

Abstract
The objective of this work was the synthesis of serum albumin targeted, Gd(III)-based magnetic resonance imaging (MRI) contrast agents exhibiting a strong pH-dependent relaxivity. Two new complexes (Gd-glu and Gd-bbu) were synthesized based on the DO3A macrocycle modified with three carboxyalkyl substituents, α-to the three ring nitrogens, and a biphenylsulfonamide arm. The sulfonamide nitrogen coordinates the Gd in a pH-dependent fashion, resulting in a decrease in the hydration state, q, as pH is increased and a resultant decrease in relaxivity (r1). In the absence of human serum albumin (HSA), r1 increases from 2.0 to 6.0 mM−1s−1 for Gd-glu and from 2.4 to 9.0 mM−1s−1 for Gd-bbu from pH 5 to 8.5 at 37°C, 0.47T, respectively. These complexes (0.2 mM) are highly bound (>98.9%) to HSA (0.69 mM), over the pH range 5 – 8.5. Binding to albumin increases the rotational correlation time and results in higher relaxivity. r1 increased 120% (pH 5) and 550% (pH 8.5) for Gd-glu and 42% (pH 5) and 260% (pH 8.5) for Gd-bbu. The increases in r1 at pH 5 were unexpectedly low for a putative slow tumbling q=2 complex. The Gd-bbu system was investigated further. At pH 5, it binds in a stepwise fashion to HSA with dissociation constants Kd1=0.65, Kd2=18, Kd3=1360 μM. The relaxivity at each binding site was constant. Luminescence lifetime titration experiments with the Eu(III) analog revealed that the inner-sphere water ligands are displaced when the complex binds to HSA resulting in lower than expected r1 at pH 5. Variable pH and temperature nuclear magnetic relaxation dispersion (NMRD) studies showed that the increased r1 of the albumin-bound q=0 complexes is due to the presence of a nearby water molecule with a long residency time (1–2 ns). The distance between this water molecule and the Gd ion changes with pH resulting in albumin-bound pH dependent relaxivity.
Keywords: pH sensitive, Gadolinium, contrast agent, serum albumin, second-sphere relaxation, activatable
Introduction
The desire to produce contrast agents that are activated by changes in physiological pH is driven by the fact that the extracellular pH of solid tumors is more acidic than that of healthy tissue.[1] Tissue ischemia, e.g. in ischemic heart disease and stroke, also results in a lower extracellular pH.[2] Thus, pH may be a valuable biomarker for the identification of tumors and ischemic tissue. There has been great interest in mapping pH change and quantifying pH in vivo using a range of imaging techniques including positron emission tomography (PET), magnetic resonance imaging and spectroscopy (MRI and MRS), and optical imaging.[1a,3]
MRI has better spatial resolution than MRS or PET, and is not limited by light scattering and absorption like optical imaging. pH-dependent MRI requires a metal-based contrast agent whose proton relaxation enhancing properties (relaxivity) change with pH. Water is imaged in MRI and the contrast agent acts as a catalyst to enhance water proton relaxation rates (1/T1, 1/T2), with relaxivity defined as the change in relaxation rate normalized to agent concentration. A change in pH can alter relaxivity by changing the hydration state of the inner- or second-coordination sphere,[4] by affecting prototropic exchange,[5] or by changing the rotational diffusion rate of the molecule.[6] For example lowering pH can result in protonation of a donor atom on a multidentate ligand opening up a site(s) for a water ligand to coordinate the metal ion and increase relaxivity.[4]
There are several examples of pH-dependent contrast agents. However quantification of pH using MRI is challenging because the MR signal change depends on both the relaxivity of the probe (pH-dependent) and its concentration. This challenge has been met through two techniques. One approach is to use two very similar contrast agents, one pH independent and one pH dependent. The MR signal intensity versus time curve of the pH independent contrast agent is used to estimate time dependent concentration of the contrast agent, and it is assumed the pH-dependent agent will have the same concentration profile.[7] The limitation of this approach is the assumption of identical pharmacokinetics of two different compounds. Another approach is to incorporate a marker that is independently quantifiable within the pH dependent contrast agent. For example F-19 MR spectroscopy has been used to quantify concentration,[8] but MR spectroscopy is limited to high concentrations and lower spatial and temporal resolution compared gadolinium enhanced MRI. Incorporation of a PET reporter like F-18 can provide quantification,[9] but this requires the use of a simultaneous MR-PET device.
We reasoned that a contrast agent with a large enough relaxivity change could identify ischemic and acidic tissue. The pH range of physiological interest is between pH 6 – 7.5. The ideal pH-dependent contrast agent would have a very high relaxivity to be detectable at low concentrations, but more importantly would have a large change in relaxivity, Δr1 from pH 6.8 to pH 7.4. If the percentage change in relaxivity is higher than the difference in concentration between ischemic and normal tissue, then low pH regions may be identified, at least qualitatively. Such a high relaxivity, high delta relaxivity pH agent could also be adapted for quantitative pH imaging using the approaches described above.
We looked to the literature to identify promising approaches. In ischemic and/or cancerous tissue, low pH is observed in the extracellular interstitial space. A pH dependent contrast agent should be small enough to rapidly cross the endothelial barrier into the extracellular space. This rules out pH-dependent nanomaterials like liposomes,[10] carbon nanotubes,[11] large dendrimers,[6b, c] and polymers.[6a, 6d] Also preferred would be contrast agents whose relaxivity increased with decreasing pH such that pathology would appear bright in an image. One class of compounds that appeared particularly promising were Gd(DO3A) derivatives with a pendant sulfonamide reported by Lowe et al., Scheme 1, where R=CF3, OMe, or Me.[4a] At high pH, the sulfonamide nitrogen is deprotonated and coordinates the Gd(III) resulting in a complex with no inner-sphere water ligands (q=0). At low pH, the sulfonamide is protonated and does not coordinate, opening up two sites for water ligation and concomitant higher relaxivity.
Scheme 1.

These compounds had several favorable features. First, relaxivity increased significantly as pH was lowered. Second, the pKa of the sulfonamide was in the appropriate range for a physiological pH sensor, and this pKa could be modulated by the choice of electronic substituent on the aromatic ring. Third, the macrocyclic DO3A core should impart high thermodynamic stability and kinetic inertness with respect to Gd decomplexation. Finally, the pendant carboxylate arms overcame a major limitation of other Gd(DO3A) derivatives, that of anion binding and quenching of relaxivity via water displacement.
Here we report two new ligands that build on the design of Lowe et al.[4a] The Gd(III) complexes are prepared and evaluated as pH dependent contrast agents. The pH dependent relaxivity, serum albumin binding, and self association of these complexes is explored. In addition, the Eu(III) complexes are synthesized to explore the effects of protein binding and anion concentration on hydration number.
Results and Discussion
Compound Strategy and Synthesis
We sought to build on the successful molecular design of Lowe et al.[4a] Their sulfonamide derivatives showed an excellent relaxivity dependence on pH brought about by a change in hydration umber from q=0 to q=2 when the sulfonamide nitrogen was protonated. Importantly, the pendant carboxylate arms appeared to prevent anion coordination to the lanthanide when q=2.[12] Anion coordination would have the effect of displacing the inner-sphere water ligands and muting the pH-dependent relaxivity effect. This was especially true for the adipate derivative (n=2, Scheme 1).
In order to increase relaxivity and the dynamic range even further for this class of compounds, we sought to make them more avid for serum albumin. Non-covalent protein binding is a well-established technique for increasing the relaxivity of paramagnetic complexes.[13] Slowing molecular tumbling increases the efficiency of nuclear relaxation by the metal ion. For q=1 complexes, the protein-bound relaxivity can be on the order of 60 mM−1s−1 at 0.5T and 30 mM−1s−1 at 1.5T.[13a, 14] Gianolio et al. showed that a q=2 complex can have an albumin-bound relaxivity of 84 mM−1s−1 at 0.5T and over 60 mM−1s−1 at 1.5T.[15] On the other hand, albumin-bound q=0 complexes tend to have much lower relaxivities. We reported q=0 complexes with albumin-bound relaxivities of 5–6 mM−1s−1 at 1.5T.[14c, 16] Thus in principal, it should be possible to obtain a pH probe whose relaxity varies by a factor of 10 from low pH to high pH at the common clinical field strength of 1.5T.
With such a large change in relaxivity (6 to 60 mM−1s−1), even small changes in pH would be amplified. For instance using a pKa of 6.7 from Lowe et al.,[4a] one predicts a relaxivity difference of 15 mM−1s−1 between pH 7.4 (normal) and pH 6.8 (ischemia, tumor); on a percentage basis this would be a 100% change in relaxivity between these two pH values.
