, whereas H-I piglets treated with vehicle displayed ischemic morphology (Figure 1A). Consistent with previously published work (Martin et al. 1997; Yang et al. 2007; Yang et al. 2010), most neurons in putamen after H-I exhibited cytoplasmic microvacuolation, eosinophilia, and nuclear pyknosis, or no longer had a distinct structure based on H&E staining. Because the number of viable neurons was not different between vehicle-treated and HET0016-treated sham piglets, results were combined into one sham group for statistical purposes. The number of viable neurons in the putamen of the H-I vehicle group was 21 ± 11% of the number in the sham-operated piglets (Figure 1B). Ischemic neuronal injury was less in groups treated with low- and high-dose HET0016 treatment. The density of viable neurons was significantly increased to 52 ± 20% by the low dose of HET0016 and to 62 ± 15% by the high dose of HET0016. No significant difference was detected between the two doses of HET0016 treatment.
Figure 1.

Effects of HET0016 on neuronal damage and neurologic deficits in piglets subjected to H-I. Piglets exposed to H-I or sham operation received vehicle, low-dose HET0016 (HET0016-L, 1 mg/kg), and high-dose HET0016 (HET0016-H, 10 mg/kg) treatment at 5 min of recovery. (A) Representative photographs of H&E-stained sections at 4 days of recovery show normal neuronal morphology (arrow head) and cytoarchitecture in putamen of sham-operated animals treated with vehicle. All H-I groups contained putaminal neurons that exhibited ischemic morphology (cytoplasmic microvacuolation, eosinophilia, and nuclear pyknosis, arrow) or that had lost distinct structure. However H-I piglets treated with low or high dose of HET0016 showed many neurons with normal morphology. Scale bar = 40 μm. (B) Quantitative results for viable putamen neurons (expressed as a percentage of the mean value of the sham+vehicle group). Data are means ± SD (n = 7 to 8 per group). *P < 0.05 versus sham-operated group; # P < 0.05 versus H-I vehicle group; ANOVA followed by the Student-Newman-Keuls test. (C) Neurologic scores during the 4-day recovery. Two-way ANOVA with repeated measured indicated a significant overall effect of treatment (P < .001) and recovery time (P < 0.001).
In caudate nucleus, neuronal injury was less severe and more variable than in putamen. The percentage of neurons with ischemic cytopathology was 69 ± 36% in the vehicle group, 87 ± 19% in the low dose of HET0016 group, 75 ± 19% in the high dose of HET0016 group. These values were not significantly different.
Sham-operated piglets treated with vehicle or HET0016 showed no neurobehavioral deficits. One day after H-I, most vehicle-treated piglets exhibited impaired consciousness, no light and/or no auditory reflexes, and low muscle tone; some showed no response to pain stimulation. Deficits diminished on subsequent days. Two-way repeated measures ANOVA indicated an overall protective effect of HET0016 treatment (P<0.001) and time (P<0.001) (Figure 1C).
Blood flow changes during and after H-I
CBF was measured in separate cohorts of piglets to determine if HET0016 post-treatment had any hemodynamic effects. Vehicle-treated sham animals (n = 4) showed relatively stable LDF over somatosensory cortex during the whole experiment, whereas LDF increased approximately 60% during the 40 min of hypoxia, declined sharply to 10% of the baseline level at the end of asphyxia, recovered to 30% above the baseline level transiently at 5 min after resuscitation, and gradually returned to the baseline level by 15 min of recovery (Figure 2A). Administration of the high-dose of HET0016 5 min after resuscitation (n = 4) or at an equivalent time in sham-operated piglets (n = 4) did not change LDF. Regional blood flow analysis in putamen with fluorescent microspheres did not reveal any significant change between 30 and 360 min of recovery compared to baseline in any group (Figure 2B). Thus, HET0016 post-treatment did not change CBF during the first 6 h of recovery.
Figure 2.

Laser-Doppler flow (LDF) over somatosensory cortex (A) and microsphere-determined blood flow in putamen (B) during baseline, hypoxia, asphyxia, and first 6 h of recovery after H-I or equivalent time in sham-operated piglets (means ± SD; n = 4 per group). Piglets were treated with vehicle or 10 mg/kg of HET0016 at 5 min of recovery.
Na+, K+-ATPase phosphorylation and activity and NR1 phosphorylation after H-I
The expression of the neuronal specific Na+,K+-ATPase α3 subunit and the more widespread α1 isoform remained unchanged with administration of HET0016 in both sham and H-I groups (Figure 3). HET0016 did not significantly change the basal level of Ser23 or Ser943 phosphorylation in sham piglets (n = 4) compared to sham piglets treated with vehicle (n = 4). Consistent with previous observations (Yang et al. 2007), H-I significantly increased both Ser23 and Ser943 phosphorylation (n = 6). The increase in Ser23 phosphorylation at 3 h after H-I was markedly reduced by the low dose of HET0016 treatment at 5 min after H-I (n = 6). In contrast, HET0016 did not inhibit the H-I–induced PKA-dependent phosphorylation of Ser943. At the same time, the activity of Na+,K+-ATPase in the putamen was markedly decreased at 3 h after H-I in the group treated with vehicle, which is consistent with previous work (Golden et al. 2001; Yang et al. 2007). Administration of HET0016 had no significant effect on Na+,K+-ATPase activity in sham-operated piglets. However, administration of HET0016 significantly increased Na+,K+-ATPase activity after H-I compared to vehicle treatment after H-I, and the level was not significantly different from the value seen in the sham group treated with vehicle.
