

Abstract
Tuberculosis (TB) is a devastating disease resulting in a death every 20 seconds. Thus, new drugs are urgently needed. Herein we report ten classes of compounds—oxazoline, oxazole, thiazoline, thiazole, pyrazole, pyridine, isoxazole, imidazo[1,2-a]pyridine, imidazo[1,2-a]pyrimidine and imidazo[1,2-c]pyrimidine—which have good (micromolar) to excellent (sub-micromolar) antitubercular potency. The 5,6-fused heteroaromatic compounds were the most potent with MIC’s as low as <0.195 μM (9 and 11). Overall, the imidazo[1,2-a]pyridine class was determined to be most promising, with potency similar to isoniazid and PA-824 against replicating Mtb H37Rv, clinically relevant drug sensitive, multi- and extensively resistant Mtb strains as well as having good in vitro metabolic stability.
Keywords: Mycobacterium tuberculosis; MDR-TB; XDR-TB; 5,6-fused heteroaromatics; imidazo[1,2-a]pyridine
1. Introduction
The battle against tuberculosis (TB, caused by the bacterium Mycobacterium tuberculosis, Mtb) has raged for millennia.1 Throughout history, TB has claimed the lives of over one billion people and currently infects one third of the world’s population. With 1.4 million deaths in 2010, TB, as a single causative agent, is the leading killer among infectious diseases.1 The spread of TB was significantly affected with the advent of several chemotherapy agents during the mid 1900s.2 However, since the 1980s TB has been on the rise. Presently, 8.8 million new cases are added annually.1 The increase in cases of TB/HIV co-infection and the spread of multiple-drug resistant TB (MDR-TB, strains that are resistant to first line drugs isoniazid and rifampin) and extensively drug resistant TB (XDR-TB, strains that are resistant to isoniazid and rifampin, as well as any fluoroquinolone and at least one of three injectable second-line drugs, such as amikacin, kanamycin, or capreomycin) are making matters worse.3 The increase in TB infection has become so alarming that the World Health Organization (WHO) declared TB a global emergency as far back as 1993.4 More than ever, there is an urgent need to develop new antitubercular drugs to the spread of TB, particularly in its hard-to-kill multidrug-resistant and latent forms where, most likely, only sustained aggressive chemotherapy with a combination of drugs will stop the disease.
Our laboratory serendipitously discovered a small molecule heterocyclic TB inhibitor based on an oxazoline scaffold through broad screening of synthetic mycobactin siderophore fragments.5 This hit was initially optimized by dehydration to an oxazole and hundreds of analogs were prepared in an effort to increase potency against Mtb and possibly refine ADME (absorption, distribution, metabolism and excretion) properties.5,6 Not previously reported was that many amides and hindered benzyl ester derivates were prepared in an effort to improve the in vitro stability to rat microsomes and simulated gastric fluid. However, very little improvement was found and nearly all the amides prepared had substantially less potency against Mtb relative to the corresponding ester (see supporting information). Therefore, we were compelled to reprioritize the risk of trying to find a new metabolically robust heterocyclic class of antitubercular compounds over the optimization of oxazoline/oxazole class. Herein we report our structure activity relationship (SAR) studies starting from the serendipitous oxazoline/oxazole benzyl ester discovery7 and leading to the identification of imidazo[1,2-a]pyridines as a new class of potent and metabolically robust antitubercular agents8,9 (see Figure 1).
Figure 1.

Various heterocyclic classes (1–14) explored for antitubercular activity.
2. Results and Discussion
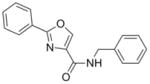
The syntheses of the oxazoline and oxazole scaffolds started by coupling of benzoyl chloride with L-serine benzyl ester followed by the dehydrative cyclization of the resulting β-hydroxy amide with bis(2-methoxyethyl)amino-sulfur trifluoride (DAST) as reported by Wipf et al.11 to form the oxazoline core (1) in 84% yield for the two steps. This oxazoline (1) was converted in 91% yield to the corresponding oxazole analog (2) by a mild BrCCl3/DBU oxidation procedure12 (Scheme 1). Hydrogenolysis of compound 2 gave the oxazole carboxylic acid intermediate which was converted to the corresponding benzyl amide (3) by an EDC-mediated coupling with benzyl amine (67% overall yield).
Scheme 1.