Lowe et al. saw a modest 2-fold increase in relaxivity for their compounds in the presence of serum albumin, but they did not report binding data for the albumin interaction.[4a] Based on the literature, it is likely that their compounds only exhibit a weak interaction with serum albumin; e.g. other hydrophilic chelates with a single aryl group have dissociation constants in the millimolar range resulting in only 10–15% of the complex being bound to protein under physiological conditions.[17] We have found that biphenyl groups impart sufficient lipophilicity to enhance albumin binding of gadolinium complexes such that >90% of the complex is bound under physiological conditions (~600 μM albumin).[13a, 14c, 18]
Based on this reasoning, the target complexes for our study were analogs of those reported by Lowe et al.[4a] and denoted as Gd-glu and Gd-bbu, Scheme 2. The synthetic strategy was similar to that reported previously but with some modifications. We first prepared the DO3A esters (g2) and (b2). Prior work utilized the dimethyl ester of 2-bromoglutarate or 2-bromoadipate. For the glutarate derivative we used benzyl-tert-butyl hydroxypentanedioate (g1) and converted it to a mesylate for the alkylation. This intermediate was available from prior studies in our lab.[19]
Scheme 2.

For the adipate derivative, we prepared (S)-dibenzyl-2-hydroxyhexanedioate (b1) using a similar procedure as Levy et al.,[19a] and then converted this to a mesylate for alkylation of cyclen. The benzyl ester provided a convenient UV visible handle for monitoring the reaction by HPLC. The best conditions for the tris-alkylation were 5 equivalents of the respective mesylate, 1 equivalent cyclen at 78 °C for 48 hr in acetonitrile with potassium carbonate as a base. The major impurity was the bis-alkylated product. The DO3A esters g2 and b2 were purified by preparative HPLC.
The biphenylsulfonamide moiety (a3) was prepared via Suzuki coupling. For alkylation of the DO3A derivatives, we converted the hydroxyl group to a mesylate (a4). Following deprotection, the ligands (b4, g4) were chelated with either GdCl3 or EuCl3. The final products were purified by preparative HPLC.
Relaxivity
The relaxivities of Gd-glu (Figure 1a and S2b) and Gd-bbu (Figure 1b and S2d) were determined as a function of pH in the range 5 to 8.5 (20 MHz, 60 MHz, 310 K). In the absence of HSA, the pH-dependent relaxivity of these two compounds was similar to what was reported by Lowe et al.[4a] Assuming that the change in relaxivity is due to deprotonation of the sulfonamide nitrogen and concomitant change in q, the pKa for this deprotonation was determined to be 6.49±0.17 for Gd-bbu and 6.58±0.07 for Gd-glu.
Figure 1.

Relaxivity vs pH at 37°C and 20 MHz for (a) 0.22 mM Gd-glu in the absence (circles) and presence of 0.79 mM HSA (triangles); (b) 0.21 mM Gd-bbu in the absence (circles) and presence of 0.69 mM HSA (triangles). Solid lines indicate fits to determine the pKa of the sulfonamide moiety.
We also noted that for Gd-bbu in the absence of protein, the solution became turbid in the pH range 5.5 to 4.5 and the onset of turbidity depended on the concentration of the compound. In this pH range the observed relaxivity appears to rise as a result of aggregation, but then decrease at pH 4.5 which may be a result of microscopic precipitation.
Relaxivity increased at all pH values when this pH titration was repeated in the presence of excess HSA. However the relaxivity increase at low pH was much less than expected (42% at 20 MHz, pH 5). Instead of rising to >60 mM−1s−1 as expected for a protein-bound q=2 complex, the relaxivity only approached 14 mM−1s−1. At high pH, the relaxivity was in the range expected for a q=0 complex bound to albumin. The relaxivity enhancement at pH 8.5 was 260% at 20 MHz for Gd-bbu. Similar behavior was observed for Gd-glu where the relaxivity in the presence of HSA was much lower than expected at pH 5. Relaxivities at 60 MHz (Figure S2a,c) were slightly lower than at 20 MHz but the shape of the r1 vs pH curve was the same as for the 20 MHz data.
This anomalously low relaxivity at low pH could arise from several possibilities. First, the affinity of the compound for HSA could be lower at lower pH, either because of a conformational change of the protein or because the phenol moiety on the compound is protonated at lower pH. This would result in fewer compounds bound at low pH and a smaller observed relaxivity increase. An alternative explanation is that the inner-sphere water molecules are displaced when the complex binds to albumin. This has been seen with other protein-bound GdDO3A derivatives, where presumably a protein side chain coordinates the Gd and displaces the water ligands.[14c, 16b, 20] Lowe et al. found that presence of the three pendant carboxylates prevented coordinating anions from displacing the inner-sphere water,[4a] and we anticipated that these pendant carboxylates would block protein side chains from binding to the metal ion. A third possibility is that the rate of inner-sphere water exchange is slow when the complex is protein bound. This is not expected given the fast water exchange of q=2 GdDO3A derivatives, but there is precedence for such an effect.[21] Finally, we noted an aggregation/precipitation phenomenon at lower pH values and this aggregation could interfere with protein binding and/or hydration state. We performed additional experiments to address these hypotheses.
pH dependent aggregation
We repeated the pH titration for Gd-bbu and performed dynamic light scattering (DLS) at each pH value. At about pH 5.5 we began to see the appearance of large aggregates (diameter, d=700 nm) with high polydispersity. In Figure 2a we show the DLS results plotted as relative scattering intensity vs pH (a relative scattering intensity of 1 indicates no aggregation). We then explored the effect of adding HSA to the aggregated Gd-bbu at pH 5, Figure 2b. Even at a low HSA: Gd-bbu ratio of 0.25, we see an immediate drop in scattering intensity. The large 700 nm aggregate (Figure 2c,d,e) disappears with addition of HSA suggesting that the HSA binding of discrete Gd-bbu breaks up the aggregation. After addition of HSA, the DLS analysis is the same as for HSA alone, with a single species of diameter 8 nm. This drop in scattering intensity and loss of aggregation at low HSA: Gd-bbu ratios is also consistent with the ability of HSA to bind several copies of Gd-bbu as will be shown below. The fact that the free energy change for HSA binding is greater than that for Gd-bbu self-association suggests that the lower than expected relaxivity at pH 5 in HSA solution is not due to some aggregation phenomenon.
Figure 2.
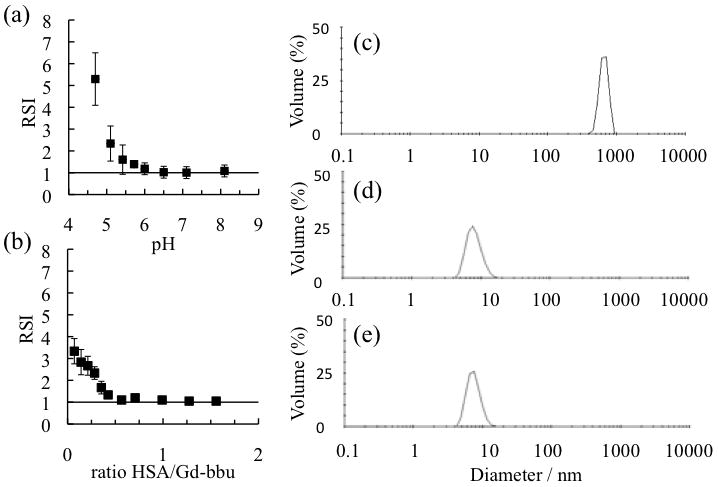
(a) Relative scattering intensity (RSI) compared to pure water as a function of pH for 0.32 mM Gd-bbu in the absence of HSA. (b) Effect of added HSA on relative scattering intensity at pH 5, 0.20 mM Gd-bbu. Data in (a) and (b) are mean ± standard deviation for triplicate experiments; solid line indicates absence of aggregation. (c) DLS data for 0.20 mM Gd-bbu at pH 4.7; (d) DLS data for HSA in HEPES buffer at pH 5; (e) DLS data for 1.5:1 HSA: Gd-bbu at pH 5. All measurements were performed at 37°C.