Figure 3.

Western blot analysis showing effects of 1 mg/kg of HET0016 treatment at 5 min of recovery on phosphorylated Ser23 and Ser943 and total protein levels of Na+,K+-ATPase (NKA) in a membrane-enriched fraction of putamen at 3 h of recovery (n = 4 to 6 per group). Synaptophysin (Syn) was used as a loading control. NKA activity was measured in contralateral putamen. Optical density (O.D.) data (means ± SD) were normalized to the sham+vehicle value. *P < 0.05 versus sham+vehicle group; # P < 0.05 versus H-I+vehicle group; one-way ANOVA followed by the Student-Newman-Keuls test.
The NMDA receptor NR1 subunit can be phosphorylated by PKC at Ser896 or by PKA at Ser897. Both sites show increased phosphorylation during early recovery from H-I in piglet striatum and may play an important role in sustaining excitotoxic damage (Guerguerian et al. 2002; Yang et al. 2007). In agreement with earlier work, H-I increased phosphorylation of NR1 at Ser897 and Ser896 without a change in overall NR1 expression (n = 6) in putamen at 3 h of recovery (Figure 4). Administration of HET0016 did not change the basal level of NR1, phospho-NR1 Ser896, or phospho-NR1 Ser897 in sham-operated piglets (n=4 each). However, administration of the low dose of HET0016 (n=6) after 5 min of recovery following H-I selectively inhibited the increase in phosphorylation of Ser896 but not Ser897. The levels of total NR1 protein were unaffected by HET0016 treatment.
Figure 4.
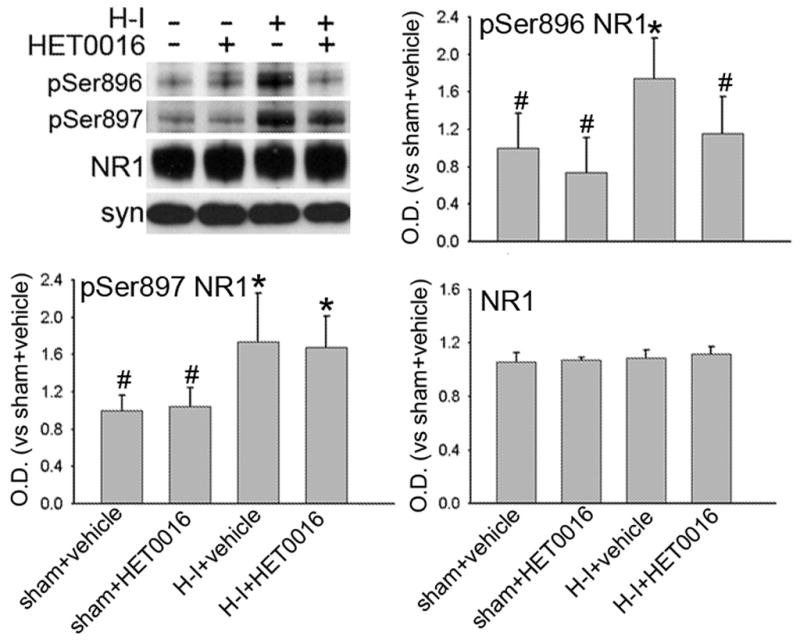
Western blot analysis showing effects of 1 mg/kg of HET0016 treatment at 5 min of recovery on phosphorylated Ser896 and Ser897 and total protein levels of NR1 in a membrane-enriched fraction of putamen at 3 h of recovery (n = 4 to 6 per group). Synaptophysin (Syn) was used as a loading control. Optical density (O.D.) data (means ± SD) were normalized to the sham+vehicle value. *P < 0.05 versus sham+vehicle group; # P < 0.05 versus H-I+vehicle group; one-way ANOVA followed by the Student-Newman-Keuls test.
Changes in Na+, K+-ATPase and NR1 phosphorylation during 20-HETE infusion
To determine if 20-HETE could directly increase phosphorylation at the PKC-sensitive sites on Na+,K+-ATPase and NR1 in the absence of ischemia, 30 μM of 20-HETE was perfused through microdialysis probes into piglet putamen. A relatively high infusate concentration was used to allow for diffusion limitation across the dialysis membrane, radial diffusion in the piglet putamen, reuptake into the phospholipid membrane, degradation, and clearance by blood flow. 20-HETE increased phosphorylation of Ser23 in Na+,K+-ATPase (Figure 5A) and Ser896 in NR1 (Figure 5B) compared to the levels in the contralateral putamen without a microdialysis probe (n = 4). No significant difference could be found in PKA-dependent Ser943 in Na+,K+-ATPase and Ser897 in NR1 in piglets treated with 20-HETE. Perfusion of microdialysis probes with vehicle did not alter the phosphorylation state at PKC- or PKA-sensitive sites compared to their contralateral levels (n = 3). The levels of total NR1 and Na+,K+-ATPase α1 and α3 protein were unaffected by 20-HETE or vehicle treatment.
