

Abstract
We have shown that the ectopic expression of Interferon Regulatory Factor (IRF-1) results in human cancer cell death accompanied by the down-regulation of the Inhibitor of Apoptosis Protein (IAP) survivin and the induction of the cyclin-dependent kinase inhibitor p21WAF1/CIP1. In this report, we investigated the direct role of p21 in the suppression of survivin. We show that IRF-1 down-regulates cyclin B1, cdc-2, cyclin E, E2F1, Cdk2, Cdk4, and results in p21-mediated G1 cell cycle arrest. Interestingly, while p21 directly mediates G1 cell cycle arrest, IRF-1 or other IRF-1 signaling pathways may directly regulate survivin in human cancer cells.
Keywords: IRF-1, survivin, p21, cell cycle, cancer
1. Introduction
The nuclear transcription factor Interferon Regulatory Factor (IRF)-1 was originally identified as a regulator of the human interferon (IFN)-β gene [1], and studies have shown its important role in both innate and adaptive immunity [2–4]. We have shown that the ectopic expression of IRF-1 promotes breast cancer cell death in vitro that is associated with the down-regulation of the inhibitor of apoptosis protein (IAP) survivin. In addition, we showed that the treatment of tumor-bearing mice with a recombinant adenovirus expressing IRF-1 resulted in the suppression of tumor growth in vivo in mouse models of breast carcinoma [5,6]. Resected tumor specimens from IRF-1 treated mice were characterized by IRF-1-positive and survivin-negative cells [6]. Delineating the direct or indirect mechanisms by which IRF-1 suppresses the expression of survivin provides insight into the regulation of survivin in cancer and facilitates the development of novel molecular targeted treatment approaches. In this current report, we begin to investigate the signaling events that are triggered by IRF-1 expression in cancer cells.
Studies have associated the induction of IRF-1 with cell cycle arrest [7,8]. In the human lung carcinoma cell line, A549, for example, infection with the measles virus resulted in IRF-1 up-regulation and G0/G1 cell cycle arrest [9]. Similarly, the activation of IRF-1 induced G1 cell cycle arrest in myc/ras-expressing NIH3T3 transformed cells that was mediated by the transcriptional down-regulation of cyclin D1 [10].
In addition, other studies have shown increased expression of both IRF-1 and the well-characterized cyclin-dependent kinase (Cdk) inhibitor p21WAF1/CIP1 (referred to as “p21” hereafter). Treatment of human hepatocellular carcinoma cells with transforming growth factor (TGF)-β1, for example, resulted in growth inhibition and the increased expression of both IRF-1 and p21 [11,12–14].
Cdk inhibitors can interact with cyclin/cyclin-dependent kinases including Cdk2 complexes that control the G1/S phase transition resulting in cell cycle arrest [12–14]. In clinical trials for the treatment of patients with malignant midgut carcinoids with a known inducer of IRF-1, interferon-alpha (IFN-α), inhibition of cell proliferation occurred via cell cycle arrest at the G1-S phase that may be mediated by the induction of p21 and IRF-1 [15]. Transfection of cells with antisense IRF-1 suppressed p21 induction and partially released cells from growth arrest [16].
While p53 is known as a primary regulator of p21 [13,17,18], other p53-independent mechanisms may promote its activity [19–21]. Direct binding of IRF-1 to the IRF-E binding site within the human p21 gene promoter, for example, has been shown [22]. In addition, IRF-1 is able to activate the p21 promoter independent of p53 during DNA damage [7].
Taken together, these studies support the contention that IRF-1 may mediate cell cycle arrest through the induction of p21. Moreover, IRF-1 mediated cell cycle arrest may be an important element in the suppression of tumor growth. In our previous studies, we showed that IRF-1 expression results in the induction of p21, and the reduced expression of survivin [6]. Survivin has been identified as the fourth top transcriptome in cancers of the breast, brain, colon, lung and melanoma [23]. Among 60 human tumor cell lines used for the NCI’s anticancer drug screening program, the highest relative levels of survivin were present in lung and breast cancer cell lines [24]. It functions to inhibit tumor cell death and several studies have shown that patients with survivin-positive tumors have significantly shorter overall survival [25–36]. Survivin disruption using dominant negative constructs, antisense survivin oligonucletides, short interfering RNA, and more recently microRNA (miRNA)-mediated RNA interference sensitizes tumor cells to apoptosis [37–44]. In this current report, we confirm the transcriptional up-regulation of p21 by IRF-1 and show that the IRF-1 induced G1 cell cycle arrest in tumor cells is mediated by p21. We evaluate the expression of Cdks in IRF-1 infected cells and show the decreased mRNA and protein levels of Cdk2, Cdk4 and cyclin E1, all involved in the G1 to S transition checkpoint. Suppression of E2F1 is also observed, and the decreased expression appears to be mediated by p21. Because p53-induced p21 and p21 alone has been shown to mediate the repression of survivin [44], we also investigated whether IRF-1-induced p21 plays a critical role in the down-regulation of survivin. We show that in p21 knockdown tumor cells, G1 cell cycle arrest is abrogated. Survivin expression, however, appears to be down-regulated by IRF-1 in both the wild-type and p21-deleted cancer cells. Collectively, these data suggest that p21 does not play a direct role in the down-regulation of survivin in IRF-1-expressing cells. When IRF-1 expression is abrogated by siRNA, however, the down-regulation of survivin is not observed suggesting that IRF-1 may directly regulate survivin. IRF-1 expression also results in the down-regulation of both cyclin B1 and cdc2 proteins, which are known to maintain survivin protein expression and stability. Taken together, IRF-1 induces p21-mediated cycle cell arrest, but other IRF-1-triggered signaling pathways promote the down-regulation of survivin.
2. Materials and methods
2.1. Cell Lines and Culture
The MDA-MB-468 and SK-BR-3 human breast cancer cell lines, the H1299 human lung adenocarcinoma, the AGS and N87 gastric cancer cell lines were purchased from the American Type Culture Collection (Manassas, VA). The MDA-MB-468 and SK-BR-3 breast cancer cells were propagated in Dulbecco’s Modified Eagle’s Medium (DMEM, BioWhittaker, Inc., Walkersville, MD) and Ham’s F-12 (Invitrogen Life Technologies, Carlsbad, CA) media at a 1:1 ratio supplemented with 10% fetal bovine serum (FBS), L-glutamine and antibiotics. The H1299, AGS, and N87 tumor cells were propagated in RPMI (BioWhittaker, Inc., Walkersville, MD) supplemented with 10% FBS, L-glutamine and antibiotics. The HCT116 p21 −/− human colon cancer cell line was generously provided by L. Zhang with the permission of B. Vogelstein and was propagated in DMEM with 10% FBS, L-glutamine and antibiotics. All cell cultures were maintained in a humidified atmosphere of 5% CO2/95% air at 37°C.
2.2. Adenoviral Infection
Construction of adenoviral vectors and infection procedures utilized to infect human cancer cell lines have been previously described [6]. Briefly, cells were washed in PBS, and serum-free Opti-MEM (Invitrogen Life Technologies, Carlsbad CA) media were added. Recombinant adenoviruses were added at the indicated multiplicities of infection (MOIs) and cells were incubated at 37°C at 5 % CO2. Previous experiments were conducted to determine the MOIs necessary to achieve similar IRF-1 protein expression in each cell line.
2.3. Immunoblotting Analysis and Antibodies
Immunoblotting was performed as has been previously described [6]. Briefly, whole cell lysates were quantified and separated by SDS–PAGE and transferred onto a nitrocellulose membrane. Loading of equal protein amounts was assessed by the staining of the nitrocellulose membranes with 0.1% Ponceau S (Sigma, St. Louis, MO) in 5% acetic acid, and by probing membranes with β-actin. Nonspecific binding was blocked with PBS-T (14 mM sodium phosphate, monobasic, monohydrate; 88 mM dibasic sodium phosphate, anhydrous; 100 mM NaCl; and 0.1% Tween 20) containing 5% nonfat milk for 1-h incubation with agitation at room temperature. Cyclin D, Cyclin E, Cdk 2, Cdk 4, Cdk 6 antibodies were purchased from Abcam Inc. (Cambridge, MA). Cdc2, cyclin B1, p21 and IRF-1 were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Anti-human survivin was purchased from R&D Systems, Inc. (Minneapolis, MN). Protein bands were visualized with Supersignal (West Pico Chemiluminescent Substrate; Pierce Biotechnology Inc., Rockford, IL) according to the manufacturer’s instructions. The bands were exposed on Kodak film (Eastman Kodak, Rochester, NY) to detect the chemiluminescence signals. Densitometric analyses of resultant protein bands were conducted using the ImageQuant TL software (Piscataway, NJ) and values were normalized against the β-actin control.