We also examined whether increasing the ionic strength would alter aggregation (Figure S3). Here we titrated up to 30 equivalents of ammonium acetate (NH4OAc) at either pH 5 (Figure S3a) or pH 6.2 (Figure S3c) and monitored the effect on DLS and relaxivity. At pH 5, NH4OAc caused a reduction in the scattering intensity but did not eliminate scattering. Ammonium acetate also caused a dramatic reduction in relaxivity, dropping r1 to 2 mM−1s−1 at high ammonium acetate ratios. This suggests that aggregation is still present but the aggregate is in a different form. The very low relaxivity suggests q=0. Interestingly, if we take this pH 5 solution with 30 equivalents ammonium acetate and begin to add HSA, the relaxivity rises from 2 to over 12 mM−1s−1 when 0.5 equivalents of HSA per Gd-bbu are added (Figure S3c). We repeated these experiments at pH 6.2 where there was no evidence of aggregation. At pH 6.2, the addition of 30 equivalents of NH4OAc has no effect on relaxivity. This latter result is consistent with the work of Lowe et al. who found that the presence of acetate did not reduce the relaxivity of their complexes.[4a]
Binding to serum albumin
The non-covalent interaction between Gd-bbu or Gd-glu and HSA was explored over the pH range 5 to 8.5. Protein-complex binding was determined by ultrafiltration across a membrane with a MW 5000 cutoff under conditions of 0.2 mM Gd complex, 0.69 mM HSA, 37 °C (ratio [HSA]/[GdL]total = 3.2).
The [Gd] in the filtrate was determined by ICP-MS and this corresponded to the concentration of the unbound complex. Under these conditions, both Gd-glu and Gd-bbu were highly bound to HSA with fraction bound ranging from 98.9 to 99.8%. Thus for the relaxivity experiments, the observed relaxivity equates to the relaxivity of the protein-bound complex.
Although the fluorophenol moiety on the sulfonamide arm may undergo deprotonation over this pH range, it does not have a measurable impact on protein binding. The anomalously low relaxivity in HSA solution at pH 5 cannot be attributed to a lack of protein binding at low pH.
We noted from the albumin titration experiments above that the protein appeared to bind multiple copies of Gd-bbu. We investigated this further by performing a full binding isotherm at pH 5, 37 °C and the results are plotted in Figure 3 as [Gd-bbu] bound per total [HSA] versus [Gd-bbu] unbound. Figure 3 shows a very strong initial binding event followed by additional binding. From the shape of the curve it is clear that the binding does not saturate at 3 equivalents of Gd-bbu bound. The data were fit to a classical stepwise thermodynamic binding model and the binding constants are listed in Table 1 as dissociation constants (Kd = 1/Ka). The initial binding constant is in the low micromolar range, 2 orders of magnitude higher affinity than the albumin-binding contrast agent MS-325.[13a] This albumin affinity is similar to that we have reported for other gadolinium complexes with biphenyl-based binding protein moieties,[14c, 18a] and is in the same range as drugs like warfarin, naproxen, and ibuprofen.[22]
Figure 3.

Binding isotherm for Gd-bbu (37 °C, pH 5, 0.80 mM) to HSA (pH 5, from 0.05 to 1.05 mM in HEPES buffer) from the ultrafiltration experiment. Solid line is the best fit to a stoichiometric binding model.
Table 1.
Stepwise stoichiometric binding constants, expressed as dissociation constants for Gd-bbu binding to HSA at 37 °C, pH 5.
| Kd1 [μM] | Kd2 [μM] | Kd3 [μM] |
|---|---|---|
| 0.65 ± 0.29 | 18.3 ± 8.4 | 1364 ± 387 |
To account for the presence of aggregation in the unbound state, we ran the binding assay in the absence of protein. In the absence of protein, 65% of the complex concentration was measured in the filtrate and presumably the remainder was aggregated. We assume that this is constant and we used this value to correct our data. Otherwise we will underestimate the amount of unbound Gd-bbu and therefore overestimate the affinity for albumin. It should be noted that this correction only has an impact on the data at high Gd-bbu:HSA ratios where there is more unbound Gd-bbu. Applying the correction has little effect on the first binding event.
Relaxivity and hydration number as a function of serum albumin concentration
In our HSA binding studies at pH 5, it was clear that Gd-bbu binds to more than one site on HSA. Our initial relaxivity studies employed an excess of HSA in order to favor protein binding. Together, the relaxivity and binding data indicate that the initial high affinity binding site is a low relaxivity site. We next sought to examine whether subsequent binding sites also displayed this anomalous low relaxivity or if relaxivity would increase as additional sites became occupied.
Figure 4 shows a plot of observed relaxivity versus the number of equivalents of HSA added. This data corresponds to the light scattering data shown above (Figure 2b). We know from the light scattering data that after 0.5 equivalents of HSA are added that there is no aggregation in solution and essentially all the Gd-bbu is bound to HSA. Addition of more HSA sees a rise in relaxivity which plateaus at 1 equivalent of HSA. Figure 4 suggests that all binding sites offer low relaxivity at pH 5. This was confirmed in an additional study where we measured relaxivity and binding as a function of pH for a 4:1 Gd-bbu:HSA solution and calculated the relaxivity due to the bound species (see supplementary material Figure S4). Even at 3 equivalents of Gd-bbu bound to HSA, we see the same relaxivity as for one equivalent bound indicating that there is no site-dependent relaxivity.
Figure 4.

(a) Relaxivity at 37°C, 0.47T, pH 5 for Gd-bbu (0.42 mM) as a function of added HSA. (b) Hydration number q of Eu-bbu at pH 5 as a function of added HSA. At [HSA]/[Eu-bbu]=0, the triangle indicates measurements made on a visible aggregate while the circle indicates measurements made on the supernatant.
We next prepared the Eu(III) analog and performed luminescence lifetime studies. The difference in luminescence decay rates when measured in H2O and D2O is proportional to the number of inner-sphere water molecule ligands, q.[23] The nature of the inner-sphere lanthanide coordination environment at pH 5 was examined as a function of HSA concentration. We used a custom-designed multi-modal confocal imaging system built by Yaseen et al.[24] to measure the luminescence lifetime of the Eu(III) excited state. The microscopic imaging device allowed us to measure Eu(III) luminescence lifetimes on a pixel-by-pixel basis across the entire field of view (~600 μm) (Figure S6a,b,c). The q values that we report in Figure 4b are averages of these local pixelwise q values, and the standard deviations reported are the standard deviations from the averaged q values. Figure 4b shows measured q as a function of HSA added at pH 5. Prior to adding HSA and at the first addition of HSA (0.22 equivalents), we observed a wide range of q values. However at the next addition of HSA (0.33 equivalents) and for subsequent HSA additions (0.5 – 2.6 equivalents), this heterogeneity in q was eliminated and q settled at 0.2±0.1 over this range.
This spatially resolved q mapping was useful for measurement in the absence of HSA where the aggregated Gd-bbu is heterogeneous. Supplemental Figure S4 shows microscopic images of the slide prior to HSA addition and indicates that parts of this large aggregate are starting to precipitate. The first two experimental points on the left of Figure 4b ([HSA]/[Eu-bbu] = 0) show two values. The higher value corresponds to q = 1.2±0.8, and this was measured in the liquid (supernatant) surrounding the visible aggregate. The lower point, with a value of q = 0.6±0.8, corresponds to the average of the solvation state within the aggregate. The large standard deviations reflect the heterogeneous nature of the soluble and insoluble aggregates. After the first addition of HSA, aggregation was still observed, explaining the low precision of the data. After the second addition of HSA, which brought the ratio of [HSA]/[Eu-bbu] to 0.33, microscopic observation showed no precipitation process and mean luminescent lifetimes throughout the sample became highly precise with q = 0.2±0.1. Additional aliquots of HSA also resulted in precise measurements of q that were unchanged. This behavior of aggregation followed by solubilization with HSA is entirely consistent with the DLS data shown above for Gd-bbu. The unchanging relaxivity data with [HSA]:[Gd-bbu] > 3 is consistent with luminescence lifetime data for Eu-bbu where q is low and does not change over this range. The q estimation is based on an empirical equation that has some uncertainty due to the quenching effects of other X-H oscillators. Thus a q value of 0.2 likely indicates no inner-sphere water ligands.
To summarize the data so far, we prepared biphenyl analogs of Ln-DO3A type complexes previously reported by Lowe et al.[4a] in order to promote albumin binding and enhance pH dependent relaxivity. In the absence of albumin, these compounds behaved as expected: low relaxivity at high pH due to coordination of sulfonamide arm and high relaxivity at lower pH when sulfonamide is protonated and q changes from 0 to 2. The biphenyl compounds show high affinity for serum albumin over the pH range 5 – 8.5. At pH 8.5, relaxivity increased by 260% upon albumin binding, but the magnitude of relaxivity was in line with what we expected for a q=0 complex bound to albumin. At low pH, the relaxivity of the albumin-bound complex increased but was much, much lower than expected for a putative q=2, albumin-bound complex. Luminescence studies on the Eu(III) analog revealed that the complex was q=0 at pH 5 when bound to albumin, and this explained the low relaxivity.