Figure 5.

Western blot analysis showing effects of microdialysis perfusion of 30 μM of 20-HETE in piglet putamen on phosphorylated and total protein levels of Na+,K+-ATPase (A) and NMDA receptor NR1 subunit (B) in a membrane-enriched fraction of putamen of piglets without H-I (n = 3 to 4 per group). Synaptophysin (Syn) was used as a loading control. Optical density (O.D.) data (means ± SD) were normalized to the value of vehicle-treated group. *P < 0.05 versus vehicle group, Student’s t-test. c: putamen contralateral to microdialysis probe; i: putamen ipsilateral to microdialysis probe; Veh: vehicle treatment.
Neuronal CYP4A localization and Erk1/2 activation after H-I
The changes in the phosphorylation of the Na+,K+-ATPase α subunit and NR1 after 20-HETE and HET0016 administration suggest that 20-HETE acts within neurons. Members of the CYP4A family that possess ω-hydroxylase activity are located in vascular smooth muscle (Dunn et al. 2008). Double immunofluoresent staining was used to determine if CYP4A is expressed in neurons in piglet putamen. Robust CYP4A immunoreactivity was observed throughout the piglet putamen and was colocalized with NeuN-positive cells (Figure 6A).
Figure 6.

(A) Double immunofluorescent staining of normal pig striatum shows co-localization of CYP4A with NeuN. (B) Double immunofluorescent staining on piglet putamen at 3 h of recovery from H-I shows co-localization of JNK1,2,3, p38 MAPK, and Erk1/2 with NeuN. Scale bar = 20 μm.
MAPKs are involved in the regulation of neuronal survival and the pathophysiological process of brain ischemia (Nozaki et al. 2001; Sawe et al. 2008). We also tested whether HET0016 alters MAPKs’ activity after H-I. JNK1/2/3-, p38-, and Erk1/2-immuno-signals were localized in striatal neurons after H-I (Figure 6B). HET0016 did not change the basal level of phospho-JNK1/2/3, phospho-p38, or phospho-Erk1/2 in sham-operated piglets (n=4 each). H-I increased phosphorylation of p38 and Erk1/2, but not phosphorylation of JNK1/2/3 in putamen at 3 h of recovery (n=6). The low dose of HET0016 (n=6) inhibited the increase in phosphorylation of Erk1/2 but not p38 (Figure 7).
Figure 7.

Western blot analysis showing effects of 1 mg/kg of HET0016 treatment at 5 min of recovery on levels of phosphorylated and total JNK1,2,3 (JNK), p38 MAPK (p38), and Erk1/2 in cytosol-enriched fraction of putamen from H-I piglets at 3 h of recovery or equivalent time in sham-operated piglets (n = 4 to 6 per group). β-actin (actin) was used as a loading control. Optical density (O.D.) data (means ± SD) were normalized to the sham+vehicle value; *P < 0.05 versus sham+vehicle group. # P < 0.05 versus H-I+vehicle group; one-way ANOVA followed by the Student-Newman-Keuls test.
Nitrosative and oxidative stress after H-I
3-Nitrotyrosine immunoreactivity and protein carbonyl formation were used as markers of nitrosative and oxidative stress (Yang et al. 2007; Yang et al. 2010). Immunoblotting of membrane-enriched extracts from putamen at 3 h after H-I indicated increased 3-nitrotyrosine immunoreactivity in vehicle-treated piglets (Figure 8A). Treatment with the low dose of HET0016 had no effect on 3-nitrotyrosine immunoreactivity in the sham group, but it significantly reduced immunoreactivity in the H-I group to levels similar to that in the sham-vehicle group. Oxyblot analysis of carbonyl groups indicated a significant increase in the membrane-enriched proteins at 3 h of recovery from H-I (Figure 8B). HET0016 significantly attenuated carbonyl formation at 3 h of recovery from H-I.
Figure 8.

Effect of 1 mg/kg of HET0016 treatment at 5 min of recovery on H-I–induced nitrative and oxidative stress in putamen at 3 h of recovery. Western blot analysis showed that HET0016 decreased H-I–induced 3-nitrotyrosine (A) and carbonyl (B) immunoreactivity on multiple protein bands in putamen of H-I piglets. Optical density (O.D.; means ± SD) was integrated over multiple protein bands for nitrotyrosine (14–191 kDa) and carbonyls (29–98 kDa). *P < 0.05 versus sham+vehicle group; #P < 0.05 versus H-I+vehicle group; one-way ANOVA followed by the Student-Newman-Keuls test.
Discussion
The present study indicates that selective inhibition of the synthesis of 20-HETE with HET0016 after resuscitation in neonatal piglets (1) protected striatal neurons from H-I injury, (2) reduced H-I-induced phosphorylation at PKC-dependent sites of NMDA receptor NR1 subunit and Na+,K+-ATPase, (3) increased Na+, K+-ATPase activity early after H-I, (4) suppressed H-I induced Erk1/2 activation, and (5) reduced nitrosative and oxidative damage to proteins. Moreover, local administration of 20-HETE in non-ischemic putamen replicated phosphorylation selectively at PKC-sensitive sites on Na+,K+-ATPase and NR1.