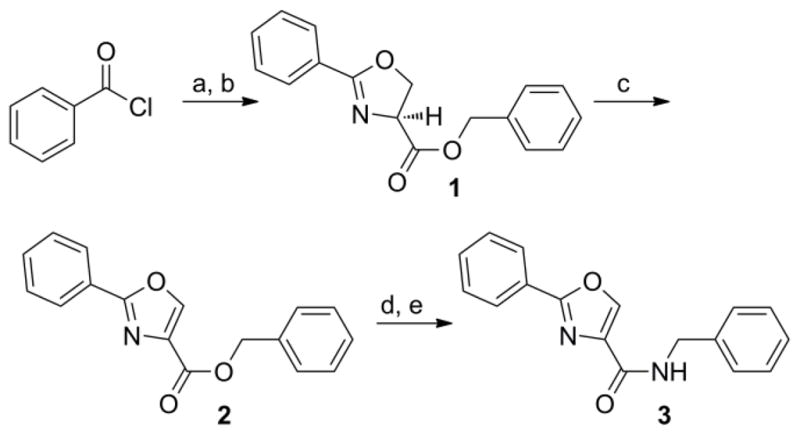
Synthesis of oxazoline (1) and oxazoles (2, 3) analogs. Reagents: (a) L-serine benzyl ester, DIPEA, CH2Cl2, 0°C to 50°C, 14 h; (b) DAST, K2CO3, −78°C, CH2Cl2, 1 h; (c) DBU, BrCCl3, CH2Cl2, 0°C to RT, 2 h; (d) H2, Pd-C, 14h; (e) Benzyl amine, EDC, DMAP, CH3CN, room temp, 14 h.
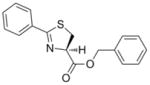
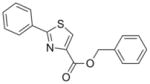
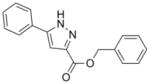
Thiazoline (4) was prepared by esterification of the corresponding carboxylic acid, which was obtained in 80% yield from condensation of benzonitrile and L-cysteine in a buffered aqueous methanol (pH 7.0) solution as reported Bergeron et al.12 (Scheme 2). Subsequent oxidation with BrCCl3/DBU gave the desired thiazole (5) in 68% yield. The pyrazole, isoxazole, pyridine and benzyl 7-methyl-2-phenylimidazo[1,2-a]pyridine-3-carboxylate analogs (6, 7, 8, and 9, respectively), as well as the N-benzyl-7-methyl-2-phenylimidazo[1,2-a]pyridine-3-carboxamide (10) and N-benzyl-2,7-dimethylimidazo[1,2-a]pyridine-3-carboxamide (12) were formed by separate EDC-mediated coupling reactions of the commercially available carboxylic acids with benzyl alcohol or benzyl amine, respectively (see supporting information) in yields ranging from −26% to 80%.
Scheme 2.

Syntheses of thiazoline (4) and thiazole (5) benzyl esters. Regents: (a) L-cysteine, NaHCO3, MeOH/H2O (pH = 7), reflux, 14 h; (b) Benzyl alcohol, EDC, DMAP, CH3CN, room temp, 14 h; (c) DBU, BrCCl3, CH2Cl2, 0°C to RT, 4 h.
Previously, we reported facile three step syntheses of various substituted N-benzyl imidazo[1,2-a]pyridine-3-carboxamides, including compound 12 that had potent antitubercular activity8. Herein we report that similar heterocycles, 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxylate (11), benzyl 2,6-dimethylimidazo[1, a]pyrimidine-3-carboxylate (13) and benzyl 2,5,7-trimethylimidazo[1,2-c]pyrimidine-3-carboxylate (14) can each be prepared in just a single step by condensation of benzyl 2-bromo-3-oxobutanoate with the appropriate 2-amino-heteroaromatic precursor demonstrating that these 5,6-fused heteroaromatic antitubercular agents can be made efficiently and cost effectively in respectable yields of 46%–75% (Scheme 3). These compounds also exhibit notable antitubercular activity against replicating and non-replicating Mtb and no toxicity to VERO cells (Table 1).
Scheme 3.