The lack of inner-sphere water at low pH when protein-bound was surprising. While most q=2 Gd(DO3A) derivatives are well known to bind coordinating anions resulting in displacement of coordinated water and reduced relaxivity,[12, 20, 25] Lowe et al. showed the presence of the pendant carboxylate groups, and especially the adipate derivative, effectively suppressed anion binding.[4a] There is also precedence for displacement of inner-sphere water from Gd(DO3A) derivatives by protein side chains.[16b, 20] We were aware of those studies and hypothesized that the pendant carboxylate groups would also suppress protein side chain coordination. In our case, it is not clear why the inner-sphere waters are displaced when the complex binds to albumin. It could be that the high local concentration of glutamate or aspartate carboxylate residues in the binding pocket results in coordination to the Gd(III). If side chain coordination is occurring, one might expect that this may not occur at different binding sites because there may not be a suitable side chain donor atom in close proximity to the Gd(III). However we found that the relaxivity did not vary for the first three binding sites. An alternate explanation is coordination of one of the pendant carboxylates to the Gd(III) ion. This would involve an 8-membered chelate ring and would not be expected to be very stable. In aqueous solution, the solvation energy of these pendant carboxylates is very high and coordination to Gd(III) is not energetically favored. In the HSA binding site, the dielectric constant will be much lower and the pendant carboxylates will be less well solvated.[26] These low dielectric conditions may favor coordination of a pendant carboxylate to Gd(III).
Regardless of the mechanism, these complexes are q=0 when bound to HSA at either high or low pH, yet there is still a pH dependent relaxivity for the albumin-bound complex. To investigate the nature of this pH dependence, we studied the field dependent relaxivity of Gd-bbu as a function of pH and temperature.
Nuclear Magnetic Relaxation dispersion (NMRD)
Figure 5a shows NMRD profiles of Gd-bbu in the absence of HSA at pH 5 and pH 8.5 at 37 °C. These profiles are consistent with our observations. At pH 5, Gd-bbu forms large aggregates and this results in an increase in the rotational correlation time of the Gd-H vector. It is well established from theory and practice that such an increase in correlation time will result in a maximum in the NMRD profile in the 20–40 MHz range.[27] At pH 8.5, where there is no evidence of aggregation, we observe a typical sigmoidal shaped curve consistent with a short correlation time.
Figure 5.

Nuclear Magnetic Relaxation Dispersions (NMRD) of Gd-bbu: (a) 0.52 mM in absence of HSA at pH 8.5 (grey triangles) and pH 5 (black triangles), 37 °C; (b) 0.39 mM at pH 8.5 with 1.4 mM HSA at 5°C (grey circles) and 37°C (black circles); (c) 0.33 mM at pH 5 with 1.3 mM HSA at 5°C (grey squares) and 37°C (black squares). Solid lines in (b) and (c) are fits to the data, see text for model and Table 2 for parameters.
In the presence of HSA, there is an increase in relaxivity at both pH values and at both temperatures (5 °C and 37 °C) and there is peak observed in the NMRD profile in the 20–30 MHz range. This peak is indicative of a long correlation time. We hypothesize that the increase in relaxivity upon binding is due to the presence of a relatively long-lived water molecule(s) or exchangeable proton(s) in close proximity to the Gd(III) ion, i.e. a long-lived second-sphere water molecule. The NMRD data were modeled to test this hypothesis.
Relaxivity can be factored into contributions arising from different classes of exchangeable protons in the different coordination spheres: inner-sphere, second-sphere, and outer-sphere. Based on the luminescence data, there are no inner-sphere water ligands when Gd-bbu is bound to HSA. This leaves second- and outer-sphere contributions to the observed relaxivity. Relaxation due to outer-sphere water molecules and exchangeable protons (water molecules or NH or OH protons from the protein) that undergo fast exchange (>109 s−1) will result in an NMRD profile that is sigmoidal in shape and lacks the high field peak. Relaxation due to a nearby long lived water molecule or exchangeable proton (second-sphere residency time τm′ > 1 ns) will result in this high field peak in the 20–30 MHz range. We assumed that the relaxivity of the former (fast exchanging protons) could be approximated by the relaxivity of Gd-bbu at pH 8.5 in the absence of protein. This is summarized in equation 1, where we define a relaxivity due to a long-lived water molecule (or other exchangeable proton(s)), r1LL.
| (1) |
| (2) |
The NMRD of the slow exchanging proton(s) was modeled using Solomon-Bloembergen-Morgan (SBM) theory.[28] Since SBM is a high field theory, we only considered data from 8 – 400 MHz; at lower fields the assumption that the Zeeman energy is much greater than the static zero field splitting breaks down and use of SBM is not appropriate.[16a]
At these higher fields, there is only one relevant spectral density term to consider. Furthermore, the lifetime of the long-lived water molecule is still much shorter than its relaxation time, τm′ ≪ T1m′, which is apparent from the fact that relaxivity is much higher at 5 °C than at 37 °C (see supplementary material, Figure S7, r1 vs temperature for ratio 4:1 Gd-bbu:HSA). Together this results in a simple analytical expression for r1LL that depends on the correlation time of the long lived water, τc, and the ratio of the number of long lived water molecules (q′) to the gadolinium-hydrogen distance, q′/r6, equation 1 (here C is a constant, equation 2, the symbols have their usual meanings[27]). The correlation time can have contributions from rotation of the complex (1/τR), electronic relaxation (1/T1e), and the exchange of the long lived water (1/τm′), equation 3. The shortest correlation time will dominate. Electronic relaxation is field dependent and can be approximated by equation 4.
| (3) |
| (4) |
We took a global analysis approach to fitting the data. While data at each pH was considered independently, we fit the 37 °C and 5 °C data together for each pH. Since, electronic relaxation of polyaminocarboxylato Gd(III) complexes is not very temperature dependent over this range,[14c, 16a, 29] we considered τv and Δ2 to be global parameters. We also assumed that the rotational correlation time of the albumin-bound complex would be much longer than lifetime of the long lived second-sphere water molecule, i.e. 1/τm′ ≫ 1/τR, and thus the relaxivity measurement would not be very sensitive to τR. The NMRD data at two temperatures was then simultaneously fit by iteratively varying 6 parameters: τm′ (37°C),τm′ (5 °C), q′/r6 (37 °C), q′/r6 (5 °C), τv and Δ2; τR was fixed at 40 ns. After this initial analysis, we found that the fitted NMRD data was rather insensitive to the value of τR, so long as τR was long (>20 ns). We also found that τv and Δ2 were strongly correlated and could not be independently determined. Ultimately we were able to reproduce the two NMRD curves (5 and 37 °C) at either pH by varying only 5 parameters, τm′ (37°C), τm′ (5 °C), q′/r6 (37 °C), q′/r6 (5 °C), and Δ2 (or τv), and the results of these fits are given in Table 2. For electronic relaxation, we could identify lower limits on τv and Δ2 and establish a linear relationship between these parameters; at pH 5, Δ2 ≥ 5×1018 s−2 with τv =9.0×Δ2 − 19.9 (units of ps). At pH 8.5, Δ2 ≥ 6×1018 s−2 with τv = 5.3×Δ2 − 9.4.
Table 2.
Molecular parameters obtained from fit of 1H NMRD data in Figure 5.
| Parameter | pH 5 | pH 8.5 |
|---|---|---|
| τR [ns] | >20a | >20a |
| τm′ (37 °C) [ns] | 1.8 ± 0.5 | 1.1 ± 0.2 |
| τm′ (5 °C) [ns] | 2.5 ± 0.6 | 1.5 ± 0.2 |
| rGd-H (37 °C) [Å] | 4.19 ± 0.14b | 4.32 ± 0.11b |
| rGd-H (5 °C) [Å] | 3.86 ± 0.05b | 4.18 ± 0.07b |
fixed in fitting at 40 ns;
assumes one long lived water molecule, i.e. two equivalent protons.
The NMRD analysis revealed that the lifetime of the long-lived water molecule is similar at pH 8.5 and at pH 5. The difference in relaxivity arises from a difference in the distance between the Gd(III) ion and this water molecule, which is shorter at pH 5. This difference in Gd-H distance might be expected. First, at high pH, the sulfonamide group is coordinated to the Gd(III) and this coordination will change the orientation of the biphenyl HSA binding group with respect to the gadolinium chelate compared to the situation at pH 5 where the sulfonamide is protonated and not coordinated. If the HSA binding pocket is the same at both pH, then the Gd chelate will be oriented differently resulting in a different distance to the long lived water. Changes in protein structure could also give rise to this change in Gd-H distance. For instance, Qiu et al. reported studies on the solvation dynamics and local rigidity in a series of reversible conformations of HSA under different pH conditions.[30] They observed that HSA undergoes a series of reversible conformational changes from acidic to basic conditions and even a change by a factor of 10 of the water exchange rate at the surface. From acidic to neutral pH, structural changes occur in domains II and III, changes in viscosity, solubility, and α-helical content in the protein were observed. As pH increases, HSA shows an increased affinity for some ligands, like warfarin.[31]
We used the simplest model to reproduce the NMRD curves, and no doubt more complicated models could be used. It is interesting to note that the data could be well reproduced with a simple isotropic model. Most NMRD data involving macromolecular systems require the Lipari-Szabo modification to account for internal motion.[32] However in our case, the correlation time is dominated by the short (~1 ns) lifetime of the long-lived water molecule and the relaxivity data is insensitive to rotational dynamics. The distances reported in Table 2 are based on one long-lived water molecule with two protons, however the adjustable parameter is q′/r6. The NMRD data cannot distinguish between a water molecule or a nearby exchangeable proton from the protein.