Synthesis of 20-HETE by CYP 4A is sensitive to PO2 (Harder et al. 1996; Gebremedhin et al. 2008). Indeed, during the perinatal period, synthesis of 20-HETE in cerebrovascular smooth muscle is postulated to serve a physiological function by producing pial arterial constriction during the transition from the placental circulation to air breathing and thereby limit the increase in oxygen delivery to the brain at birth (Ohata et al. 2010). Likewise, 20-HETE synthesis might serve to limit CBF recovery during the reoxygenation after asphyxia. However, we found no evidence that HET0016 altered CBF during the first 6 h following H-I. Selective 20-HETE synthesis inhibitors have been shown to be protective following focal cerebral ischemia in adult animals (Miyata et al. 2005; Omura et al. 2006; Poloyac et al. 2006; Tanaka et al. 2007; Renic et al. 2009). However, administration of the inhibitor at reperfusion was able to reduce infarct volume without affecting LDF (Renic et al. 2009). In addition, superfusion of HET0016 over the pial surface did not significantly attenuate the loss of pial arteriolar dilation during focal cerebral ischemia (Cao et al. 2009). Thus, protection during reperfusion from focal ischemia and from global H-I in immature brain appears to be independent of CBF. Protection of hippocampal slice cultures from oxygen-glucose deprivation by HET0016 is also consistent with a non-vascular mechanism (Renic et al. 2011). The neuronal localization of CYP4A in naïve piglet putamen in our study, in post-ischemic cortical neurons of adult rat (Omura et al. 2006), and in neurons in hippocampal slice cultures (Renic et al. 2011) suggest that protection by 20-HETE synthesis inhibition may mediated by activation of a signaling cascade pathway in neurons.
PKC appears to play a critical role in 20-HETE–induced signal transduction. For example, PKC is involved in the mechanism by which 20-HETE inhibits opening of calcium-activated K+ channels and activates voltage-operated Ca2+ channels in vascular smooth muscle and cardiac myocytes (Lange et al. 1997; Sun et al. 1999; Zeng et al. 2010). Moreover, suppression of renal Na+,K+-ATPase by 20-HETE is also PKC-dependent (Nowicki et al. 1997). Na+,K+-ATPase plays an important role in maintaining ion gradients, osmotic balance, cell volume regulation, resting membrane potential, neuronal excitability, and the Na+-coupled reuptake of glutamate. PKC can phosphorylate Na+,K+-ATPase and decrease its activity in a cooperative manner with PKA (Cheng et al. 1997; Therien and Blostein 2000; Blanco 2005). Our previous work has indicated that the suppressive effect of dopamine D1 receptor activation on Na+,K+-ATPase during H-I is associated with PKA-dependent phosphorylation of Ser943 (Yang et al. 2007). Here, we show that 20-HETE can lead to phosphorylation of Ser23 in Na+,K+-ATPase in the putamen of piglets and that HET0016 can reduce the H-I induced phosphorylation at this PKC-sensitive site and limit the H-I–induced decrease in Na+,K+-ATPase activity. These findings are in agreement with a previous report in renal tubular cells showing 20-HETE suppression of activity associated with Ser23 phosphorylation (Nowicki et al. 1997). Because either a D1 antagonist or a 20-HETE synthesis inhibitor can improve striatal Na+,K+-ATPase activity after H-I in the present model, both Ser23 and Ser943 phosphorylation appear to be required for much of the observed suppression of activity. Together, these observations are consistent with protein studies showing that the PKC-mediated suppression of Na+,K+-ATPase activity requires Ser23 (Logvinenko et al. 1996) and that PKA-dependent phosphorylation of Ser943 modulates the PKC-dependent regulation of enzyme activity (Cheng et al. 1997).
Our previous work has shown that H-I could produce PKC-dependent phosphorylation of NR1 after reoxygenation (Guerguerian et al. 2002; Yang et al. 2007; Mueller-Burke et al. 2008). In the present work, 20-HETE was found to be capable of producing phosphorylation at the PKC-sensitive site, Ser896, on NR1, and HET0016 alleviated H-I–induced phosphorylation at this site. Phosphorylation is an important mechanism for the regulation of NMDA receptor function. Although It is not precisely known how phosphorylation at Ser896 modulates NMDA receptor channel activity, studies have shown that hypoxia increases NMDA receptor Ca2+ influx into cultured cortical neurons through a mechanism involving PKC-mediated phosphorylation of NR1 Ser896 (Bickler et al. 2004). PKC is thought to be involved in recruitment of NMDA receptors to the cell surface through exocytosis and potentiate its function (Carroll and Zukin 2002; Barria 2007). In addition, PKC is associated with rapid delivery of NMDA receptors to the neuronal dendritic shafts and spines (Lan et al. 2001). These findings imply that PKC-dependent phosphorylation of NR1 might augment downstream signal cascades that result from NMDA receptor activation after H-I. Therefore, the neuroprotection of HET0016 may rely, in part, on the alleviation of H-I induced PKC-dependent NR1 phosphorylation.