2.4. Cell Cycle Analysis
Cells were incubated with BrdU (BD Pharmingen, San Diego, CA) for three hours prior to being harvested with 0.25% trypsin, rinsed in PBS, and fixed in 70% ultra cold ETOH while vortexing. Cells were then incubated with FITC-anti BrdU and stained with propidium iodide according to the manufacturer’s instructions (BD Pharmingen, San Jose, CA). Cell cycle was assessed by flow cytometric analysis using a Becton Dickinson FACSort (Becton Dickinson, St Louis, MO). For PI staining alone, cells were harvested using 0.25% trypsin, rinsed in PBS, and fixed in 80% ultra cold ETOH. Cells were then washed and resuspended in PBS prior to treatment with 200 μg/mL DNase free RNase A followed by 30 μg/mL propidium iodide (BD Pharmingen) for 30 minutes at room temperature. A minimum of 10,000 cells per sample were recorded using the Beckman Coulter XL-MCL™ (Beckman Coulter, Inc., Fullerton, CA) flow cytometer. Cellular debris and doublets were excluded. Cells were then assayed for cell cycle arrest and these data were analyzed via ModFit LT™ software (Verity Software House, Topsham, ME). Percentages of cells in each phase of the cell cycle were calculated. Cell cycle analyses were also repeated in triplicate using MDA-MB-468 breast cancer cells either uninfected or infected with Ad-Ψ5 or Ad-IRF-1 at a MOI of 10. Average values and standard deviation statistical analyses were computed.
2.5. Transient Transfection of siRNA
Transient transfection of siRNA for human IRF-1 was described previously [46].
2.6. shRNA Plasmid Vector Construction
shRNA specific for human p21 was designed according to the manufacturer’s protocol for designing shRNA hairpins encoded by the shRNA expression vector pSilencer CMV 4.1 (Ambion, Inc., Austin, TX). The target sequence was attained from a predesigned and verified siRNA purchased from Ambion (pre-designed and validated siRNA ID #1621). The 55 nucleotide oligo encoding the human p21 specific shRNA was designed by utilizing Ambion’s online tool. The sense and antisense oligonucleotides were annealed and ligated to the BamHI and Hind III sites of pSilencer 4.1-CMV (Ambion) to form the neomycin selectable vector pSilencer 4.1-CMV-p21 shRNA. The inserts were confirmed by sequencing. The pSilencer 4.1-CMV-NEG containing non-specific shRNA was utilized as a negative control (Ambion Co., Austin, TX).
Stable clones were generated by transfecting the plasmids using FuGene (Roche, Basel, Switzerland) per the manufacturer’s protocol followed by the selection of clones with 600 μg/mL of G418 (Mediatech, Inc., Herndon, VA). At least 10 independent clones were isolated using sterile cloning rings and these were subsequently propagated. The stable cell lines were then screened for p21 knockdown following stimulation with 1000 U/ml of IFN-γ for 24 hours.
2.7. Quantitative real time RT-PCR
Primers were designed using <http://frodo.wi.mit.edu/> by entering the cDNA sequence for each gene of interest from the NCBI website into the Primer3 input screen and selecting against a human mispriming library. Primer sequences were synthesized by Invitrogen (Carlsbad, CA): β-actin forward primer agaaaatctggcaccacacc, reverse primer ccatctcttgctcgaagtcc; p21/CDKN1A forward primer gacaccactggagggtgact, reverse primer ggattagggcttcctcttgg; Cdk2 forward primer ttgtcaagctgctggatgtc, reverse primer cgagtcaccatctcagcaaa; Cdk4 forward primer ctgaccgggagatcaaggta, reverse primer ctggtcggcttcagagtttc. RNA was extracted from 4 × 106 cells cultured in 100 mm × 20 mm tissue culture plates using TRIzol® (Invitrogen, Carlsbad, CA) and chloroform. The RNA was then purified by using the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Samples were prepared using the Stratagene 1-step Brilliant II SYBR Green QRT-PCR kit (Cedar Creek, TX). RT-PCR was performed with the Stratagene Mx3000P. Relative quantities were assessed in comparison with the ROX reference dye and a β-actin (Invitrogen, Carlsbad, CA) housekeeping normalizer. The RT2Profiler™ PCR Array: Human Cell Cycle was purchased from SuperArray (Frederick, MD) and the manufacturer’s instructions were followed including the use of the corresponding first strand cDNA synthesis kit and the master mix from SuperArray. RNA from Ad-Ψ5 infected cells was used as a control.
2.8. Statistics
Data are presented as means ± standard error of the mean or standard deviation with representative experiments depicted in each figure. Comparisons between values were analyzed using analysis of variance (ANOVA) for comparison between the uninfected, Ad-Ψ5 vector control, and the Ad-IRF-1 infected cell cohorts. Student’s t test was utilized to compare the mean values between cell cohorts, i.e., Ad-IRF-1 and Ad-IRF-1 + p21. Standard deviations were computed for cell cycle analyses of MDA-MB-468 cells either uninfected or infected with Ad-Ψ5 or Ad-IRF-1.
3. Results
3.1. IRF-1 Induces G1 Arrest in Human Breast Cancer Cells
Having shown that the ectopic expression of IRF-1 resulted in tumor cell death and the induction of p21 expression in MDA-MB-468 breast cancer cells [6], cell cycle arrest was evaluated in each of the cell cohorts by FITC-BrdU labeling and propidium iodide staining. The sub-G0/G1 (apoptotic) fraction was gated out before analysis. Bivariate staining and the assessment of the G1, S, and G2 fractions are shown in Fig. 1A. The multiplicities of infection (MOI) of 2.5, 5, and 10 were selected to determine the effect of the level of IRF-1 expression on cell cycle progression (Fig. 1B). The resultant increase in IRF-1 expression with increased MOI is also shown (Fig. 1C). There is a marked decrease in the S phase fraction in the Ad-IRF-1 infected cells compared with the uninfected and Ad-Ψ5 infected control cell cohorts. The cell cycle arrest that is observed in the Ad-IRF-1 infected cells occurs in a dose-dependent manner. There is a slight increase in the G2 fraction in the Ad-IRF-1 infected cells (Fig. 1B) that is similar to that observed in the Ad-Ψ5 infected control cell cohort, hence, this increase is unlikely related to IRF-1 expression. These experiments were also repeated in triplicate using a MOI of 10 (Table 1). Collectively, these data show a significant increase in the G1 fraction in the Ad-IRF-1 infected cells compared with the uninfected cell cohort. The increase in the G1 fraction in the Ad-IRF-1 infected cell cohort is highlighted when compared to the corresponding Ad-Ψ5 control cells, thus, further confirming that IRF-1 causes G1 arrest.
Figure 1.

IRF-1 induces a dose-dependent G1 arrest in human breast cancer. (A) MDA-MB-468 human breast cancer cells were either not infected (NI), or infected with the control Ad-Ψ5 or Ad-IRF-1 recombinant adenoviruses at varying multiplicities of infection (MOI) and cell cycle arrest at 24 h was evaluated by FITC-BrdU labeling and propidium iodide staining. An example of the bivariate cell cycle output data is shown. Increases in BrdU are associated with S phase while PI staining indicates either one or two copies of DNA corresponding with G1 or G2 phases. Percentage of cells in each phase can then be calculated. (B) MDA-MB-468 cells were either not infected (NI) or infected with either Ad-Ψ5 or Ad-IRF-1 at the indicated MOIs. Cells were fixed in ETOH, permeabilized, and stained with FITC-αBrdU and propidium iodide (PI). Cell cycle analysis was then performed and percentages of cells in each phase, G1, S, and G2 are shown as assessed by two dimensional analyses as seen in A. (C) MDA-MB-468 cells were either not infected (NI) or infected with either Ad-Ψ5 or Ad-IRF-1 at the indicated MOIs. Infected cells were harvested 24 h after infection, and immunoblotting was conducted as described in Materials and Methods.
Table 1. IRF-1 induces G1 arrest in MDA-MB-468 human breast cancer cells.