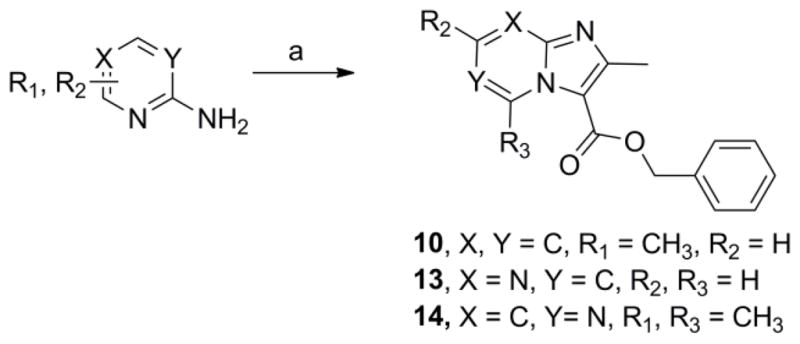
One step syntheses of benzyl 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxylate (10), benzyl 2-methylimidazo[1,2-a]pyrimidine-3-carboxylate (13) and benzyl 2,5,7-trimethylimidazo[1,2-c]pyrimidine-3-carboxylate (14). Reagents: (a) Benzyl 2-bromo-3-oxobutanoate, NaHCO3, 1,2-dimethoxyethane, 110°C, 24–36 h.
Table 1.
Activity of various heterocyclic scaffolds (1–14) against replicating Mtb H37Rv in various media, against non-replicating Mtb —LORA and cytotoxicity in VERO cells
| Compound | Mol. Wt. | Calc. ClogP* | MIC mean ±/− SD (μM) [MIC range] against replicating Mtb in medium: | LORA (μM) | VERO IC50 (μM) | ||
|---|---|---|---|---|---|---|---|
| GAS | GAST | 7H12 | |||||
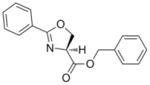 1 |
281.31 | 2.76 | 4.9 ± 1.7 [2.9–6.2] | 4.4 [5.7–3.7] | 16.6 ± 11.1 [3.8–24.0] | 63 | >128 |
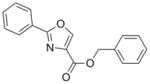 2 |
279.29 | 3.47 | 5.7± 2.3 [0.5–7.9] | 0.49 [<1–0.5] | 3.9± 1.0 [3.0–5.0] | 37 | 121 |
 3 |
278.31 | 3.70 | >65 [23.1–>128] | 59 | >40 [19.5–>50] | NT | NT |
 4 |
297.37 | 2.94 | 12.9± 4.8 [9.1–18.3] | 3.5 | 11.6± 5.1 [7.7–17.4] | NT | >128 |
 5 |
295.36 | 4.10 | 6.0± 4.0 [1.5–8.5] | 6.2 | 7.2± 3.0 [4.0–9.8] | NT | >128 |
 6 |
278.31 | 4.54 | 3.5± 1.4 [1.8–4.3] | >64 | >38 [15.0–>50] | NT | >128 |
 7 |
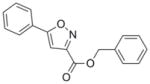
279.29 | 4.30 | 2.2± 1.0 [1.1–2.9] | NT | 0.8± 0.01 [0.7–0.8] | 28.0± 1.3 [26.9–29.4] | >50 |
 8 |
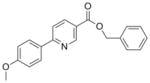
319.35 | 4.71 | 1.9±0.6 [1.5–2.5] | NT | 9.5±3.2 [5.8–11.9] | NT | NT |
 9 |
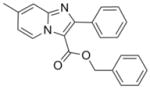
342.39 | 5.89 | <0.195 | NT | <0.195 | 21.7± 0.8 [21.1–22.6] | >50 |
 10 |
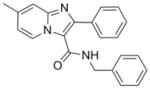
341.41 | 5.30 | 0.6±0.1 [0.5–0.7] | NT | 2.9±0.1 [2.8–3.0] | >50 | >50 |
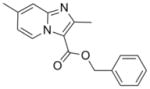 11 |
280.32 | 4.07 | <0.195 | NT | <0.195 | 34.8± 3.4 [31.3–38.2] | >50 |
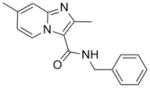 12 |
279.34 | 3.60 | 0.7 [0.4–1.1] | 2.0 [<0.5–3.4] | 2.3 [1.9–2.7] | 54 | >128 |
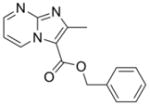 13 |
267.28 | 2.40 | 2.3±0.5 [1.7–2.7] | NT | 4.7±1.1 [3.2–5.2] | NT | >50 |
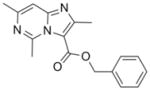 14 |
295.34 | 3.40 | 1.1±0.3 [0.9–1.4] | NT | 5.3±0.5 [5.0–5.9] | NT | >50 |
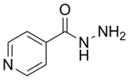 Isoniazid (INH) |
137.14 | −0.67 | 0.22±0.01 [0.23–0.22] | NT | 0.37±0.12 [0.23–0.46] | >512 | >128 |
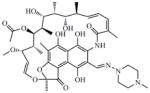 Rifampin (RMP) |
822.94 | 6.04 | 0.09±0.01 [0.08–0.1] | 0.2 | 0.04±0.01 [0.03–0.05] | 2.6 | 113 |
Calculated ClogP by ChemDraw version 12.0; SD = standard deviation; MIC = Minimum Inhibitory Concentration required to inhibit the growth of 90% of organisms; NT = Not tested; GAS = Glycerol-alanine-salts media; GAST = Iron deficient glycerol-alanine-salts with Tween 80 media; 7H12 = 7H9 broth base media with BSA, casein hydrolysate, catalase, palmitic acid; LORA = Low oxygen recovery assay using the Mtb H37Rv luxAB luminescence strain; VERO = African green monkey kidney cell line to evaluate toxicity. Values reported are the average of three individual measurements.