The MAPKs are a family of intracellular signaling molecules, including three major members: ERK (including ERK1/2), p38 MAPK, and JNK (including JNK1, JNK2, and JNK3). ERK, p38, and JNK represent three different signaling cascades that transduce a broad range of extracellular stimuli into diverse intracellular responses by both transcriptional and non-transcriptional regulation (Johnson and Lapadat 2002). All the MAPKs, activated by phosphorylation via different upstream MAPK kinases, regulate a diverse set of functions affecting cell fate, including cell proliferation and differentiation, adaptation to the stress, and apoptosis (Widmann et al. 1999). The MAPKs regulate neuronal survival and are involved in brain ischemia (Nozaki et al. 2001; Sawe et al. 2008). JNK and p38 MAPK are detrimental in ischemic brain, and inhibition of these two kinases can impede apoptosis (Barone et al. 2001; Guan et al. 2006). Erk1/2 activation was found in putamen of H-I piglet brain, which is consistent with a previous report of activated Erk1/2 in neonatal H-I rat brain (Wang et al. 2003). Although it is still not clear how 20-HETE activates Erk1/2, previous work has indicated that this effect is independent of PKC activation. For instance, 20-HETE can activate the raf/MEK/Erk pathway through epithelial growth factor receptor and c-Src in cultured renal epithelial cells (Akbulut et al. 2009). The precise role of activated Erk1/2 in ischemic brain is still debatable (Sawe et al. 2008), however, some evidence suggests that Erk1/2 may exacerbate neuronal injury. For example, activated Erk1/2 immuno-signals are mainly located in injured neurons early after H-I (Wang et al. 2003); Erk1/2 exaggerates inflammation via interleukin 1β up-regulation (Wang et al. 2004); and inhibition of Erk1/2 phosphorylation induced the NF-κB-regulated anti-apoptotic pathway (Lu et al. 2010). Therefore, further evidence is needed in the future to clarify the role of Erk1/2 activation in H-I piglet brain.
Synthesis of 20-HETE is associated with increased superoxide production in aortic endothelial cells (Cheng et al. 2008), cerebral arteries (Dunn et al. 2008), and hippocampal slice cultures exposed to oxygen-glucose deprivation (Renic et al. 2011). Here, we found evidence of increased nitrosative and oxidative stress that was alleviated by the 20-HETE synthesis inhibitor. Therefore, whether the reduced oxidative stress is directly related to reduced synthesis of 20-HETE or secondary to downstream effects of 20-HETE on PKC-dependent alterations of NMDA receptors, Na+,K+-ATPase activity, or voltage-operated Ca2+ channels in neurons requires more detailed study in neuronal systems.
A limitation of the present study is that we did not measure tissue concentrations of 20-HETE after H-I and demonstrate that HET0016 inhibited an increase in 20-HETE concentration. Although HET0016 is a specific inhibitor of 20-HETE synthesizing enzyme relative to other eicosanoid enzymes and reduces 20-HETE level in rat brain (Miyata et al. 2001; Renic et al. 2009), we cannot exclude an off-target effect of HET0016.
H-I encephalopathy in term newborns produces chronic neurological disability in survivors. Our study demonstrates that treatment with two different doses of the selective 20-HETE synthesis inhibitor HET0016 after H-I reduces neuronal injury in selectively vulnerable putamen and accelerates neurologic recovery in a large animal model of neonatal H-I encephalopathy. One limitation of the present study is that the time course of phosphorylation changes and the efficacy of delayed treatment were not determined. Nevertheless, the present study demonstrates that 20-HETE plays an important role in intensifying neuronal injury in the selectively vulnerable putamen and leads to the marked suppression of Na+,K+-ATPase activity and to nitrative and oxidative protein alterations. These adverse effects of 20-HETE may be mediated by PKC-dependent phosphorylation of Na+,K+-ATPase α and NMDA receptor NR1subunits. In addition, Erk1/2 activation may also be involved in the pathophysiological process. Therefore, 20-HETE is an important mediator that contributes to oxidative stress and neurodegeneration in global cerebral ischemia in immature brain. Therapeutic use of a 20-HETE synthesis inhibitor may provide a new strategy for reducing brain damage after neonatal H-I injury.
Acknowledgments
This work was supported by NIH grants NS060703 (R.C.K.), HL059996 (D.R.H., R.J.R., R.C.K), HL033833 (D.R.H) and HL092105 (D.R.H.), and by an American Heart Association-Phillips Resuscitation Research Fellow Award (Z.-J.Y.) The authors have no competing financial interest.