MDA-MB-468 human breast cancer cells were either not infected (NI), or infected with the control Ad-Ψ5 or Ad-IRF-1 recombinant adenoviruses at multiplicities of infection (MOI) of 10 and cell cycle arrest at 24 h was evaluated by FITC-BrdU labeling and propidium iodide staining as described in Materials and Methods. Cell cycle analyses were conducted in triplicate and percentages of cells in each phase were assessed. Data are presented as mean values and standard deviation of the means was computed for each data set.
| Treatment | G1 % | S % | G2 % |
|---|---|---|---|
| NI | 35.48 ± 0.35 | 35.22 ± 0.34 | 29.29 ± 0.66 |
| Ad-Ψ5 | 36.38 ± 0.92 | 33.81 ± 1.72 | 29.81 ± 1.87 |
| Ad-IRF-1 | 60.31 ± 1.60 | 24.35 ± 2.07 | 15.34 ± 1.46 |
3.2. IRF-1 Regulates Cyclin and Cyclin Dependent Kinase Expression
The ectopic expression of IRF-1 effectively induced the expression of the cyclin-dependent kinase inhibitor p21 and G1 cell cycle arrest. The expression of other cell cycle related genes was evaluated in the MDA-MB-468/Ad-IRF-1 infected breast cancer cells utilizing a cell cycle specific gene array that uses real-time RT-PCR. We report the results of the screening array that were subsequently confirmed by either quantitative real-time RT-PCR, Western immunoblotting, or both, in Table 2. p21 is seen to be markedly increased in the cell cycle specific gene array. Interestingly, p21 is known to effectively inhibit Cyclin E/Cdk2 complexes that lead to G1 arrest, and both Cyclin E1 and Cdk2 appear to be down-regulated at the transcriptional level. p21 has also been shown to inhibit Cyclin D/Cdk4 or 6 complexes [48]. While neither cyclin D1 nor cyclin D2 appear to be transcriptionally down-regulated, the down-regulation of Cdk4 is observed. Reduced expression of Cdk2 and Cdk4 mRNA was confirmed by quantitative real-time RT-PCR (Fig. 2A), and the suppression of Cdk2 and Cdk4 is statistically significant with p<0.05 by ANOVA. Furthermore, by Western immunoblotting, we show that ectopic IRF-1 expression results in decreased protein expression of Cdk2, Cdk4, and Cyclin E1 (Fig. 2B).
Table 2. Transcriptional targets of ectopic IRF-1 expression in MDA-MB-468 breast cancer cells.
MDA-MB-468 breast cancer cells were either uninfected (NI), or infected with Ad-Ψ5 or Ad-IRF-1 at a MOI of 25. Cells were harvested 24 h post infection. qRT-PCR was performed as has been described in Materials and Methods. RT2 Profiler™ PCR array was utilized to determine the transcriptional targets of ectopic IRF-1 expression. A list of genes and the fold change is presented.
| Gene | Comparative difference vs. Ad-Ψ5 | Fold increase or decrease vs. Ad-Ψ5 |
|---|---|---|
| Cyclin B1 | 0.64 | −1.57 |
| Cyclin D1 | 1.46 | 1.46 |
| Cyclin D2 | 1.10 | 1.10 |
| Cyclin E1 | 0.30 | −3.35 |
| CDC2 | 0.79 | −1.27 |
| CDK 2 | 0.31 | −3.24 |
| CDK 4 | 0.25 | −4.01 |
| CDK 6 | 1.70 | 1.70 |
| p21 | 23.34 | 23.34 |
| survivin | 0.61 | −1.64 |
Figure 2.

Ectopic expression of IRF-1 suppresses Cdk2, Cdk4, and Cyclin E1 expression. (A) MDA-MB-468 breast cancer cells were either uninfected (NI), or infected with the Ad-Ψ5 control or Ad-IRF-1 adenovirus at MOI 25. Lysates were harvested at 24 hours post infection. Quantitative real-time RT-PCR was performed as described in Materials and Methods, and relative quantities of cdk2 and cdk4 were computed by utilizing the MxPro™ software. These experiments were repeated twice with comparable results. * denotes p<0.05 by ANOVA. (B) MDA-MB-468 breast cancer cells were either uninfected or infected with Ad-Ψ5 control or Ad-IRF-1 at a MOI of 10. Cellular lysates were prepared and Western immunoblotting was conducted as has been described in Materials and Methods. Blots were probed for Cdk2, Cdk4, cyclin E, and β-actin.
3.3. IRF-1 Induces p21-Independent of p53 Status in Tumor Cells
Previously, we showed that the ectopic expression of IRF-1 induces p21 protein expression in MDA-MB-468 and SK-BR-3 p53 mutant human breast cancer cell lines [6]. Using quantitative real-time RT-PCR, we show that p21 up-regulation occurs at the transcriptional level and a marked increase in p21 mRNA in MDA-MB-468/Ad-IRF-1 infected breast cancer cells is observed (Fig. 3A). Repetition of these experiments showed an average increase of 28 fold up-regulation of p21 mRNA upon the ectopic expression of IRF-1 when compared to the normalized uninfected (NI) control cell cohorts. Similar increases in p21 mRNA were also observed in the p53 null human non-small-cell lung cancer cell line, H1299, when compared to the normalized no infection control (NI, Fig. 3A). To further confirm that p21 up-regulation by IRF-1 is independent of p53 status, an additional p53 mutated gastric cancer cell line, N87, as well as the p53 wild-type gastric cancer cell line, AGS, were infected to express IRF-1. Similar to the MDA-MB-468 and H1299 cancer cell lines, an increase in p21 protein expression was observed in the N87 gastric cancer cell line (Fig. 3B). A baseline level of p21 expression is observed in the p53 wild-type AGS gastric cell line that is slightly increased by the ectopic expression of IRF-1. These findings are similar to the results that we reported for the wild-type p53 breast cancer cell line MCF-7 [6]. These data are consistent with the function of IRF-1 as a nuclear transcription factor with regulatory elements in the promoter region of p21.
Figure 3.

IRF-1 induces the upregulation of p21 mRNA and protein expression in cancer cells. (A) MDA-MB-468 human breast cancer cells and H1299 human non-small-cell lung cancer cells were not infected (NI), or infected with the control Ad-Ψ5 vector or Ad-IRF-1 adenoviruses at MOI 25. 24 h post infection, cell pellets were lysed with TRIzol® and RNA was purified with the Qiagen RNeasy kit. Quantitative real time RT-PCR was performed as described in Materials and Methods. Relative quantities of p21 mRNA were computed by MxPro™ quantitative PCR software (Stratagene, La Jolla, CA). * denotes p<0.005 by ANOVA. (B) MDA-MB-468 and H1299 cancer cell lines were infected as described in A. AGS and N87 gastric cancer cells were infected at MOI 50. 24 h post infection, cells were harvested and immunoblotting was performed as described in Materials and Methods.
3.4. p21 mediates IRF-1 induced G1 arrest but not Down-Regulation of Survivin
To determine the direct effect of the induction of p21 on cell cycle arrest in cancer cells that express IRF-1, we used p21 siRNA to abrogate the induced increase in p21 expression. We confirmed that siRNA to p21 was effective in inhibiting p21 expression in the human non-small-cell lung cancer H1299 cell line infected with Ad-IRF-1 (Fig. 4A). Western immunoblotting of H1299/Ad-IRF-1 infected cell lysates show the upregulation of p21 that is subsequently reduced by approximately 26-fold when cells are transfected with p21 siRNA. The scrambled control siRNA, Si Neg, was ineffective in suppressing p21 expression. We furthered these results by evaluating cell cycle effects of IRF-1 in the absence of or in the presence of p21 siRNA by propidium iodide staining. Similar to the MDA-MB-468 breast cancer cells, the ectopic expression of IRF-1 in H1299 cells induces a G1 cell cycle arrest (Fig. 4B). Suppression of p21 using siRNA profoundly abrogated the G1 arrest in the H1299 cell line. p21 siRNA decreased the G1 fraction to a level comparable to the uninfected and the Ad-Ψ5 infected control cell cohorts. In addition, the S fraction was increased in the p21 siRNA transfected/Ad-IRF-1 infected cells that was comparable to the uninfected and the Ad-Ψ5 infected cells. Collectively, p21 siRNA abrogated the effect of IRF-1 on G1 arrest in H1299 cells.
Figure 4.

p21 mediates IRF-1 induced G1 arrest in human non-small-cell lung cancer. (A) H1299 lung cancer cells were either untransfected or transfected with a control siNeg, or siRNA to p21. Untransfected cells treated with the transfection reagent (T.R.) siPORT™ Amine are included as an additional control. 24 hours post transfection the cells were either not infected, or infected with Ad-Ψ5 or Ad-IRF-1 recombinant replication defective adenoviruses at a MOI of 25. 24 hours post infection, cells were harvested and immunoblotting was performed as described in Materials and Methods. (B) H1299 cells were treated as in A, and 24 hours post infection, cells were fixed in 80% ETOH, permeabilized, and stained with propidium iodide. Cells were then assayed for cell cycle arrest and these data were analyzed via ModFit LT™ software. ANOVA analysis was utilized to compare the means between no infection NI, Ad-Ψ5, and Ad-IRF-1, while the Student’s t test compared means between Ad-IRF-1 and Ad-IRF-1 + p21 siRNA (* = p<0.05 for both analyses). These experiments were conducted in triplicate. (C) H1299 cells were transfected as in A, and subsequently infected with Ad-Ψ5, Ad-IRF-1, or Ad-p21 at a MOI of 25. Cells were harvested 24 h post infection and immunoblotting was conducted as has been described in Materials and Methods. These experiments were repeated twice with comparable results.