The Gd-glu system showed similar relaxivity data when bound to HSA. Although we did not perform luminescence studies on the Eu(III) analog or perform NMRD, it is highly likely that the relatively low relaxivity observed at low pH is due to displacement of inner-sphere water molecules as well. Interestingly the Gd-glu system had a U-shaped pH dependence when bound to HSA, see Figure 1a. HSA undergoes a neutral to basic (N→B) conformational transition from pH 7 to pH.[33] It is likely that this results in a change in the internuclear distance between the Gd(III) ion and the long lived water. However, the Gd-bbu relaxivity is not sensitive to this N→B transition.
The majority of water molecules hydrating proteins have residency times on the order of 10s of picoseconds, but it is well established that some water molecules can have longer residency times.[34] The 1–2 ns lifetime of the long-lived water molecule observed here is in accord with a water molecule hydrogen bonded to the surface of a protein. Interestingly, other albumin-bound q=0 complexes show similar behavior.[14c, 16a] The combination of NMRD and q=0 complexes could be used to probe the dynamics of protein hydration.
Conclusion
Two new pH-dependent MR contrast agents were synthesized with high affinity for serum albumin. The agents were designed to no inner-sphere water ligands at high pH (low relaxivity). Pendant carboxylate groups were included to prevent anion binding to the complex at low pH, and it was hoped, to prevent displacement of inner-sphere water ligands by protein sidechains. However when the complex was bound to HSA at pH 5, the water ligands were displaced and the relaxivity was substantially lower than anticipated. These results suggest that the [Gd(DO3A)(H2O)2] moiety is not suitable for use in protein targeted contrast agents due to water displacement upon protein binding. Despite being q=0 while bound to HSA over the pH range 5 – 8.5, a pH-dependent relaxivity was still observed. This was due to a long lived water molecule or other exchangeable proton in close proximity to the Gd(III) ion. As the pH decreased, the distance between this water and the Gd(III) ion decreased resulting in a pH-dependent relaxivity increase.
Experimental Section
Instrumentation
Ultrafiltration units (UFC3LCC00, regenerated cellulose membrane of 5000 Da nominal molecular weight cutoff) were obtained from Millipore Corp. (Bedford, MA). All gadolinium concentrations were determined by ICP-MS on an Agilent 7500a system. 1H NMR and 13C NMR spectra were recorded on a Varian 500 NMR system equipped with a 5 mm broadband probe. Longitudinal relaxation times T1 were measured using the inversion recovery method on Bruker Minispecs mq20 (20 MHz) and mq60 (60 MHz). The 1/T1 NMRD profiles were measured on a Stelar Spinmaster fast field cycling NMR relaxometer (2.35 × 10-4 − 0.47 T; 1H Larmor frequencies: 0.01–20 MHz) equipped with a VTC90 temperature control unit, on Bruker Minispecs (0.71 T (30 MHz), 0.94 T (40 MHz), and 1.41 T (60 MHz)), on a Bruker Avance-200 console connected to 2.35 T (100 MHz) and 4.7 T (200 MHz) cryomagnets, and on a Bruker DRX-400 (9.4 T, 400 MHz). Microwave irradiation was carried out using a Personal Chemistry Emrys Optimizer Microwave Synthesizer. Purifications were performed using two methods. Method A: Normal phase chromatography on a Combiflash Companion/TS (Teledyne ISCO, 120 g RediSep Rf silica cartridge) using A: Hexane, B: Ethyl acetate, flow rate 85 mL/min over 15 min. Method B: preparative HPLC on a Rainin, Dynamax (column: 250 mm Kromasil C18) using A: 0.1% TFA in water, B: 0.1% TFA in MeOH, flow rate 15 mL/min over 15 min. HPLC purity analysis (both UV and MS detection) were carried out on an Agilent 1100 system (column: Phenomenex Luna, C18(2) 100×2 mm) with UV detection at 220, 254 and 280 nm using a gradient of 20 mM ammonium formate (pH 6.8) with 5% (9:1 ACN: 20 mM ammonium formate) to 95% (9:1 ACN: 20 mM ammonium formate), flow rate 0.8 mL/min over 15 min.
Luminescence
Luminescence measurements were collected using the confocal portion of a custom-designed multi-modal microscope.[21] Briefly, a continuous-wave diode laser (λ = 532 nm, B&W Tek) provides excitation light that is temporally gated by an electro-optical modulator (ConOptics, Danbury, CT) with extinction ratio ~200 at 532 nm. The excitation beam passes through several conditioning optics, including a beam expander with pinhole spatial filter, polarizer, shutter, dichroic mirror, scan lens, and tube lens and a 20x-magnification objective lens (XLumPlan FL, Olympus, NA = 0.95). Using customized control software and galvanometric scanners (Cambridge Technology, Inc. Lexington, MA), the excitation beam is guided to selected locations in the ~600 um field of view. The emitted luminescence was de-scanned and collected using an avalanche photodiode photon counting module (APD, SPCM-AQRH-10, Perkin Elmer, Waltham, MA), sampled at 50 MHz with a high speed DIO card (National Instruments, Austin, TX). Data were processed using custom-written software in C and MATLAB (Mathworks, Natick, MA). Detected luminescent photons were binned into 50 us-long bins, yielding time-dependent phosphorescence decay profiles. Using a nonlinear least squares fitting routine, the resulting time courses were fit with a single-exponential function. A sample’s luminescence lifetime is equal to its fitted profile’s calculated time constant.
Chemicals
Human serum albumin (HSA) (Fraction V Powder 96–99% albumin, containing fatty acids) was purchased from Sigma Chemical Co. (St. Louis, Mo.). The synthesis of ligands was carried out as shown in Scheme 2. (4-(benzyloxy)-3-fluorophenyl)boronic acid was purchased from Frontier Scientific, Inc. and (S)-2-aminohexanedioic acid (98%) was purchased from Alfa Aesar, A Johnson Matthey Company. Other reagents were supplied by Aldrich Chemical Co., Inc., and were used without further purification. Solvents (HPLC grade) were purchased from various commercial suppliers and used as received.
4-bromo-N-(2-hydroxyethyl)benzenesulfonamide (a2)
Chlorotrimethylsilane (1.30 g, 12.0 mmol) was added to a stirred mixture of 2-aminoethanol (a1) (665 mg, 10.89 mmol) and Et3N (1.29 mg, 13.0 mmol) in CH2Cl2 (50 ml) cooled to 0–5 °C in an ice bath and monitored by TLC (CH2Cl2/MeOH/Et3N 92:8 stained with ninhydrin) for completeness. After 1 h, the mixture was warmed to RT under stirring and the mixture was concentrated in vacuo. 4-bromobenzene-1-sulfonyl chloride (3.05 g, 12.0 mmol) was then added and Et3N (1.31 g, 13.0 mmol) in CH3CN (50 mL) cooled to 0–5 °C in an ice bath and monitored by TLC (hexane/ethyl acetate 80:20) for completeness. After 1 h, the mixture was warmed to RT under stirring. After 2 h, the mixture was concentrated in vacuo and purified using normal phase chromatography (method A) to give (a2) (2.02 g, 7.19 mmol, yield 66%). 1H NMR (CDCl3, 500 MHz, 303 K): δ =7.95 (4H), 7.75 (NH), 4.44 (OH), 3.85 (2H), 3.74 (2H) ppm; 13C NMR (CDCl3, 125 MHz, 303 K):δ = 139.6, 133.2, 130.7, 127.9, 61.4, 48.4 ppm. LC/MS (ESI+): C8H10BrNO3S (m/z) calc: 281.15 [MH+]; found: 281.1 (MH+).