Abbreviations used
- 20-HETE
20-hydroxyeicosatetraenoic acid
- CYP
cytochrome
- P450 H-I
hypoxia-ischemia
- NMDA
N-methyl-D-aspartate
- PKC
protein kinase C
- CBF
cerebral blood flow
- MAPK
mitogen-activated protein kinase
- Erk1/2
extracellular signal-regulated kinase 1/2
- HET0016
N-hydroxy-N′-(4-n-butyl-2-methylphenyl) formamidine
- PKA
protein kinase A
- LDF
laser-Doppler flow
- PBS
phosphate-buffered saline
References
- Agnew DM, Koehler RC, Guerguerian AM, Shaffner DH, Traystman RJ, Martin LJ, Ichord RN. Hypothermia for 24 hours after asphyxic cardiac arrest in piglets provides striatal neuroprotection that is sustained 10 days after rewarming. Pediatr Res. 2003;54:253–262. doi: 10.1203/01.PDR.0000072783.22373.FF. [DOI] [PubMed] [Google Scholar]
- Akbulut T, Regner KR, Roman RJ, Avner ED, Falck JR, Park F. 20-HETE activates the Raf/MEK/ERK pathway in renal epithelial cells through an EGFR- and c-Src-dependent mechanism. Am J Physiol Renal Physiol. 2009;297:F662–670. doi: 10.1152/ajprenal.00146.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, Legos JJ, Erhardt JA, Ohlstein EH, Hunter AJ, Harrison DC, Philpott K, Smith BR, Adams JL, Parsons AA. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev. 2001;21:129–145. doi: 10.1002/1098-1128(200103)21:2<129::aid-med1003>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Barria A. Subunit-specific NMDA receptor trafficking to synapses. Elsevier Academic Press; 2007. pp. 203–206. [DOI] [PubMed] [Google Scholar]
- Berkowitz ID, Gervais H, Schleien CL, Koehler RC, Dean JM, Traystman RJ. Epinephrine dosage effects on cerebral and myocardial blood flow in an infant swine model of cardiopulmonary resuscitation. Anesthesiology. 1991;75:1041–1050. doi: 10.1097/00000542-199112000-00017. [DOI] [PubMed] [Google Scholar]
- Bickler PE, Fahlman CS, Ferriero DM. Hypoxia increases calcium flux through cortical neuron glutamate receptors via protein kinase C. J Neurochem. 2004;88:878–884. doi: 10.1046/j.1471-4159.2003.02203.x. [DOI] [PubMed] [Google Scholar]
- Blanco G. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin Nephrol. 2005;25:292–303. doi: 10.1016/j.semnephrol.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Cao S, Wang LC, Kwansa H, Roman RJ, Harder DR, Koehler RC. Endothelin rather than 20-HETE contributes to loss of pial arteriolar dilation during focal cerebral ischemia with and without polymeric hemoglobin transfusion. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1412–1418. doi: 10.1152/ajpregu.00003.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Zukin RS. NMDA-receptor trafficking and targeting: implications for synaptic transmission and plasticity. Trends Neurosci. 2002;25:571–577. doi: 10.1016/s0166-2236(02)02272-5. [DOI] [PubMed] [Google Scholar]
- Cheng J, Ou JS, Singh H, Falck JR, Narsimhaswamy D, Pritchard KA, Jr, Schwartzman ML. 20-hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2008;294:H1018–1026. doi: 10.1152/ajpheart.01172.2007. [DOI] [PubMed] [Google Scholar]
- Cheng XJ, Hoog JO, Nairn AC, Greengard P, Aperia A. Regulation of rat Na(+)-K(+)-ATPase activity by PKC is modulated by state of phosphorylation of Ser-943 by PKA. Am J Physiol. 1997;273:C1981–1986. doi: 10.1152/ajpcell.1997.273.6.C1981. [DOI] [PubMed] [Google Scholar]
- Dixon G, Badawi N, Kurinczuk JJ, Keogh JM, Silburn SR, Zubrick SR, Stanley FJ. Early developmental outcomes after newborn encephalopathy. Pediatrics. 2002;109:26–33. doi: 10.1542/peds.109.1.26. [DOI] [PubMed] [Google Scholar]
- Dunn KM, Renic M, Flasch AK, Harder DR, Falck J, Roman RJ. Elevated production of 20-HETE in the cerebral vasculature contributes to severity of ischemic stroke and oxidative stress in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;295:H2455–2465. doi: 10.1152/ajpheart.00512.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebremedhin D, Yamaura K, Harder DR. Role of 20-HETE in the hypoxia-induced activation of Ca2+-activated K+ channel currents in rat cerebral arterial muscle cells. Am J Physiol Heart Circ Physiol. 2008;294:H107–120. doi: 10.1152/ajpheart.01416.2006. [DOI] [PubMed] [Google Scholar]
- Glenny RW, Bernard S, Brinkley M. Validation of fluorescent-labeled microspheres for measurement of regional organ perfusion. J Appl Physiol. 1993;74:2585–2597. doi: 10.1152/jappl.1993.74.5.2585. [DOI] [PubMed] [Google Scholar]
- Golden WC, Brambrink AM, Traystman RJ, Martin LJ. Failure to sustain recovery of Na,K-ATPase function is a possible mechanism for striatal neurodegeneration in hypoxic-ischemic newborn piglets. Brain Res Mol Brain Res. 2001;88:94–102. doi: 10.1016/s0169-328x(01)00032-8. [DOI] [PubMed] [Google Scholar]