Hypophosphorylated retinoblastoma (Rb) protein binds and inhibits proteins including the E2F family transcription factors that promote cells to enter the S-phase [48–49]. Upon Rb phosphorylation by Cdks, Rb releases E2F proteins that subsequently assist proliferation and progression from G1 to S phase. p21 has been shown to prevent cyclin/Cdk complexes from phosphorylating Rb [50]. Western immunoblotting shows that E2F1 is decreased in H1299 cells infected with both Ad-p21 and Ad-IRF-1 (Fig. 4C). The transfection reagent or the Si Neg controls did not affect the observed decrease in E2F1. p21 siRNA restores E2F1 expression in Ad-IRF-1 infected cells that appears to confirm that p21 mediates the decrease in E2F1 protein by ectopic expression of IRF-1. Cyclin D1 was also evaluated and similar to the MDA-MB-468/Ad-IRF-1 infected cells (Fig. 2 and Table 2) a change in the expression of cyclin D1 was not observed in H1299 cancer cells (Fig. 4C).
The direct effect of the induction of p21 on cell cycle arrest in MDA-MB-468 breast cancer cells that express IRF-1 was also assessed by generating p21 knockdown MDA-MB-468 clones by transfection with p21 shRNA. The control clone denoted as c4 (Fig. 5A) was generated by stable transfection with the vector expressing non-specific shRNA and was renamed p21Neg. IFN-γ has been shown to upregulate p21 [51]. Since differences in p21 protein were difficult to assess at baseline, we cultured the different clones with IFN-γ. While several p21 knockdown clones were confirmed, the most profound reduction in p21 was observed in the p21c11 clone (Fig. 5A). To investigate the direct effect of p21 in mediating G1 cell cycle arrest in Ad-IRF-1 infected MDA-MB-468 breast cancer cells, the p21c11 clone was infected with a higher MOI of Ad-IRF-1 to achieve IRF-1 expression comparable to the p21Neg/Ad-IRF-1 control cells. As seen in Fig. 5B, there is a marked suppression of p21 protein in p21c11 versus p21Neg control cells. Similar to the H1299 non-small-lung cancer cell line, E2F1 is decreased by IRF-1 expression in the control p21Neg cells, but decreased expression is not observed in the p21c11 cells (Fig. 5B). Furthermore, G1 cell cycle arrest was observed in the control p21-positive cells that express IRF-1, but G1 arrest was not observed in the p21 knockdown p21c11 cells (Fig. 5C). Similar results were obtained using the p21c15 clone (data not included).
Figure 5.

p21 mediates IRF-1 induced G1 arrest but not suppression of survivin in breast cancer cells. (A) MDA-MB-468 cells were stably transfected with either control scrambled shRNA (p21Con.) or shRNA to p21. Clones were expanded under G418 selection. Independent clones were selected and stimulated with 1000 U/mL of IFNγ for 24 hours. Cells were harvested and Western immunoblotting was performed as described in Materials and Methods. (B) p21 control and p21c11 clones from A were either not infected or infected with Ad-Ψ5 or Ad-IRF-1 at MOIs of 5 and 10 respectively resulting in equivalent IRF-1 expression. 24 hours post infection, cells were harvested and immunoblotting was conducted as described in Materials and Methods. (C) p21 control and p21c11 clones were either NI or were infected with Ad-Ψ5 or Ad-IRF-1 at MOI of 10 and 25 respectively. 48 hours post infection, cells were harvested and fixed in 80% ETOH. Cells were subsequently permeabilized, stained with PI, and cell cycle analysis was conducted. ANOVA analysis within each of the uninfected, Ad-Ψ5, or Ad-IRF-1 cell cohorts was utilized to compare means, while the Student’s t test compared the means between the p21 control and p21c11 cell clones infected with the same virus. ‡ = p<0.001 by ANOVA * = p<0.05 by Student’s t test. (D) Parental MDA-MB-468 control cells, p21Neg control cells, and p21c11 knockdown cells were either not infected (NI) or infected with Ad-Ψ5 or Ad-IRF-1. Cells were harvested at the 24 and 48 h time points. IRF-1, p21, survivin, and β-actin were assessed by Western immunoblotting as described in Materials and Methods.
We have previously shown that the ectopic expression of IRF-1 in breast cancer cells resulted in the induction of p21 and reduced levels of survivin [6]. Furthermore, Lohr et al. showed that the suppression of survivin in HCT116 human colon cancer cells was mediated by p21 [45]. To determine whether p21 played a direct role in the down-regulation of survivin in Ad-IRF-1 infected cells, we evaluated the expression of survivin in the control and p21 knockdown cell cohorts that were either uninfected or infected with Ad-Ψ5 or Ad-IRF-1 adenoviruses. Reduction in survivin protein expression are usually seen at the 36 h time point, and the most profound decrease was observed at 48 h post IRF-1 infection in MDA-MB-468 breast cancer cells [6]. Despite the absence of expression at 24 h (Fig. 5D), IRF-1 induced p21 expression is seen in the shRNA knockdown clones at the 48 h time point, although expression is less than the control cell cohorts (Fig. 5D). Survivin expression is decreased in all three cohorts at the 48 h time point. Since p21 was virtually undetectable at the 24 h time point and less detectable than the control cell cohorts at the 48 h time point, it is conceivable that this low level of expression of p21 plays a role in the down-regulation of survivin. It should be noted that survivin expression is slightly higher in the MDA-MB-468 cells infected with IRF-1 at the 48 h time point when compared with the p21c11 clone infected with IRF-1. Taken together, these data do not support the contention that p21 plays a critical role in survivin regulation.
To further evaluate the role of p21 in survivin expression, we utilized IFN-γ to assess the expression of p21, IRF-1, and survivin in the MDA-MB-468 breast cancer cells. As seen in Fig. 6A, IFN-γ induced IRF-1 expression and suppressed the protein expression of survivin in IFN-γ treated cells. We also evaluated survivin expression in MDA-MB-468 breast cancer cells transfected with siRNA against IRF-1. Suppression of survivin was abrogated in siIRF-1 transfected cells when compared with lipofectamine and siNull control transfected cells suggesting that IRF-1 may play a direct role in down-regulating survivin.
Figure 6.
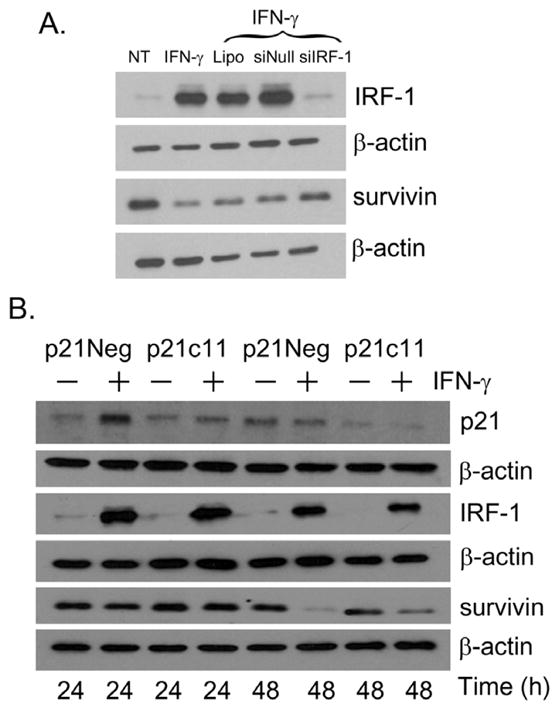
p21-independent suppression of Survivin in human breast cancer cells. (A) MDA-MB-468 cells were either not transfected or transfected with a control siNeg, or siRNA to IRF-1. Untransfected cells were treated with the transfection reagent Lipofectamine (Lipo). 24 h post transfection, the cells were either not treated (NT) or cultured with 1000 U/mL of IFN-γ. 24 h post treatment, cells were harvested and immunoblotting was performed as described in Materials and Methods. (B) p21 psilencer control and p21c11 p21 knockdown MDA-MB-468 clones were either untreated or cultured with 1000 U/mL of IFNγ. Cells were harvested at the 24 and 48 h time points and Western immunoblotting was conducted as has been described in Materials and Methods.