4′-(benzyloxy)-3′-fluoro-N-(2-hydroxyethyl)-[1,1′-biphenyl]-4-sulfonamide (a3)
a2 (48.1 mg, 0.17 mmol) and (4-(benzyloxy)-3-fluorophenyl)boronic acid (49.85 mg, 0.202 mmol) were dissolved in ethanol (5 ml) in a microwave vial. Pd(PPh3)2Cl2 (12 mg, 0.017 mmol) and Et3N (347.5 mg, 3.44 mmol) were added, and the reaction mixture was irradiated in the microwave synthesizer at 100°C for 30 min. After the reaction was cooled to RT, the product was filtered, the filtrate was concentrated, and the crude mixture was purified using normal phase chromatography (method A) to give (a5) (28.1 mg, 0.07 mmol, yield 42%). 1H NMR (CDCl3, 500 MHz, 303 K): δ = 7.75 (4H), 7.45 (NH), 7.40 (2H), 7.35 (2H), 7.22 (3H), 7.18 (1H), 5.21 (2H), 3.57 (OH), 2.98 (4H) ppm. LC/MS (ESI+): C21H20FNO4S (m/z) calc: 402.12 [MH+]; found: 402.1 (MH+).
(S)-dibenzyl 2-hydroxyhexanedioate (b1)
(S)-2-aminohexanedioic acid (1.00 g, 6.2 mmol) was dissolved in water (8 mL). Concentrated HCl (0.977 mL, 12.1 mmol) was added. A solution of sodium nitrite (2.48 g, 35.98 mmol) dissolved in water (8 mL) was then added very slowly (3 ml/h) at 0–5°C. The solution was stirred overnight. After acidification (pH 2), the product was extracted with ethyl acetate and dried in vacuo, giving a mixture of 2-hydroxyhexanedioic acid and 5-carboxy-δ-lactone: LC/MS (ESI+): diacid C6H10O5 (m/z) calc: 163.06 [MH+]; found: 163.1 (MH+); lactone C6H8O4 (m/z) calc: 145.05 [MH+]; found: 145.1 (MH+). Then, a solution of 1N KOH (6.2 g, 6.2 mmol) was added in a single portion to a stirred solution of the mixture dissolved in THF (10 mL) and heated at 40°C for 2 h. The reaction was then concentrated to a solid in vacuo (55°C, <30 Torr), dried under vacuum overnight and acid was not purified and used crude in the next step. LC/MS (ESI+): C6H10O5 (m/z) calc: 163.06 [MH+]; found: 163.1 (MH+). (S)-2-hydroxyhexanedioic acid (613 mg, 3.78 mmol) was suspended and stirred in DMF (8 mL), and benzyl bromide (1.29 g, 7.56 mmol) was added at RT. After stirring 8 h, the mixture was concentrated in vacuo and was purified by preparative HPLC (method B, gradient of 5 to 65% solvent B, monitoring at 220 nm). Fractions containing the product were concentrated to give 230 mg of b1 (0.67 mmol, yield 11%). 1H NMR (CDCl3, 500 MHz, 303 K): δ = 7.3 (10H), 5.2 (2H), 5.1 (2H), 4.2 (1H), 2.4 (4H), 1.8 (4H) ppm; 13C NMR (CDCl3, 500 MHz, 303 K): δ = 174 (CO), 173 (CO), 135, 128 (aryl), 77.2, 67, 33.6, 20.3 ppm. LC/MS (ESI+): C20H22O5 (m/z) calc: 343.15 [MH+]; found: 343.1 (MH+).
(2S,2′S,2″S)-hexabenzyl 2,2′,2″-(1,4,7,10-tetraazacyclododecane-1,4,7-triyl) tri-hexane-dioate (b2)
Methanesulfonyl chloride (184 mg, 1.60 mmol) was added to a stirred mixture of b1 (500 mg, 1.46 mmol) and Et3N (177 mg, 1.75 mmol) in CH2Cl2 anhydrous (30 mL) cooled to 0–5 °C in an ice bath and monitored by HPLC for completeness. After the addition was complete, the mixture was warmed to RT under stirring. After 1 h, the mixture was concentrated in vacuo and purified using normal phase chromatography (method A) to give b5 (580 mg, 1.38 mmol). LC/MS (ESI+): C21H24O7S (m/z) calc: 421.13 [MH+]; found: 421.1 (MH+). The mesylate b5 (840 mg, 2.0 mmol), as a solution in anhydrous CH3CN (50 mL), was added to a stirred mixture of cyclen (1,4,7,10-tetraazacyclododecane) (86 mg, 0.5 mmol) and dry potassium carbonate (553 mg, 4.0 mmol) in CH3CN (50 mL) preheated to 78 °C, with monitoring by HPLC for completeness. After 48 h, the reaction mixture was cooled to RT, filtered with a 200 nm syringe filter to remove potassium salts and concentrated in vacuo. The mixture was then purified by preparative HPLC (method B, gradient of 5 to 95% solvent B, monitoring at 220 nm). Fractions containing the product were concentrated to give 201 mg of b2 (0.18 mmol, 35% conversion of cyclen). 1H NMR (CDCl3, 500 MHz, 303 K): δ = 7.3 (30H), 5.2 (12H), 4.4 (3H), 3.3 (broad) ppm; 13C NMR (CDCl3, 125 MHz, 303 K): δ = 167, 164, 135, 128, 66.2, 45.7, 33.3, 30.1, 20.0 ppm. LC/MS (ESI+): C68H80N4O12 (m/z) calc: 1145.59 [MH+]; found 1146.4 (MH+).
hexabenzyl 2,2′,2″-(10-(2-(4′-(benzyloxy)-3′-fluoro-[1,1′-biphenyl]-4-yl sulfonamido) ethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)trihexanedioate (b3)
Methanesulfonyl chloride (91.9 mg, 0.80 mmol) was added to a stirred mixture of a5 (293 mg, 0.73 mmol) and Et3N (96 mg, 0.95 mmol) in CH2Cl2 anhydrous (20 mL). After the addition was complete, the mixture was warmed to RT under stirring. After 1 h, the mixture was concentrated in vacuo and purified using normal phase chromatography (method A) to give a4 (312 mg, 0.65 mmol). LC/MS (ESI+): C22H22FNO6S2 (m/z) calc: 480.10 [MH+]; found: 480.5 (MH+). The mesylate a4 (128 mg, 0.27 mmol), as a solution in anhydrous CH3CN (30 mL), was added to a stirred mixture of b2 (254 mg, 0.22 mmol) and dry potassium carbonate (92 mg, 0.67 mmol) in CH3CN (50 mL) preheated to 78°C, monitoring by HPLC for completeness. After 12 h, the reaction mixture was cooled to RT, filtered with a 200 nm syringe filter to remove potassium salts and concentrated in vacuo. The mixture was then purified by preparative HPLC (method B, gradient of 5 to 95% solvent B). Fractions containing the product were concentrated to give 275 mg of b3 (0.18 mmol, yield 82%). 1H NMR (CDCl3, 500 MHz, 303 K): δ = 7.5–7.92 (42H), 5.2–5.4 (12H), 3.21 (3H), 3.45 (3H) ppm; 13C NMR (CDCl3, 125 MHz, 303 K): δ = 20.5, 21.9, 25.3, 29.1, 29.7, 36.1, 38.2, 41.4, 66.7, 71.4, 114.4, 115.1, 116.7, 129.2, 135.7, 173.4 ppm. LC/MS (ESI+): C89H98FN5O15S (m/z) calc: 1528.68 [MH+]; found 1528.8 (MH+).
2,2′,2″-(10-(2-(3′-fluoro-4′-hydroxy-[1,1′-biphenyl]-4-ylsulfonamido)ethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)trihexanedioic acid (b4)
10% Pd on carbon (3.00 g) was added to a methanol solution (20 mL) of b3. The mixture was subjected to hydrogen bubbling and stirred for 12 h and monitored by HPLC for completeness. The mixture was then filtered through a fine frit, and the filtrate was concentrated in vacuo. LC/MS (ESI+): C40H56FN5O15S (m/z) calc: 898.36 [MH+]; found 898.4 (MH+).
(2S,2′S,2″S)-5-tribenzyl-1-tri-tert-butyl-2,2′,2″-(1,4,7,10-tetraazacyclododecane-1,4,7-triyl) tripentanedioate (g2)
Methanesulfonyl chloride (214 mg, 1.87 mmol) was added to a stirred mixture of g1 (500 mg, 1.70 mmol) and Et3N (192 mg, 1.90 mmol) in CH2Cl2 anhydrous (30 mL) cooled to 0–5 °C in an ice bath and monitored by analytical HPLC for completeness. After the addition was complete, the mixture was warmed to RT under stirring. After 1 h, the mixture was concentrated in vacuo and purified using normal phase chromatography (method A) to give the mesylate form of g1 (525 mg, 1.41 mmol). LC/MS (ESI+): C17H24O7S (m/z) calc: 373.13 [MH+]; found 373.4 (MH+). The mesylate (360 mg, 0.964 mmol), as a solution in anhydrous CH3CN (25 mL), was added under N2 to a stirred mixture of cyclen (1,4,7,10-tetraazacyclododecane) (42 mg, 0.24 mmol) and dry potassium carbonate (268 mg, 1.93 mmol) in CH3CN (20 ml) preheated to 80 °C, monitoring by analytical HPLC for completeness. After 48 h, the reaction mixture was cooled to RT, filtered with a 200 nm syringe filter to remove potassium salts and concentrated in vacuo. The mixture was then purified by preparative HPLC (method B). Fractions containing the product were concentrated to give 86.6 mg of g2 (0.08 mmol, 36% conversion of cyclen). LC/MS (ESI+): C56H80N4O12, (m/z) calc: 1001.59 [MH+]; found 1002.3 (MH+).