Previously, we spent considerable synthetic effort to develop an understanding of the SAR of the oxazoline/oxazole benzyl ester scaffold (1 and 2) by preparing nearly a thousand analogs and screening them for activity against Mtb H37Rv (ATCC #27294) by the microplate alamar blue assay (MABA).13 We anticipated that various benzyl amides (like 3) would not only retain potency seen with the benzyl esters but also improve the in vitro metabolic properties of the oxazoline and oxazole scaffolds. However, such modifications were not effective (see supporting information). Next, we explored the closely related thiazoline/thiazole scaffolds (4 and 5) and found that their MIC values (13 and 6 μM, respectively, in GAS13 media) were similar to those of the oxazoline/oxazoles (5 and 6 μM for 1 and 2, respectively, Table 1). The thiazoline/thiazole scaffolds (4 and 5) were also found to be non-toxic to VERO14 cells with IC50 values >128 μM, but, unfortunately, they showed no improved in vitro metabolic stability (see supporting information). The pyrazole scaffold (6) gave the most ambiguous screening results showing good activity in GAS media (3.5 μM, average of three tests), but weak potency in 7H1213 media (>38 μM, average of three tests). It is possible that the potency of the pyrazoles might be carbon source dependant, an issue experienced and described by Pethe et al. at Novartis,15 more protein bound in the 7H12 media than the GAS or just not very soluble. Nonetheless, we purposely screened our compounds against Mtb H37Rv in at least two different types of assay media as GAS media contains glycerol-alanine-salts while the 7H12 uses palmitic acid as its carbon source. Additionally, we periodically performed assays in GAST16 media to rule out possible iron dependence. Use of GAST, an iron deficient media containing glycerol-alanine-salts with Tween 80, often causes a shift in antibacterial MIC values of iron binding siderophores17 and hydroxamate-substituted cephalosporin derivatives.18 Though originally modeled after the oxazoline core of the mycobactin class of siderophores,17 all of the heterocyclic classes screened appear to be non-iron dependant. The isoxazole and pyridine scaffolds (7 and 8) showed good potency against Mtb H37Rv (2.2 and 1.9 μM, respectively in the GAS media) that was comparable to the activity of the analogous oxazoline/oxazole and thiazoline/thiazole scaffolds, but concerns about metabolic stability of the constituent benzyl ester lingered. The isoxazole scaffold (7) also has been well documented as a potent antitubercular scaffold by Kozikowski et al.19
At this juncture we were fortunate to have made an agreement allowing for the access and screening of compounds from the Dow AgroScience (DAS) chemical bank against Mtb. This allowed for a deepening of our SAR studies around our oxazole and thiazole scaffolds, as well as exploration of other heterocyclic classes including various ethyl imidazo[1,2-a]pyridine-3-carboxylates that showed moderate activity with MIC values of 41 to 86 μM (see supporting information). These initial imidazo[1,2-a]pyridines screened from DAS lacked the benzyl ester that was found to improve potency in the oxazoline/oxazole scaffolds we previously explored.5 Gratifyingly, when the benzyl ester analog (11) was made, a dramatic >2 orders of magnitude increase in potency was observed from an MIC of ~65 μM as the ethyl ester (79, in supporting information) to MIC <0.195 μM as the corresponding benzyl ester (11). In comparison, the benzyl ester oxazole (2), with an MIC range of 0.5–6 μM, was within an order of magnitude as potent as the corresponding ethyl ester analog (81, in supporting information) with an MIC of 8–20 μM. Therefore, simple