Survivin expression was also evaluated in the p21 knockdown MDA-MB-468 clones. Prior to culture with IFN-γ, survivin expression was comparable in both the p21Neg control and p21c11 p21 knockdown cell cohorts (Fig. 6B). Despite similar expression of IRF-1 and marked suppression of p21 expression in the p21cll clone compared with the p21Neg clone, survivin expression was reduced at the 48 h time point in both the p21Neg and p21c11 clones cultured with IFN-γ (Fig. 6B).
In addition, to further confirm that survivin expression could be decreased by IRF-1 in the complete absence of p21 protein, we utilized the HCT116 human colon cancer cell line that has p21 deleted by homologous recombination. In these HCT116 p21−/− cells, the p21 gene is deleted at both alleles and there should be a complete absence of p21 protein expression. We confirmed the absence of p21 in HCT116 p21−/− cancer cells by western immunoblotting in uninfected, Ad-Ψ5, or Ad-IRF-1 infected cells (Fig. 7A). In the absence of p21, the ectopic expression of IRF-1 resulted in the reduced expression of survivin (Fig. 7B). Collectively, these data show that p21 does not play a major role in the down-regulation of survivin in these cancer cells.
Figure 7.
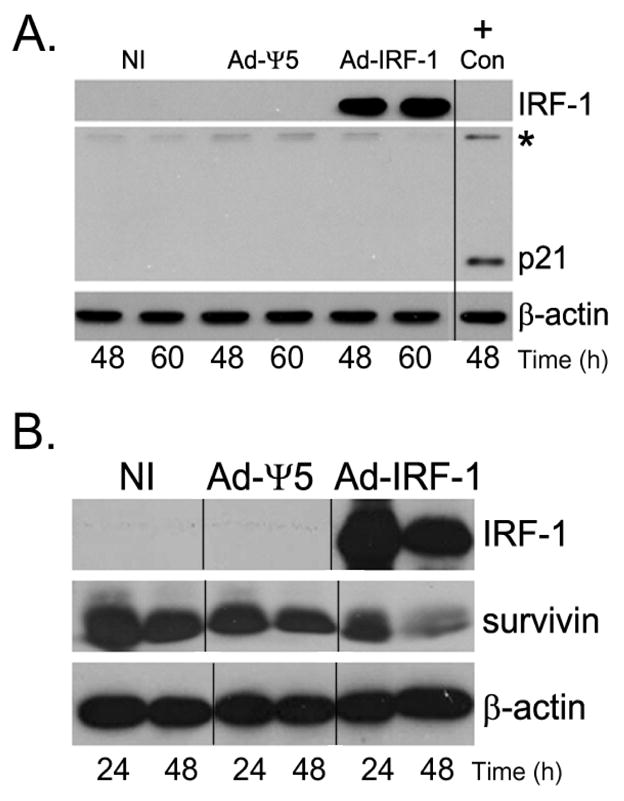
Survivin expression is down-regulated in Ad-IRF-1 infected HCT116 p21−/− human colon cancer cells. (A) HCT 116 p21−/− cells were infected at MOI of 10 and cells were harvested at the indicated time points post infection. IRF-1, p21, and β-actin were assessed by immunoblotting as described in Materials and Methods. On a separate gel run at the same time HCT116 wild type cell lysates were assessed for p21 expression and used as a positive control. * marks a non-specific band that was used to align the separate blots run and stained at the same time. Molecular weight markers were also used for alignment. A p21 band is absent in the HCT116 p21−/− cells regardless of infection. (B) Survivin is decreased by the ectopic IRF-1 expression in HCT116 p21−/− cells. HCT116 p21−/− cells were either uninfected or infected with Ad-Ψ5 or Ad-IRF-1. Cells were harvested at the indicated time points post infection and IRF-1, survivin, and β-actin were assessed by immunoblotting as described in Materials and Methods.
3.5. Reduction in Cdc2 in Ad-IRF-1 Infected Breast Cancer Cells
p21 is not a critical regulator of survivin expression in these IRF-1-expressing cancer cells, hence, we investigated the expression of molecules that have been shown to maintain survivin expression and stability [52–53]. The mitotic kinase complex cdc2-cyclin B1 has been shown to be required for survivin phosphorylation on the Thr34 residue [54]. To investigate whether cdc-2 and cyclin B1 are down-regulated in IRF-1 infected cells, MDA-MB-468 and SK-BR-3 human breast cancer cells were harvested at varying time points post infection. Cdc-2 expression is comparable in both the uninfected or Ad-Ψ5 infected cell cohorts. Cdc-2 expression is reduced in the Ad-IRF-1-infected cells, and the most profound decrease in expression is observed at the 48 h post infection time point (Fig. 8A). There is an over three-fold decrease in cdc-2 expression in the MDA-MB-468/Ad-IRF-1 and SK-BR-3/Ad-IRF-1 infected cells when compared with the uninfected and Ad-Ψ5 infected cell cohorts at the 48 h time point post infection (Fig. 8A and Fig. 8B). We also investigated whether cyclin B1 is present in these cells. Cyclin B1 is highly expressed in various tumors [55–58] and oncogenes such as c-myc and H-ras have been shown to induce the cyclin B1 promoter that may contribute to chromosomal instability [59–60]. There is a decrease in the protein level of cyclin B1 at the 48 h time point in the MDA-MB-468/Ad-IRF-1 cells when compared with the Ad-Ψ5 infected control cell cohort. A greater than a five-fold decrease in cyclin-B1 in the SK-BR-3/Ad-IRF-1 infected cells is observed at the 36 h post infection time point when compared with the uninfected and the SK-BR-3/Ad-Ψ5 infected control cells. Taken together, cyclin B1 and cdc2, both of which are known to be involved in survivin expression and activation, are decreased in breast cancer cells that express IRF-1.
Figure 8.

Ectopic expression of IRF-1 results in the down-regulation of cdc-2 and cyclin B1 in human breast cancer cells. (A) The human breast cancer cell line, MDA-MB-468, was either uninfected (NI) or infected with the Ad-Ψ5 or Ad-IRF-1 as has been previously described. Cells were harvested at various time points post infection and Western immunoblotting was conducted as has been described in Materials and Methods. (B) The SK-BR-3 human breast cancer cell line was either uninfected (NI) or infected with the Ad-Ψ5 or Ad-IRF-1 as has been previously described.
4. Discussion
Previously, we reported that the ectopic expression of IRF-1 in human breast cancer cells results in tumor cell death and the suppression of tumor growth in xenogenic mouse models of breast carcinoma that was associated with the reduced expression of the IAP survivin. The cyclin dependent kinase inhibitor, p21, was upregulated in both the Ad-IRF-1 infected p53 mutant MDA-MB-468 and SK-BR-3 human breast cancer cell lines [6].
p21 is known to induce G1 cell cycle arrest and has been shown to be upregulated by IFN γ [48]. IRF-1 binding sites have also been identified in the promoter region of p21 [22]. Moreover, IRF-1 and p21 upregulation have been associated in many studies of cell cycle arrest [61,62]. The precise signaling mechanisms that result in cell cycle arrest, however, have not been fully delineated. Given these observations, in this current report, we begin to investigate the signaling events triggered by IRF-1 and determine whether IRF-1 induces cell cycle arrest via a p21-mediated mechanism.
To confirm and characterize the cell cycle arrest associated with the ectopic expression of IRF-1 and subsequent p21 upregulation, double staining with PI and FITC-αBrdU in uninfected, control Ad-Ψ5, or Ad-IRF-1 infected BrdU-pulsed cancer cells was conducted. Indeed, we show that a G1 cell cycle arrest results from the ectopic expression of IRF-1 in both MDA-MB-468 breast cancer cells and H1299 non-small-cell lung cancer cells. Moreover, our studies clearly confirm the transcriptional upregulation of p21 in these cancer cells by IRF-1. Increased expression of p21 in observed in the MDA-MB-468, H1299, and N87 cancer cells that are all p53 mutated or deleted, hence, increased expression of p21 is independent of p53 status.
We also evaluated the expression of cyclins and cyclin dependent kinases. Ad-IRF-1 infected MDA-MB-468 breast cancer cells had reduced protein levels of Cdk2, Cdk4, and cyclin E. Transcriptional repression of Cdk2 and Cdk4 were also confirmed by quantitative real time RT-PCR.