(2S,2′S,2″S)-2,2′,2″-(1,4,7,10-tetraazacyclododecane-1,4,7-triyl)tripentanedioic acid (g3)
a4 was prepared following the same condition described for synthesis of b3. a4 (90.0 mg, 0.19 mmol) as a solution in anhydrous CH3CN (15 mL) was added under N2 to a stirred mixture of g3 (125 mg, 0.13 mmol) and dry potassium carbonate (52 mg, 0.38 mmol) in CH3CN (15 mL) preheated to 78°C, monitoring by HPLC for completeness. After 12 h, the reaction mixture was cooled to RT, filtered with a 200 nm syringe filter to remove potassium salts and concentrated in vacuo. The mixture was then purified by preparative HPLC (method B). Fractions containing the product were concentrated to give 152 mg of g3 (0.11 mmol, 86%). LC/MS (ESI+): C77H98FN5O15S (m/z) calc: 1384.69 [MH+]; found 1384.7 (MH+).
2,2′,2″-(10-(2-(3′-fluoro-4′-hydroxy-[1,1′-biphenyl]-4-ylsulfonamido)ethyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)tripentanedioic acid (g4)
10% Pd/C (3.00 g) was added to a methanol solution (20 mL) of g3 (115 mg, 0.08 mmol). The mixture was subjected to hydrogen bubbling and stirred for 12 h. HPLC indicated the completion of the reaction. The mixture was then filtered through a fine frit, and the filtrate was concentrated in vacuo and coevaporated with acetonitrile in order to azeotrope out residual water and to remove any remaining methanol. Then a cocktail of 5% triisopropylsilane, 5% anisole and 90% TFA was added to the product, stirred for 4 h and monitored by analytical HPLC for completeness. LC/MS (ESI+): C37H50FN5O15S (m/z) calc: 856.31 [MH+]; found 856.3 (MH+).
(Ln-bbu and Ln-glu)
Lanthanide complexes were prepared in aqueous solution following reaction of the compound b4 and compound g4 respectively with hydrated LnCl3 at pH 6 (18 h, 50 °C), then raised at pH 9 (30 min). The purification of Ln-bbu and Ln-glu was carried out through preparative HPLC with neutral ammonium acetate buffer or pure water/acetonitrile eluant. Each purification method gave a satisfactory LC/MS trace of the final compounds Ln-bbu and Ln-glu with expected masses. LC/MS (ESI+): C37H47FGdN5O15S (Gd-glu) (m/z) calc: 1011.21 [MH+]; found: 1011.0; C40H53FGdN5O15S (Gd-bbu) (m/z) calc: 1053.26 [MH+]; found: 1053.25; C40H53EuFN5O15S (Eu-bbu) (m/z) calc: 1048.26 [MH+]; found: 1048.25; see supplementary material for purity analyses.
Preparation of 12.0% (w/v) Human Serum Albumin (HSA)
Lyophilized HSA was dissolved in a 50 mM HEPES buffer to generate a 12.0% (w/v) HSA solution. A molecular weight of 66 435 Da was used to estimate % (w/v) to a molar concentration. The protein concentration [HSA] was determined by measuring its absorbance at 280 nm for four dilutions. The linear regression on absorbance vs dilution gives a slope of [HSA] × ε, where ε is the molar absorptivity of HSA (35700 M−1cm−1).
Ultrafiltration Measurements of Binding of Gd-bbu to HSA
Gd-bbu/HSA samples ranging from 0.051 to 1.0 mM HSA in 0.4 mM Gd-bbu were made by combining appropriate amounts of 12.0% (w/v) HSA and 0.8 mM Gd-bbu. Aliquots (400 μL) of these samples were placed in 5 kDa ultrafiltration units, incubated at 37 °C for 20 min, and then centrifuged at 3500 g for 7 min. The filtrates from these ultrafiltration units were used to determine the free concentration of Gd-bbu of each of the samples. Duplicate aliquots were processed for each concentration sample of Gd-bbu in 4.5% (w/v) HSA. Concentrations of Gd-bbu/HSA samples and ultrafiltrates were determined by measuring the Gd concentration using ICP-MS.
Supplementary Material
Acknowledgments
L. M. acknowledges the Swiss National Science Foundation for a fellowship. Fabrice Yerly is thanked for providing the program VISUALISEUR/OPTIMISEUR, version 2.3.7 (2010), for least-squares fitting of the NMRD data and the pKa experiment. We thank Zhaoda Zhang for helpful discussions regarding the synthesis. This work was supported by Award Numbers R01EB009062 and R21EB009738 from the National Institute of Biomedical Imaging and Bioengineering and R01NS057476 from the National Institute of Neurological Disorders and Stroke.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.a) Helmlinger G, Yuan F, Dellian M, Jain RK. Nat Med. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]; b) Swietach P, Vaughan-Jones RD, Harris AL. Cancer Metastasis Rev. 2007;26:299–310. doi: 10.1007/s10555-007-9064-0. [DOI] [PubMed] [Google Scholar]
- 2.Tomlinson FH, Anderson RE, Meyer FB. Stroke. 1993;24:2030–2039. doi: 10.1161/01.str.24.12.2030. discussion 2040. [DOI] [PubMed] [Google Scholar]
- 3.a) Buxton RB, Wechsler LR, Alpert NM, Ackerman RH, Elmaleh DR, Correia JA. J Cereb Blood Flow Metab. 1984;4:8–16. doi: 10.1038/jcbfm.1984.2. [DOI] [PubMed] [Google Scholar]; b) Garcia-Martin ML, Herigault G, Remy C, Farion R, Ballesteros P, Coles JA, Cerdan S, Ziegler A. Cancer Res. 2001;61:6524–6531. [PubMed] [Google Scholar]; c) Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM. J Magn Reson Imaging. 2002;16:430–450. doi: 10.1002/jmri.10181. [DOI] [PubMed] [Google Scholar]; d) Zhang X, Lin Y, Gillies RJ. J Nucl Med. 2010;51:1167–1170. doi: 10.2967/jnumed.109.068981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Lowe M, Parker D, Reany O, Aime S, Botta M, Castellano G, Gianolio E, Pagliarin R. J Am Chem Soc. 2001;123:7601–7609. doi: 10.1021/ja0103647. [DOI] [PubMed] [Google Scholar]; b) Woods M, Kiefer GE, Bott S, Castillo-Muzquiz A, Eshelbrenner C, Michaudet L, McMillan K, Mudigunda SDK, Ogrin D, Tircsó G, Zhang S, Zhao P, Sherry AD. J Am Chem Soc. 2004;126:9248–9256. doi: 10.1021/ja048299z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Kalman FK, Woods M, Caravan P, Jurek P, Spiller M, Tircsó G, Kiraly R, Brücher E, Sherry AD. Inorg Chem. 2007;46:5260–5270. doi: 10.1021/ic0702926. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang S, Wu K, Sherry AD. Angew Chem Int Ed. 1999 [PubMed] [Google Scholar]
- 6.a) Aime S, Botta M, Geninatti Crich S, Giovenzana G, Palmisano G, Sisti M. Chem Commun. 1999:1577–1578. doi: 10.1021/bc990056m. [DOI] [PubMed] [Google Scholar]; b) Ali MM, Woods M, Caravan P, Opina ACL, Spiller M, Fettinger JC, Sherry AD. Chemistry. 2008;14:7250–7258. doi: 10.1002/chem.200800402. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Laus S, Ruloff R, Toth E, Merbach A. Chemistry. 2003;9:3555–3566. doi: 10.1002/chem.200204612. [DOI] [PubMed] [Google Scholar]; d) Mikawa M, Miwa N, Bräutigam M, Akaike T, Maruyama A. J Biomed Mater Res. 2000;49:390–395. doi: 10.1002/(sici)1097-4636(20000305)49:3<390::aid-jbm12>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 7.a) Garcia-Martin ML, Martinez GV, Raghunand N, Sherry AD, Zhang S, Gillies RJ. Magn Reson Med. 2006;55:309–315. doi: 10.1002/mrm.20773. [DOI] [PubMed] [Google Scholar]; b) Raghunand N, Howison C, Sherry AD, Zhang S, Gillies RJ. Magn Reson Med. 2003;49:249–257. doi: 10.1002/mrm.10347. [DOI] [PubMed] [Google Scholar]