ethyl esters do not appear to impart the sub-micromolar potency seen with the benzyl esters (see supporting information). Next, when the N-benzyl-2,7-dimethylimidazo[1,2-a]pyridine-3-carboxamide (12) was synthesized and assessed (MIC of 0.7–2.3 μM), we confirmed that extended scaffold SAR studies beyond esters had been appropriate. Indeed, the 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxamide scaffold displayed both excellent antitubercular potency, greatly improved in vitro metabolic stability and pharmacokinetic properties relative to the oxazoline/oxazole scaffolds (as reported previously in separate publications).5,8 The chemical dissimilarity between the oxazole and imidazo[1,2-a]pyridine scaffolds suggested that these compounds likely inhibited different targets, a notion that was confirmed by microarray analysis of Mtb H37Rv treated with these compounds which showed different transcriptional signatures (ref 8 and unpublished results). Imidazo[1,2-a]pyridine (12) had excellent in vitro stability in rat liver microsomes with 90% of compound remaining after a 15-min incubation and 100% remaining after a 15-min incubation in simulated gastric fluid20 compared to oxazole (2) where only 11% remained in microsomes and 0.6% remained in the simulated gastric fluid after a 15-min incubation. In 2009, various functionalized 2-aryl-3-amino-imidazo[1,2-a]pyridines were reported21 as in vitro Mtb glutamine synthetase inhibitors, but this publication neglected to give an assessment of these compounds versus whole cell Mtb H37Rv making it hard to compare these scaffolds for potential similarities. Previous data8 suggested a mechanism of action distinct from inhibition of glutamine synthesis since transcriptional profiling experiments of Mtb H37Rv treated with compound 12 had suggested an effect on energy metabolism. Additionally, a set of polyfunctional imidazo[1,2-a]pyridines was recently disclosed22 which displayed a broader spectrum of antibacterial activity than that which was published for our 2,7-dimethylimidazo[1,2-a]pyridine-3-carboxamide scaffold.8 In comparison, we reported our class to be active against only select mycobacteria strains (M. kansasii, M. bovis BCG, and M. avium, MIC’s 1.3, 0.3, 1.3 μM, respectively) while no activity (MIC > 50 μM) was seen against the Gram positive (S. aureus) or Gram negative (E. coli) organisms whereas the reported polyfunctional imidazo[1,2-a]pyridines did have potency against both the Gram positive and Gram negative organisms (MIC’s of 0.5 and 1 μg/mL, respectively). The potency of these compounds against M. avium further suggested that the target was unlikely glutamine synthetase since glutamine synthetase inhibitors were found to have considerably poorer efficacy against M. avium due to the finding that this organism secretes only small quantities of this protein.23
Comparison of the MIC ranges (taken from multiple screens) of the various scaffold analogs to one another showed a positive improvement in activity with the 5,6-fused heteroaromatics (9–14), displaying potency closer to that of the first line antitubercular drug isoniazid (MIC = 0.2–0.5 μM) and significantly better than the previous explored classes (1–8) that have low micromolar potency (Figure 2). The 5,6-fused heteroaromatics were non-toxic to the VERO cells (IC50 >50 μM) and some had moderate activity in the simulated latent TB assay (LORA16) with compound 9 having the lowest MIC of 22 μM (isoniazid was >512 μM). Again, the 5,6-fused heteroaromatics were prepared in just a single synthetic step with the exception of compounds 10 and 12.
Figure 2.

Comparison of the MIC range against Mtb H37Rv of the various heteroaromatic scaffolds prepared (MIC in μM).