To determine the role of IRF-1-induced p21 in cell cycle arrest, p21 knockdown MDA-MB-468 breast cancer and H1299 lung cancer cell lines were generated. G1 cell cycle arrest was abrogated in IRF-1-expressing p21-knockdown clones, indicating that p21 plays an important role in mediating G1 arrest in these cancer cells. By immunoblotting E2F1 that promotes cells to enter the S-phase [48–49] is reduced in IRF-1-expressing cells and abrogation of p21 restores E2F1 expression, thus, supporting p21-mediated repression of E2F1.
Our previous studies showed that the ectopic expression of IRF-1 in human breast cancer cells resulted in the induction of p21 and the down-regulation of survivin in both the p53 mutant MDA-MB-468/Ad-IRF-1 and SK-BR-3/Ad-IRF-1 human breast cancer cell lines [6]. We utilized RNA interference technology to evaluate the role of p21 expression in mediating the down-regulation of survivin. Despite the marked decrease in p21 protein in p21c11 cells compared with the p21control cell cohort, survivin expression appeared to be decreased in p21c11 MDA-MB-468 p21 knockdown cells when cultured with either IFN-γ that induces IRF-1 expression or direct expression with Ad-IRF-1. Furthermore, the ectopic expression of IRF-1 results in the suppression of survivin in the human colon cancer cell line, HCT116 p21−/−, where the p21 gene is deleted at both alleles. Interestingly, we observed an increase in survivin expression in MDA-MB-468 breast cancer cells that were transfected with siRNA to IRF-1. IRF-1 may, thus, play a direct role in the down-regulation of survivin. This observation warrants further investigation.
Taken together, p21 does not appear to directly mediate survivin down-regulation in this context. Other studies have shown that p53-induced suppression of survivin was mediated by p21 [45]. It has also been shown that p21 was strongly induced in HCT116 p53−/− or HCT116 p53+/+ human colon cancer cells that were treated with lexatumumab, that binds with TRAIL-R2, in combination with the histone deacetylase (HDAC) inhibitor, Suberoylanilide hydroxamic acid (SAHA) [63]. While survivin expression was decreased in the SAHA treated p21+/+ clone, the expression of survivin remained constant in the p21−/− clone supporting the contention that p21 is required for the suppression of survivin in these cells [63]. It appears that other independent or overlapping signaling pathway(s) may exist to down-regulate survivin and promote cell death in Ad-IRF-1-expressing cancer cells.
Phosphorylation on Thr34 via the mitotic kinase complex cdc2-cyclin B1 is required to maintain survivin expression and stability in cancers cells [54]. Mutations in the Thr34 residue abrogates survivin phosphorylation and results in tumor cell apoptosis in vitro [54] and the inhibition of tumor growth in vivo [64,65]. Phosphorylation of cdc2 may regulate survivin by increasing its protein stability at mitosis [52]. The cdc2-cyclin B1 kinase complex is activated via a series of phosphorylation and dephosphorylation pathways. Cdc2 is phosphorylated on Thr161 by cdc2-activating kinase (CAK) and dephosphorylated on Thr 14 by cdc25c [66,67]. This active cdc-2 bound to cyclin B1 is now able to phosphorylate survivin. We evaluated the expression of these signaling molecules by Western immunoblotting. Our results show that cdc-2 is indeed down-regulated in Ad-IRF-1 infected MDA-MB-468 and SK-BR-3 breast cancer cells. Similarly, other studies have shown that the anti-HIV proviral transcription factor, tetra-O-methyl nordihydroguaiaretic acid (M4N), was able to cause growth arrest and apoptosis in malignant cells by the suppression of Cdc2 and survivin [68]. In contrast to our studies, however, the protein level of cyclin B1 remained constant.
We observed that cyclin B1 was down-regulated in breast cancer cells that express IRF-1. Studies show that the high expression of cyclin B1 is associated with an aggressive phenotype and it is an independent prognostic factor in breast cancer [69]. In addition, studies specifically targeting cyclin B1 show that its depletion results in the inhibition of the kinase activity of Cdc2, induction of G2/M cell cycle arrest, the suppression of tumor cell proliferation, and apoptosis [70].
Taken together, our data show that IRF-1 promotes tumor cell death, the suppression of tumor growth, and cell cycle arrest. While IRF-1 induction of p21 results in G1 cell cycle arrest in the cancer cell lines that we have evaluated, p21 does not appear to directly mediate the observed down-regulation of survivin. We did observe reduced expression of both cdc-2 and cyclin B1 which are known to regulate survivin expression. Moreover, our data suggest that IRF-1 may directly regulate survivin. These results warrant further investigation. Other mechanisms may be mediated by IRF-1 to promote decreased expression of survivin. We are actively investigating these signaling pathways. The precise delineation of these IRF-1 signaling events will facilitate the identification of molecules that may be selectively targeted to down-regulate survivin and promote tumor cell death, thus, leading to novel molecular therapeutic strategies for the treatment of patients with cancer.
Acknowledgments
Grant support was provided by NIH grant CA098403 and the Susan G. Komen for the Cure grant BCTR0708040 (J.H. Yim) and the Elsa U. Pardee Foundation (E. Pizzoferrato). We also thank Emily Petrack for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fujita T, Sakakibara J, Yoshiaki S, Miyamoto M, Kimura Y, Taniguchi T. Evidence for a nuclear factor(s), IRF-1, mediating induction and silencing properties to human IFN-B gene regulatory elements. EMBO. 1988;7:3397–3405. doi: 10.1002/j.1460-2075.1988.tb03213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogasawara K, Hida S, Azimi N, Tagaya Y, Sato T, Yokochi-Fukuda T, Waldmann TA, Taniguchi T, Taki S. Requirement for IRF-1 in the microenvironment supporting development of natural killer cells. Nat. 1988;391:700–703. doi: 10.1038/35636. [DOI] [PubMed] [Google Scholar]
- 3.Yim JH, Wu SJ, Casey MJ, Norton JA, Doherty GM. IFN regulatory factor-1 gene transfer into an aggressive, nonimmunogenic sarcoma suppresses the malignant phenotype and enhances immunogenicity in syngeneic mice. J Immunol. 1997;158:1284–1292. [PubMed] [Google Scholar]
- 4.Yim JH, Wu SJ, Lowney JK, Vander Velde TL, Doherty GM. Enhancing in vivo tumorigenicity of B16 melanoma by overexpressing interferon regulatory factor-2: resistance to endogenous IFN-gamma. J Interferon Cytokine Res. 1999;19:723–729. doi: 10.1089/107999099313569. [DOI] [PubMed] [Google Scholar]
- 5.Kim PK, Armstrong M, Liu Y, Yan P, Bucher B, Zuckerbraun BS, Gambotto A, Billiar TR, Yim JH. IRF-1 expression induces apoptosis and inhibits tumor growth in mouse mammary cancer cells in vitro and in vivo. Oncogene. 2004;23:1125–1135. doi: 10.1038/sj.onc.1207023. [DOI] [PubMed] [Google Scholar]
- 6.Pizzoferrato E, Liu Y, Gambotto A, Armstrong MJ, Stang MT, Gooding WE, Alber SM, Shand SH, Watkins SC, Storkus WJ, Yim JH. Ectopic Expression of Interferon Regulatory Factor-1 Promotes Human Breast Cancer Cell Death and Results in Reduced Expression of Survivin. Cancer Res. 2004;64:8381–8388. doi: 10.1158/0008-5472.CAN-04-2223. [DOI] [PubMed] [Google Scholar]
- 7.Tanaka N, Ishihara M, Lamphier MS, Nozawa H, Matsuyama T, Mak TW, Aizawa S, Tokino T, Oren M, Taniguchi T. Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nat. 1996;382:816–818. doi: 10.1038/382816a0. [DOI] [PubMed] [Google Scholar]