- Guan QH, Pei DS, Zong YY, Xu TL, Zhang GY. Neuroprotection against ischemic brain injury by a small peptide inhibitor of c-Jun N-terminal kinase (JNK) via nuclear and non-nuclear pathways. Neuroscience. 2006;139:609–627. doi: 10.1016/j.neuroscience.2005.11.067. [DOI] [PubMed] [Google Scholar]
- Guerguerian AM, Brambrink AM, Traystman RJ, Huganir RL, Martin LJ. Altered expression and phosphorylation of N-methyl-D-aspartate receptors in piglet striatum after hypoxia-ischemia. Brain Res Mol Brain Res. 2002;104:66–80. doi: 10.1016/s0169-328x(02)00285-1. [DOI] [PubMed] [Google Scholar]
- Harder DR, Gebremedhin D, Narayanan J, Jefcoat C, Falck JR, Campbell WB, Roman R. Formation and action of a P-450 4A metabolite of arachidonic acid in cat cerebral microvessels. Am J Physiol. 1994;266:H2098–2107. doi: 10.1152/ajpheart.1994.266.5.H2098. [DOI] [PubMed] [Google Scholar]
- Harder DR, Narayanan J, Birks EK, Liard JF, Imig JD, Lombard JH, Lange AR, Roman RJ. Identification of a putative microvascular oxygen sensor. Circ Res. 1996;79:54–61. doi: 10.1161/01.res.79.1.54. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Lange A, Gebremedhin D, Narayanan J, Harder D. 20-Hydroxyeicosatetraenoic acid-induced vasoconstriction and inhibition of potassium current in cerebral vascular smooth muscle is dependent on activation of protein kinase C. J Biol Chem. 1997;272:27345–27352. doi: 10.1074/jbc.272.43.27345. [DOI] [PubMed] [Google Scholar]
- Logvinenko NS, Dulubova I, Fedosova N, Larsson SH, Nairn AC, Esmann M, Greengard P, Aperia A. Phosphorylation by protein kinase C of serine-23 of the alpha-1 subunit of rat Na+,K(+)-ATPase affects its conformational equilibrium. Proc Natl Acad Sci U S A. 1996;93:9132–9137. doi: 10.1073/pnas.93.17.9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu K, Liang CL, Liliang PC, Yang CH, Cho CL, Weng HC, Tsai YD, Wang KW, Chen HJ. Inhibition of extracellular signal-regulated kinases 1/2 provides neuroprotection in spinal cord ischemia/reperfusion injury in rats: relationship with the nuclear factor-kappaB-regulated anti-apoptotic mechanisms. J Neurochem. 2010;114:237–246. doi: 10.1111/j.1471-4159.2010.06747.x. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Brambrink A, Koehler RC, Traystman RJ. Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. J Comp Neurol. 1997;377:262–285. doi: 10.1002/(sici)1096-9861(19970113)377:2<262::aid-cne8>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Brambrink AM, Price AC, Kaiser A, Agnew DM, Ichord RN, Traystman RJ. Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol Dis. 2000;7:169–191. doi: 10.1006/nbdi.2000.0282. [DOI] [PubMed] [Google Scholar]
- Marumo T, Eto K, Wake H, Omura T, Nabekura J. The inhibitor of 20-HETE synthesis, TS-011, improves cerebral microcirculatory autoregulation impaired by middle cerebral artery occlusion in mice. Br J Pharmacol. 2010;161:1391–1402. doi: 10.1111/j.1476-5381.2010.00973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata N, Taniguchi K, Seki T, Ishimoto T, Sato-Watanabe M, Yasuda Y, Doi M, Kametani S, Tomishima Y, Ueki T, Sato M, Kameo K. HET0016, a potent and selective inhibitor of 20-HETE synthesizing enzyme. Br J Pharmacol. 2001;133:325–329. doi: 10.1038/sj.bjp.0704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata N, Seki T, Tanaka Y, Omura T, Taniguchi K, Doi M, Bandou K, Kametani S, Sato M, Okuyama S, Cambj-Sapunar L, Harder DR, Roman RJ. Beneficial effects of a new 20-hydroxyeicosatetraenoic acid synthesis inhibitor, TS-011 [N-(3-chloro-4-morpholin-4-yl) phenyl-N′-hydroxyimido formamide], on hemorrhagic and ischemic stroke. J Pharmacol Exp Ther. 2005;314:77–85. doi: 10.1124/jpet.105.083964. [DOI] [PubMed] [Google Scholar]
- Mueller-Burke D, Koehler RC, Martin LJ. Rapid NMDA receptor phosphorylation and oxidative stress precede striatal neurodegeneration after hypoxic ischemia in newborn piglets and are attenuated with hypothermia. Int J Dev Neurosci. 2008;26:67–76. doi: 10.1016/j.ijdevneu.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthalif MM, Benter IF, Karzoun N, Fatima S, Harper J, Uddin MR, Malik KU. 20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin-dependent protein kinase II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Proc Natl Acad Sci U S A. 1998;95:12701–12706. doi: 10.1073/pnas.95.21.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicki S, Chen SL, Aizman O, Cheng XJ, Li D, Nowicki C, Nairn A, Greengard P, Aperia A. 20-Hydroxyeicosa-tetraenoic acid (20 HETE) activates protein kinase C. Role in regulation of rat renal Na+,K+-ATPase. J Clin Invest. 1997;99:1224–1230. doi: 10.1172/JCI119279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki K, Nishimura M, Hashimoto N. Mitogen-activated protein kinases and cerebral ischemia. Mol Neurobiol. 2001;23:1–19. doi: 10.1385/MN:23:1:01. [DOI] [PubMed] [Google Scholar]