Curious as to whether there would be potency against multi-drug resistant (MDR) and extensively drug resistant (XDR) Mtb strains, three compounds typical of their class—an oxazole (2), a thiazole (5) and an imidazo[1,2-a]pyridine (11), as well as the bactericidal nitroimidazole PA-82424 were screened against a panel of clinical drug sensitive and drug resistant strains (Table 2). We were pleased to find that the potency determined in replicating Mtb H37Rv (in the 7H9 media) was retained and at times improved against the drug sensitive, MDR- and XDR-Mtb strains screened for all three classes. Oxazole (2) had good potency in the lower micromolar range (MIC = 1–9 in μM) against these clinical strains, while the related thiazole (5) was significantly less potent having MIC values from 4 to 34 μM. The imidazo[1,2-a]pyridine (11) had potency better than that of PA-824, a clinical candidate, with MIC values of <0.04 μM against all of the clinical strains with the exception of one MDR strain (MIC = 0.6 μM) that is resistant to isoniazid, rifampin, ethambutol and rifabutin. Interestingly, there was a drug sensitive strain that showed resistance to PA-824 (MIC > 14 μM) but was nonetheless inhibited by the three compound classes. This finding was surprising but may be explained by the high mutation frequency previously found for this nitroimidazole compound.25 Imidazo[1,2-a]pyridine benzyl ester analog (11) was also slightly more potent against the clinical strains than that which was reported for imidazo[1,2-a]pyridine benzyl amide analog (12) which had MIC’s of 0.07 to 2.3 μM (against a panel of twelve MDR- and XDR-Mtb strains).8
Table 2.
MDR- and XDR-Mtb activity of compounds 2, 5, 11 and control PA-824 in μM (MIC in μg/mL).
| Compound MIC μM (μg/mL) | ||||
|---|---|---|---|---|
| 2 | 5 | 11 | PA-824 | |
| Drug sensitive Mtb clinical strain #1 | 4.4–8.9 (1.25–2.5) | 33.9 (10) | <0.04 (<0.01) | 0.45–0.86 (0.16–0.31) |
| Drug sensitive Mtb clinical strain #2 | 2.2–4.4 (0.6–1.25) | 8.5 (2.5) | <0.04 (<0.01) | >13.9 (>5) |
| MDR-TB resistant to HREZSKP | 8.9 (2.5) | 33.9 (10) | <0.04 (<0.01) | 0.45–0.86 (0.16–0.31) |
| MDR-TB resistant to HREKP | 1.1 (0.3) | 4.2 (1.25) | <0.04 (<0.01) | 0.45–0.86 (0.16–0.31) |
| MDR-TB resistant to HRERb | 8.9 (2.5) | 33.9 (10) | 0.57 (0.16) | 0.45–0.86 (0.16–0.31) |
| XDR-TB resistant to HRESKO | 4.4 (1.25) | 16.9 (5) | <0.04 (<0.01) | 0.86 (0.31) |
| XDR-TB resistant to HREKO | 8.9 (2.5) | 16.9 (5) | <0.04 (<0.01) | 0.22 (0.08) |
Almost no growth though up to 0.07 μM (0.02 μg/mL). MIC = Minimum Inhibitory Concentration required to inhibit the growth of 90% of organisms; MICs were done in 7H9/glucose/glycerol/BSA/0.05% Tween 80 and the average of three individual measurements. Abbrevations: H = isoniazid, R = rifampin, E = ethambutol, Z = pyrazinamide, S = streptomycin, K = kanamycin, P = para-aminosalicylic acid, Rb = rifabutin, O = ofloxacin.
Conclusion
Herein we report ten classes of compounds with good (micromolar) to excellent (sub-micromolar) antitubercular potency, suggesting that there are new antitubercular compound classes yet to be found. Three 5,6-fused heteroaromatic scaffolds (imidazo[1,2-a]pyridine, imidazo[1,2-a]pyrimidine, and imidazo[1,2-c]pyrimidine) were prepared in just one synthetic step and each is amenable to further elaboration. In particular, the imidazo[1,2-a]pyridine class emerged as the most promising by having potency similar to isoniazid and PA-824 against replicating Mtb H37Rv, clinically relevant drug sensitive, MDR- and XDR-Mtb strains (demonstrated by compound 11), as well as good in vitro metabolic stability (demonstrated by compound 12). It is our hope that the introduction of these various antitubercular compound classes will lower the barrier towards discovery of new antitubercular agents and inspire industry to join in the fight to combat TB.
Supplementary Material
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, NIAID and by Grant 2R01AI054193 from the National Institutes of Health (NIH) and in part by intermediates provided from DAS. We would like to thank the University of Notre Dame, especially the Mass Spectrometry and Proteomics Facility (Bill Boggess, Michelle Joyce, Nonka Sevova), which is supported by the grant CHE-0741793 from the NIH. We thank Prof. Jennifer DuBois for regular and lasting scientific discussions. The excellent technical assistance of Baojie Wan and Yuehong Wang with antitubercular assays at UIC is greatly appreciated.