- 8.Prost S, Bellamy CO, Cunningham DS, Harrison DJ. Altered DNA repair and dysregulation of p53 in IRF-1 null hepatocytes. FASEB J. 1998;12:181–188. doi: 10.1096/fasebj.12.2.181. [DOI] [PubMed] [Google Scholar]
- 9.Yokota S, Okabayashi T, Yokosawa N, Fujii N. Growth Arrest of Epithelial Cells during Measles Virus Infection Is Caused by Upregulation of Interferon Regulatory Factor 1. J Virol. 2004;78:4591–4598. doi: 10.1128/JVI.78.9.4591-4598.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kröger A, Stirnweiss A, Pulverer JE, Klages K, Grashoff M, Reimann J, Hauser H. Tumor Suppression by IFN Regulatory Factor-1 is mediated by transcriptional down-regulation of cyclin D1. Cancer Res. 2007;67:2972–2981. doi: 10.1158/0008-5472.CAN-06-3564. [DOI] [PubMed] [Google Scholar]
- 11.Xiong Y, Hannon G, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nat. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 12.Gartel AL, Serfas MS, Tyner AL. p21-negative regulatory of the cell cycle. Proc Am Soc Exp Biol Med. 1996;213:138–149. doi: 10.3181/00379727-213-44046. [DOI] [PubMed] [Google Scholar]
- 13.Niculescu AB, 3rd, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21(Cip/Waf1) at both the G1/S and the G2/M cell cycle transitions:pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998;18:629–643. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogryzko VV, Wong P, Howard BH. WAF1 Retards S-Phase Progression Primarily by Inhibition of Cyclin-Dependent Kinases. Mol Cell Biol. 1997;17:4877–4882. doi: 10.1128/mcb.17.8.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oberg K. Interferon in the management of neuroendocrine GEP-tumors:a review. Digestion. 2000;62(Suppl 1):92–97. doi: 10.1159/000051862. [DOI] [PubMed] [Google Scholar]
- 16.Miyazaki M, Sakaguchi M, Akiyama I, Sakaguchi Y, Nagamori S, Huh N. Involvement of Interferon Regulatory Factor 1 and S100C/A11 in Growth Inhibition by Transforming Growth Factor B1 in Human Hepatocellular Carcinoma Cells. Cancer Res. 2004;64:4155–4161. doi: 10.1158/0008-5472.CAN-03-2750. [DOI] [PubMed] [Google Scholar]
- 17.Dulic V, Stein GH, Far DF, Reed SI. Nuclear accumulation of p21Cip1 at the onset of mitosis: a role at the G2/M-phase transition. Mol Cell Biol. 1998;18:546–557. doi: 10.1128/mcb.18.1.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cayrol C, Flemington EK. The Epstein-Barr virus bZIP transcription factor Zta causes G0/G1 cell cycle arrest through induction of cyclin-dependent kinase inhibitors. EMBO. 1996;15:2748–2759. [PMC free article] [PubMed] [Google Scholar]
- 19.Biggs JR, Kudlow JE, Kraft AS. The role of the transcription factor Sp1 in regulating the expression of the WAF1/CIP1 gene in U937 leukemic cells. J Biol Chem. 1996;271:901–906. doi: 10.1074/jbc.271.2.901. [DOI] [PubMed] [Google Scholar]
- 20.Somasundaram K, Zhang H, Zeng YX, Houvras Y, Peng Y, Zhang H, Wu GS, Licht JD, Weber BL, El-Deiry WS. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/Cip1. Nat. 1997;389:187–190. doi: 10.1038/38291. [DOI] [PubMed] [Google Scholar]
- 21.Zeng YX, Somasundaram K, el-Deiry WS. AP2 inhibits cancer cell growth and activates p21/WAF1/CIP1 expression. Nat Genet. 1997;15:78–82. doi: 10.1038/ng0197-78. [DOI] [PubMed] [Google Scholar]
- 22.Coccia EM, Del Russo N, Stellacci E, Orsatti R, Benedetti E, Marziali G, Hiscott J, Battistini A. Activation and repression of the 2–5A synthetase and p21 gene promoters by IRF-1 and IRF-2. Oncogene. 1999;18:2129–2137. doi: 10.1038/sj.onc.1202536. [DOI] [PubMed] [Google Scholar]
- 23.Velculescu VE, Madden SL, Zhang L, Lash AE, Yu J, Rago C, Lal A, Wang CJ, Beaudry GA, Ciriello KM, Cook BP, Dufault MR, Ferguson AT, Gao Y, He TC, Hermeking H, Hiraldo SK, Hwang PM, Lopez MA, Luderer HF, Mathews B, Petroziello JM, Polyak K, Zawel L, Zhang W, Zhang X, Zhou W, Haluska FG, Jen J, Saraswati S, Landes GM, Riggins GJ, Vogelstein B, Kinzler KW. Analysis of human transcriptome. Nat Genet. 1999;23:387–388. doi: 10.1038/70487. [DOI] [PubMed] [Google Scholar]
- 24.Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC. IAP-Family Protein Survivin Inhibits Caspase Activity and Apoptosis Induced by Fas (CD95), Bax, Caspases, and Anticancer Drugs. Cancer Res. 1998;58:5315–5320. [PubMed] [Google Scholar]
- 25.Kawasaki H, Altieri DC, Lu CD, Toyoda M, Tenjo T, Tanigawa N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998;58:5071–5074. [PubMed] [Google Scholar]
- 26.Monzó M, Rosell R, Felip E, Astudillo J, Sanchez JJ, Maestre J, Martin C, Font A, Barnadas A, Abad A. A novel anti-apoptosis gene:Re-expression of survivin mRNA as a prognosis marker in non-small-cell lung cancers. J Clin Oncol. 1999;17:2100–2104. doi: 10.1200/JCO.1999.17.7.2100. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka K, Iwamoto S, Gon G, Nohara T, Iwamoto M, Tanigawa N. Expression of survivin and its relationship to loss of apoptosis in breast carcinomas. Clin Cancer Res. 2000;6:127–134. [PubMed] [Google Scholar]
- 28.Kato J, Kuwabara Y, Mitani M, Shinoda N, Sato A, Toyama T, Mitsui A, Nishiwaki T, Moriyama S, Kudo J, Fujii Y. Expression of survivin in esophageal cancer:correlation with prognosis and response to chemotherapy. Int J Cancer. 2001;95:92–95. doi: 10.1002/1097-0215(20010320)95:2<92::aid-ijc1016>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 29.Chakravarti A, Noll E, Black PM, Finkelstein DF, Finkelstein DM, Dyson NJ, Loeffler JS. Quantitatively determined survivin expression levels are of prognostic value in human gliomas. J Clin Oncol. 2002;20:1063–1068. doi: 10.1200/JCO.2002.20.4.1063. [DOI] [PubMed] [Google Scholar]
- 30.Kajiwara Y, Yamasaki F, Hama S, Yahara K, Yoshioka H, Sugiyama K, Arita K, Kurisu K. Expression of survivin in astrocytic tumors:correlation with malignant grade and prognosis. Cancer. 2003;97:1077–1083. doi: 10.1002/cncr.11122. [DOI] [PubMed] [Google Scholar]
- 31.Schlette EJ, Medeiros LJ, Goy A, Lai R, Rassidakis GZ. Survivin expression predicts poorer prognosis in anaplastic large-cell lymphoma. J Clin Oncol. 2004;22:1682–1688. doi: 10.1200/JCO.2004.10.172. [DOI] [PubMed] [Google Scholar]
- 32.Tonini G, Vincenzi B, Santini D, Scarpa S, Vasaturo T, Malacrino C, Coppola R, Magistrelli P, Borzomati D, Baldi A, Antinori A, Caricato M, Nuzzo G, Picciochi A. Nuclear and cytoplasmic expression of survivin in 67 surgically resected pancreatic cancer patients. Br J Cancer. 2005;92:2225–2232. doi: 10.1038/sj.bjc.6602632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barnes N, Haywood P, Flint P, Knox WF, Bundred NJ. Survivin expression in in situ and invasive breast cancer relates to COX-2 expression and DCIS recurrence. Br J Cancer. 2006;94:253–258. doi: 10.1038/sj.bjc.6602932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinnis AR, Luckett JC, Walker RA. Survivin is an independent predictor of short-term survival in poor prognostic breast cancer patients. Br J Cancer. 2007;96:639–645. doi: 10.1038/sj.bjc.6603616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paydas S, Ergin M, Erdogans S, Seydaoglu G, Yavuz S, Disel U. Thrombospondin-1 (TSP-1) and Survivin (S) expression in non-Hodkin’s lymphomas. Leuk Res. 2008;32:243–250. doi: 10.1016/j.leukres.2007.06.024. [DOI] [PubMed] [Google Scholar]