- 8.Gianolio E, Napolitano R, Fedeli F, Arena F, Aime S. Chem Commun. 2009:6044–6046. doi: 10.1039/b914540k. [DOI] [PubMed] [Google Scholar]
- 9.Frullano L, Catana C, Benner T, Sherry AD, Caravan P. Angew Chem Int Ed Engl. 2010;49:2382–2384. doi: 10.1002/anie.201000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Løkling KE, Skurtveit R, Bjørnerud A, Fossheim SL. Magn Reson Med. 2004;51:688–696. doi: 10.1002/mrm.20009. [DOI] [PubMed] [Google Scholar]
- 11.Hartman KB, Laus S, Bolskar RD, Muthupillai R, Helm L, Toth E, Merbach AE, Wilson LJ. Nano Lett. 2008;8:415–419. doi: 10.1021/nl0720408. [DOI] [PubMed] [Google Scholar]
- 12.Supkowski RM, Horrocks WD., Jr Inorg Chem. 1999;38:5616–5619. doi: 10.1021/ic990597n. [DOI] [PubMed] [Google Scholar]
- 13.a) Caravan P, Cloutier NJ, Greenfield MT, McDermid SA, Dunham SU, Bulte JW, Amedio JC, Jr, Looby RJ, Supkowski RM, Horrocks WD, Jr, McMurry TJ, Lauffer RB. J Am Chem Soc. 2002;124:3152–3162. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]; b) Lauffer RB. Magn Reson Med. 1991;22:339. doi: 10.1002/mrm.1910220237. [DOI] [PubMed] [Google Scholar]
- 14.a) Aime S, Chiaussa M, Digilio G, Gianolio E, Terreno E. J Biol Inorg Chem. 1999;4:766–774. doi: 10.1007/s007750050349. [DOI] [PubMed] [Google Scholar]; b) Avedano S, Tei L, Lombardi A, Giovenzana GB, Aime S, Longo D, Botta M. Chem Commun. 2007:4726–4728. doi: 10.1039/b714438e. [DOI] [PubMed] [Google Scholar]; c) Dumas S, Jacques V, Sun WC, Troughton JS, Welch JT, Chasse JM, Schmitt-Willich H, Caravan P. Invest Radiol. 2010;45:600–612. doi: 10.1097/RLI.0b013e3181ee5a9e. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jacques V, Dumas S, Sun WC, Troughton JS, Greenfield MT, Caravan P. Invest Radiol. 2010;45:613–624. doi: 10.1097/RLI.0b013e3181ee6a49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gianolio E, Giovenzana GB, Longo D, Longo I, Menegotto I, Aime S. Chemistry. 2007;13:5785–5797. doi: 10.1002/chem.200601277. [DOI] [PubMed] [Google Scholar]
- 16.a) Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M, McMurry TJ. Inorg Chem. 2007;46:6632–6639. doi: 10.1021/ic700686k. [DOI] [PubMed] [Google Scholar]; b) Zech S, Sun WC, Jacques V, Caravan P, Astashkin AV, Raitsimring AM. Chem Phys Chem. 2005;6:2570–2577. doi: 10.1002/cphc.200500250. [DOI] [PubMed] [Google Scholar]
- 17.a) Ou MH, Tu CH, Tsai SC, Lee WT, Liu GC, Wang YM. Inorg Chem. 2006;45:244–254. doi: 10.1021/ic050329r. [DOI] [PubMed] [Google Scholar]; b) Vander Elst L, Maton F, Laurent S, Seghi F, Chapelle F, Muller RN. Magn Reson Med. 1997;38:604–614. doi: 10.1002/mrm.1910380415. [DOI] [PubMed] [Google Scholar]
- 18.a) Dumas S, Troughton JS, Cloutier NJ, Chasse JM, McMurry TJ, Caravan P. Aus J Chem. 2008;61:682–686. [Google Scholar]; b) Zhang Z, Greenfield MT, Spiller M, McMurry TJ, Lauffer RB, Caravan P. Angew Chem Int Ed Engl. 2005;44:6766–6769. doi: 10.1002/anie.200502245. [DOI] [PubMed] [Google Scholar]
- 19.a) Levy SG, Jacques V, Zhou KL, Kalogeropoulos S, Schumacher K, Amedio JC, Scherer JE, Witowski SR, Lombardy R, Koppetsch K. Org Proc Res Dev. 2009;13:535–542. [Google Scholar]; b) Overoye-Chan K, Koerner S, Looby RJ, Kolodziej AF, Zech SG, Deng Q, Chasse JM, McMurry TJ, Caravan P. J Am Chem Soc. 2008;130:6025–6039. doi: 10.1021/ja800834y. [DOI] [PubMed] [Google Scholar]; c) Spuentrup E, Ruhl KM, Botnar RM, Wiethoff AJ, Buhl A, Jacques V, Greenfield MT, Krombach GA, Gunther RW, Vangel MG, Caravan P. Circulation. 2009;119:1768–1775. doi: 10.1161/CIRCULATIONAHA.108.826388. [DOI] [PubMed] [Google Scholar]
- 20.Aime S, Gianolio E, Terreno E, Giovenzana GB, Pagliarin R, Sisti M, Palmisano G, Botta M, Lowe MP, Parker D. J Biol Inorg Chem. 2000;5:488–497. doi: 10.1007/pl00021449. [DOI] [PubMed] [Google Scholar]
- 21.Zech SG, Eldredge HB, Lowe MP, Caravan P. Inorg Chem. 2007;46:3576–3584. doi: 10.1021/ic070011u. [DOI] [PubMed] [Google Scholar]
- 22.a) O’Reilly RA. J Clin Invest. 1967;46:829–837. doi: 10.1172/JCI105582. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Honore B, Brodersen R. Mol Pharmacol. 1984;25:137–150. [PubMed] [Google Scholar]
- 23.a) Horrocks WD, Jr, Sudnick DR. J Am Chem Soc. 1979;101:334–340. [Google Scholar]; b) Beeby A, Clarkson IM, Dickins RS, Faulkner S, Parker D, Royle L, de Sousa AS, Williams JAG, Woods M. J Chem Soc Perk Trans 2. 1999:493–504. [Google Scholar]
- 24.Yaseen MA, Srinivasan VJ, Sakadzic S, Wu W, Ruvinskaya S, Vinogradov SA, Boas DA. Opt Express. 2009;17:22341–22349. doi: 10.1364/OE.17.022341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terreno E, Botta M, Boniforte P, Bracco C, Milone L, Mondino B, Uggeri F, Aime S. Chemistry. 2005;11:5531–5537. doi: 10.1002/chem.200500129. [DOI] [PubMed] [Google Scholar]
- 26.Schutz CN, Warshel A. Proteins. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 27.a) Caravan P. Chem Soc Rev. 2006;35:512–523. doi: 10.1039/b510982p. [DOI] [PubMed] [Google Scholar]; b) Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chem Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 28.a) Solomon I. Phys Rev. 1955;99:559–565. [Google Scholar]; b) Bloembergen N, Morgan LO. J Chem Phys. 1961;34:842–850. [Google Scholar]
- 29.Powell DH, Ni Dhubhghaill OM, Pubanz D, Helm L, Lebedev YS, Schlaepfer W, Merbach AE. J Am Chem Soc. 1996;118:9333–9346. [Google Scholar]
- 30.Qiu W, Zhang L, Okobiah O, Yang Y, Wang L, Zhong D, Zewail AH. J Phys Chem B. 2006;110:10540–10549. doi: 10.1021/jp055989w. [DOI] [PubMed] [Google Scholar]
- 31.a) van der Giesen WF, Wilting J. Biochem Pharmacol. 1983;32:281–285. doi: 10.1016/0006-2952(83)90556-7. [DOI] [PubMed] [Google Scholar]; b) Wanwimolruk S, Birkett DJ. Biochim Biophys Acta. 1982;709:247–255. doi: 10.1016/0167-4838(82)90467-8. [DOI] [PubMed] [Google Scholar]
- 32.Lipari G, Szabo A. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 33.Peters TJ. All About Albumin: Biochemistry, Genetics, and Medical Applications. Academic Press; San Diego: 1996. [Google Scholar]
- 34.Halle B. Philos Trans R Soc Lond B Biol Sci. 2004;359:1207–1224. doi: 10.1098/rstb.2004.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.