- Ohata H, Gebremedhin D, Narayanan J, Harder DR, Koehler RC. Onset of pulmonary ventilation in fetal sheep produces pial arteriolar constriction dependent on cytochrome p450 omega-hydroxylase activity. J Appl Physiol. 2010;109:412–417. doi: 10.1152/japplphysiol.01090.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Tanaka Y, Miyata N, Koizumi C, Sakurai T, Fukasawa M, Hachiuma K, Minagawa T, Susumu T, Yoshida S, Nakaike S, Okuyama S, Harder DR, Roman RJ. Effect of a new inhibitor of the synthesis of 20-HETE on cerebral ischemia reperfusion injury. Stroke. 2006;37:1307–1313. doi: 10.1161/01.STR.0000217398.37075.07. [DOI] [PubMed] [Google Scholar]
- Poloyac SM, Zhang Y, Bies RR, Kochanek PM, Graham SH. Protective effect of the 20-HETE inhibitor HET0016 on brain damage after temporary focal ischemia. J Cereb Blood Flow Metab. 2006;26:1551–1561. doi: 10.1038/sj.jcbfm.9600309. [DOI] [PubMed] [Google Scholar]
- Renic M, Kumar SN, Gebremedhin D, Florence MA, Gerges NZ, Falck JR, Harder DR, Roman RJ. Protective effect of 20-HETE inhibition in a model of oxygen-glucose deprivation in hippocampal slice cultures. Under revision. 2011 doi: 10.1152/ajpheart.00340.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renic M, Klaus JA, Omura T, Kawashima N, Onishi M, Miyata N, Koehler RC, Harder DR, Roman RJ. Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2009;29:629–639. doi: 10.1038/jcbfm.2008.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas-Zeballos M, Zeballos GA, Gootman PM. A sterotaxic atlas of the developing swine (Sus scrofa) forebrain. Vol. 2. Plenum Press; New York: 1986. pp. 887–906. [Google Scholar]
- Sawe N, Steinberg G, Zhao H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J Neurosci Res. 2008;86:1659–1669. doi: 10.1002/jnr.21604. [DOI] [PubMed] [Google Scholar]
- Sun CW, Falck JR, Harder DR, Roman RJ. Role of tyrosine kinase and PKC in the vasoconstrictor response to 20-HETE in renal arterioles. Hypertension. 1999;33:414–418. doi: 10.1161/01.hyp.33.1.414. [DOI] [PubMed] [Google Scholar]
- Szatkowski M, Attwell D. Triggering and execution of neuronal death in brain ischaemia: two phases of glutamate release by different mechanisms. Trends Neurosci. 1994;17:359–365. doi: 10.1016/0166-2236(94)90040-x. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Omura T, Fukasawa M, Horiuchi N, Miyata N, Minagawa T, Yoshida S, Nakaike S. Continuous inhibition of 20-HETE synthesis by TS-011 improves neurological and functional outcomes after transient focal cerebral ischemia in rats. Neurosci Res. 2007;59:475–480. doi: 10.1016/j.neures.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Therien AG, Blostein R. Mechanisms of sodium pump regulation. Am J Physiol Cell Physiol. 2000;279:C541–566. doi: 10.1152/ajpcell.2000.279.3.C541. [DOI] [PubMed] [Google Scholar]
- Van Oosterhout MF, Willigers HM, Reneman RS, Prinzen FW. Fluorescent microspheres to measure organ perfusion: validation of a simplified sample processing technique. Am J Physiol. 1995;269:H725–733. doi: 10.1152/ajpheart.1995.269.2.H725. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhu C, Qiu L, Hagberg H, Sandberg M, Blomgren K. Activation of ERK1/2 after neonatal rat cerebral hypoxia-ischaemia. J Neurochem. 2003;86:351–362. doi: 10.1046/j.1471-4159.2003.01838.x. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Wu DC, Huang FP, Yang GY. Inhibition of MEK/ERK 1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia. Brain Res. 2004;996:55–66. doi: 10.1016/j.brainres.2003.09.074. [DOI] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Yang ZJ, Carter EL, Torbey M, Martin LJ, Koehler RC. Sigma receptor ligand 4-phenyl-1-(4-phenylbutyl)-piperidine modulates neuronal nitric oxide synthase/postsynaptic density-95 coupling mechanisms and protects against neonatal ischemic degeneration of striatal neurons. Exp Neurol. 2010;221:166–174. doi: 10.1016/j.expneurol.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZJ, Ni X, Carter EL, Kibler K, Martin LJ, Koehler RC. Neuroprotective effect of acid-sensing ion channel inhibitor psalmotoxin-1 after hypoxia-ischemia in newborn piglet striatum. Neurobiol Dis. 2011;43:446–454. doi: 10.1016/j.nbd.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZJ, Torbey M, Li X, Bernardy J, Golden WC, Martin LJ, Koehler RC. Dopamine receptor modulation of hypoxic-ischemic neuronal injury in striatum of newborn piglets. J Cereb Blood Flow Metab. 2007;27:1339–1351. doi: 10.1038/sj.jcbfm.9600440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q, Han Y, Bao Y, Li W, Li X, Shen X, Wang X, Yao F, O’Rourke ST, Sun C. 20-HETE increases NADPH oxidase-derived ROS production and stimulates the L-type Ca2+ channel via a PKC-dependent mechanism in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2010;299:H1109–1117. doi: 10.1152/ajpheart.00067.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]