- 36.Wu YK, Chen KT, Kuo YB, Huang YS, Chan EC. Quantitative detection of survivin in malignant pleural effusion for the diagnosis and prognosis of lung cancer. Cancer Lett. 2009;273:331–335. doi: 10.1016/j.canlet.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Wu W, Tahir SK, Kroeger PE, Rosenberg SH, Cowsert LM, Bennett F, Krajewski S, Krajewska M, Welsh K, Reed JC, Ng SC. Down-regulation of survivin by antisense oligonucleotides increases apoptosis, inhibits cytokinesis and anchorage-independent growth. Neoplasia. 2000;2:235–241. doi: 10.1038/sj.neo.7900091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Olie RA, Simões-Wüst AP, Baumann B, Leech SH, Fabbro D, Stahel RA, Zangemeister-Wittke U. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res. 2000;60:2805–2809. [PubMed] [Google Scholar]
- 39.Xia C, Xu Z, Yuan X, Uematsu K, You L, Li K, Li L, McCormick F, Jablons DM. Induction of apoptosis in mesothelioma cells by antisurvivin oligonucleotides. Mol Cancer Ther. 2002;1:687–694. [PubMed] [Google Scholar]
- 40.Tran J, Master Z, Yu JL, Rak J, Dumont DJ, Kerbel RS. A role for survivin in chemoresistance of endothelial cells mediated by VEGF. PNAS. 2002;99:4349–4354. doi: 10.1073/pnas.072586399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fulda S, Debatin KM. Sensitization for anticancer drug-induced apoptosis by the chemopreventive agent resveratrol. Oncogene. 2004;23:6702–6711. doi: 10.1038/sj.onc.1207630. [DOI] [PubMed] [Google Scholar]
- 42.Uchida H, Tanaka T, Sasaki K, Kato K, Dehari H, Ito Y, Kobune M, Miyagishi M, Taira K, Tahara H, Hamada H. Adenovirus-mediated transfer of siRNA against survivin induced apoptosis and attenuated tumor cell growth in vitro and in vivo. Mol Ther. 2004;10:162–171. doi: 10.1016/j.ymthe.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 43.Wang Z, Sampath J, Fukuda S, Pelus LM. Disruption of the inhibitor of apoptosis protein survivin sensitizes Bcr-abl-positive cells to STI571-induced apoptosis. Cancer Res. 2005;65:8224–8232. doi: 10.1158/0008-5472.CAN-05-0303. [DOI] [PubMed] [Google Scholar]
- 44.Liu Q, Fu H, Xing R, Tie Y, Zhu J, Sun Z, Zheng X. Survivin knockdown combined with apoptin overexpression inhibits cell growth significantly. Cancer Biol Ther. 2008;7:1053–1060. doi: 10.4161/cbt.7.7.6100. [DOI] [PubMed] [Google Scholar]
- 45.Lohr K, Moritz C, Contente A, Dobbelstein M. p21/CDKN1A mediates negative regulation of transcription by p53. J Biol Chem. 2003;35:32507–16. doi: 10.1074/jbc.M212517200. [DOI] [PubMed] [Google Scholar]
- 46.Stang MT, Armstrong MJ, Watson GA, Sung KY, Liu Y, Ren B, Yim JH. Interferon regulatory factor-1-induced apoptosis mediated by a ligand-independent fas-associated death domain pathway in breast cancer cells. Oncogene. 2007;26:6420–6430. doi: 10.1038/sj.onc.1210470. [DOI] [PubMed] [Google Scholar]
- 47.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 48.Johnson D, Schneider-Broussard R. Role of E2F in cell cycle control and cancer. Front Biosci. 1998;3:d447–448. doi: 10.2741/a291. [DOI] [PubMed] [Google Scholar]
- 49.Sun A, Bagella L, Tutton S, Romano G, Giordano A. From G0 to S phase: a view of the roles played by the retinoblastoma (Rb) family members in the Rb-E2F pathway. J Cell Biochem. 2007;102:1400–1404. doi: 10.1002/jcb.21609. [DOI] [PubMed] [Google Scholar]
- 50.Boulaire J, Fotedar A, Fotedar R. The functions of the cdk-cyclin kinase inhibitor p21WAF1. Pathol Biol. 2000;48:190–202. [PubMed] [Google Scholar]
- 51.Harvat B, Jetten AM. Gamma-interferon induces an irreversible growth arrest in mid-G1 in mammary epithelial cells which correlates with a block in hyperphosphorylation of retinoblastoma. Cell Growth Differ. 1996;7:289–300. [PubMed] [Google Scholar]
- 52.O’Connor DS, Wall NR, Porter AC, Altieri DC. A p34cdc2 survival checkpoint in cancer. Cancer Cell. 2002;2:43–54. doi: 10.1016/s1535-6108(02)00084-3. [DOI] [PubMed] [Google Scholar]
- 53.Park R, Chang CC, Liang YC, Chung Y, Henry RA, Lin E, Mold DE, Huang RC. Systemic Treatment with Tetra-O-Methyl Nordihydroguaiaretic Acid Suppresses the Growth of Human Xenograft Tumors. Clin Cancer Res. 2005;11:4601–4609. doi: 10.1158/1078-0432.CCR-04-2188. [DOI] [PubMed] [Google Scholar]
- 54.O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio PC, Altieri DC. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. PNAS. 2000;97:13103–13107. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dong Y, Sui L, Watanabe Y, Sugimoto K, Tokuda M. Clinical relevance of cyclin B1 overexpression in laryngeal squamous cell carcinoma. Cancer Lett. 2002;177:13–19. doi: 10.1016/s0304-3835(01)00770-4. [DOI] [PubMed] [Google Scholar]
- 56.Hassan KA, Ang KK, El-Naggar AK, Story MD, Lee JI, Liu D, Hong WK, Mao L. Cyclin B1 overexpression and resistance to radiotherapy in head and neck squamous cell carcinoma. Cancer Res. 2002;62:6414–6417. [PubMed] [Google Scholar]
- 57.Takeno S, Noguchi T, Kikuchi R, Uchida Y, Yokoyama S, Muller W. Prognostic value of cyclin B1 in patients with esophageal squamous cell carcinoma. Cancer. 2002;94:2874–2881. doi: 10.1002/cncr.10542. [DOI] [PubMed] [Google Scholar]
- 58.Li JQ, Kubo A, Wu F, Usuki H, Fujita J, Bandoh S, Masaki T, Saoo K, Takeuchi H, Kobayashi S, Imaida K, Maeta H, Ishida T, Kuriyama SS. Cyclin B1, unlike cyclin G1, increases significantly during colorectal carcinogenesis and during later metastasis to lymph nodes. Int J Oncol. 2003;22:1101–10. [PubMed] [Google Scholar]
- 59.Yin XY, Grove L, Datta NS, Katula K, Long MW, Prochownik EV. Inverse regulation of cyclin B1 by c-Myc and p53 and induction of tetraploidy by cyclin B1 overexpression. Cancer Res. 2001;61:6487–6493. [PubMed] [Google Scholar]
- 60.Santana C, Ortega E, Garcia-Carranca A. Oncogenic H-ras induces cyclin B1 expression in a p53-independent manner. Mutat Res. 2002;508:49–58. doi: 10.1016/s0027-5107(02)00172-0. [DOI] [PubMed] [Google Scholar]
- 61.Harper JW, Adami G, Wein N, Keyomarsi K, Elledge S. The p21 cdk- interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 62.Harper JW, Elledge SJ, Keyomarsi K, Dynlacht BM, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E, Fox MP, Wei N. Inhibition of Cyclin-dependent Kinases by p21. Mol Biol Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nawrocki ST, Carew JS, Douglas L, Cleveland JL, Humphreys R, Houghton JA. Histone Deacetylase Inhibitors Enhance Lexatumumab-Induced Apoptosis via a p21Cip1-Dependent Decrease in Survivin Levels. Cancer Res. 2007;67:6987–6994. doi: 10.1158/0008-5472.CAN-07-0812. [DOI] [PubMed] [Google Scholar]
- 64.Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibition of melanoma tumor growth in vivo by survivin targeting. PNAS. 2001;98:635–640. doi: 10.1073/pnas.230450097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mesri M, Wall NR, Li J, Kim RW, Altieri DC. Cancer gene therapy using a survivin mutant adenovirus. J Clin Invest. 2001;108:981–990. doi: 10.1172/JCI12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pines J. Cell cycle: Checkpoint on the nuclear frontier. Nat. 1999;397:104–105. doi: 10.1038/16344. [DOI] [PubMed] [Google Scholar]
- 67.Porter LA, Donoghue DJ. Cyclin B1 and CDK1: nuclear localization and upstream regulators. Progress in Cell Cycle Res. 2003;5:335–347. [PubMed] [Google Scholar]
- 68.Chang CC, Heller JD, Kuo J, Huan RCC. Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. PNAS. 2004;101:13239–13240. doi: 10.1073/pnas.0405407101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aaltonen K, Amini RM, Heikkilä P, Aittomäki K, Tamminen A, Nevanlinna H, Blomqvist C. High cyclin B1 expression is associated with poor survival in breast cancer. Br J Cancer. 2009;100:1055–1060. doi: 10.1038/sj.bjc.6604874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yuan J, Kramer A, Eckerdt F, Roller M, Kaufmann M, Strebhardt K. Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene. 2004;23:5843–5852. doi: 10.1038/sj.onc.1207757. [DOI] [PubMed] [Google Scholar]