Footnotes
Experimental procedures and analytical data for compounds (1-14) can be found as well as additional SAR, metabolism studies and a description of the assays used.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Global tuberculosis control WHO report. 2011. WHO/HTM/TB/2011.16. [Google Scholar]
- 2.Snider DE, Jr, Raviglione M, Kochi A. Global Burden of Tuberculosis, Tuberculosis: Pathogenesis, Protection, and Control. ASM Press; Washington, D.C.: 1994. [Google Scholar]
- 3.Sacchettini JC, Rubin EJ, Freundlich JS. Nat Rev Microbiol. 2008;6:41–52. doi: 10.1038/nrmicro1816. [DOI] [PubMed] [Google Scholar]
- 4.Tuberculosis: A global emergency. World Health Forum. 1993;14:438. [PubMed] [Google Scholar]
- 5.Moraski GC, Chang M, Villegas-Estrada A, Franzblau S, Möllmann U, Miller MJ. Eur J Med Chem. 2010;45:1703–1716. doi: 10.1016/j.ejmech.2009.12.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moraski GC, Franzblau SG, Miller MJ. Heterocycles. 2009;80:977–988. doi: 10.3987/COM-09-S(S)69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller MJ, Hu J. 6,403,623 B1. US Patent. 2002
- 8.Moraski GC, Markley LD, Hipskind PA, Boshoff H, Cho S, Franzblau SG, Miller MJ. Med Chem Lett. 2011;2:466–470. doi: 10.1021/ml200036r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller MJ, Moraski GC, Markley LD, Davis GE. 2011/057145 A2. WO. 2011
- 10.Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR. Org Lett. 2000;2:1165–1168. doi: 10.1021/ol005777b. [DOI] [PubMed] [Google Scholar]
- 11.Williams DR, Lowder PD, Gu YG, Brooks DA. Tet Lett. 1997;38:331–334. [Google Scholar]
- 12.Bergeron RJ. 6,559,315 B1. US Patent. 2003
- 13.Collins L, Franzblau SG. Antimicrob Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE. Proc Nat Acad Sci USA. 2000;97:1252–1257. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pethe K, Sequeira PC, Agarwalla S, Rhee K, Kuhen K, Phong WY, Patel V, Beer D, Walker JR, Duraiswamy J, Jiricek J, Keller TH, Chatterjee A, Tan MP, Ujjini M, Roa SPS, Camacho L, Bifani P, Mak PA, Ma I, Barnes SW. Nature Commun. 2010;57:1–8. doi: 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. Antimicrob Agents Chemother. 2007;51:1380–1385. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minnick AA, McKee JA, Dolence EK, Miller MJ. Antimicrob Agents Chemother. 1992;36:840–50. doi: 10.1128/aac.36.4.840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller MJ, Zhao G, Vakulenko S, Franzblau S, Möllmann U. Org Biomol Chem. 2006;4:4178–4185. doi: 10.1039/b612475e. [DOI] [PubMed] [Google Scholar]
- 19.Lilienkampf A, Pieroni M, Wan B, Wang Y, Franzblau SG, Kozikowski AP. J Med Chem. 2010;53:678–88. doi: 10.1021/jm901273n. [DOI] [PubMed] [Google Scholar]
- 20.Simulated gastric fluid and simulated intestinal fluid. In The United States Pharmacopeia 23, The National Formulary 18. The United States Pharmacopeial Convention, Inc; Rockville, MD: 1995. p. 2053. [Google Scholar]
- 21.Odell LR, Nilsson MT, Gising J, Lagerlund O, Muthas D, Nordqvist A, Karlen A, Larhed M. J Med Chem Lett. 2009;19:4790–4793. doi: 10.1016/j.bmcl.2009.06.045. [DOI] [PubMed] [Google Scholar]
- 22.Al-Tel TH, Al-Qawasmeh RA. Eur J Med Chem. 2010;45:5848–5855. doi: 10.1016/j.ejmech.2010.09.049. [DOI] [PubMed] [Google Scholar]
- 23.Harth G, Horwitz MA. J Exp Med. 1999;189:1425–36. doi: 10.1084/jem.189.9.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. Nature. 2000;405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 25.Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, Daniels L, Dick T, Pang SS, Barry CE. Proc Natl Acad Sci USA. 2006;103:431–6. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.