

Abstract
NOXO1β is a cytosolic protein which, in conjunction with NOXA1, regulates generation of reactive oxygen species (ROS) by the Nox1 enzyme complex. NOXO1β is targeted to membranes through an N-terminal PX domain. We have used nuclear magnetic resonance spectroscopy to solve the structure of the NOXO1β PX domain and surface plasmon resonance to assess phospholipid specificity. The solution structure of the NOXO1β PX domain shows greatest similarity to the PI3K-C2α PX domain with regard to the positions and types of residues that are predicted to interact with phosphatidylinositol phosphate (PtdInsP) head groups. Surface plasmon resonance experiments identify PtdIns(4,5)P2 and PtdIns(3,4,5)P3 as preferred targets of NOXO1β PX. These findings contrast with previous lipid overlay experiments which show strongest binding to monophosphorylated PtdInsP and phosphatidylserine. Our data suggest that localized membrane accumulation of PtdIns(4,5)P2 or PtdIns(3,4,5)P2 may serve to recruit NOXO1β and activate Nox1.
Keywords: NMR spectroscopy, reactive oxygen species, Nox1, protein-lipid interactions, phosphatidylinositol
Introduction
Reactive oxygen species generated by NADPH Oxidase 1 (Nox1) are involved in cell death/survival 1, tumor angiogenesis 2, and motility/invasiveness 3. Nox1 is a member of the NOX/DUOX family of oxidases, integral membrane proteins with the ability to catalyze transfer of electrons from NADPH to O2 with formation of superoxide. Full activation involves association of the membrane-integral Nox1/p22phox heterodimer with cytosolic proteins Rac1, NOXO1, and NOXA1. Association of NOXA1 with Nox1 is likely the principal activating step, but NOXA1 has low intrinsic affinity for Nox1 and no membrane-targeting properties. Rather, NOXA1 is stabilized at the oxidase through association with Rac1, which targets membranes via C-terminal prenyl-/geranylgeranylation and poly-basic motifs, and NOXO1, which targets p22phox via tandem SH3 domains and targets membranes via an N-terminal PX domain 4, 5. The activated complex is thus similar to the activated phagocytic oxidase complex, which includes association of membrane-integral Nox2 and p22phox with cytosolic proteins p47phox (a NOXO1 homolog), p67phox (a NOXA1 homolog), p40phox, and Rac.
NOXO1 exists as four splice variants, α-δ, which differ by insertions/deletions within the PX domain 6. NOXO1β is the major variant, while NOXO1γ is abundant in testis. PX domains are ~130 residue domains that function in protein- or phospholipid-targeting. Structures of the p40phox and Grd19p PX domains bound to short-acyl chain PtdIns(3)P identify a consensus PtdIns binding pocket and show an Arg residue coordinating the D3 phosphate 7, 8. Similar pockets are present in all structures of PtdIns-binding PX domains. Variations in the size of the pocket and in the identity of residues that contact the PtdIns head group appear to dictate specificity of a given PX domain for a given phosphorylated PtdIns. Protein-lipid overlay analyses of NOXO1β suggest a broad preference for anionic phosphatidylinositols, including PtdIns(3,5)P2, PtdIns(3)P, PtdIns(4)P, and PtdIns(5)P. Cell localization studies show that NOXO1β is constitutively membrane-bound and that this localization is dependent on the PX domain 9–11. The constitutive membrane localization, and hence activation of Nox1, by NOXO1β is consistent with constitutive ROS production observed in HEK293 cells transfected with murine Nox1, NOXA1, and NOXO1 12. In contrast, the p47phox PX domain shows preference for PtdIns(3,4)P2 which is generated by PI-3K in stimulated cells 13; phagocytic Nox2, which is regulated in part by colocalization with p47phox, is not active in unstimulated macrophages or neutrophils. Notably, studies in which human, rather than murine, Nox1, NOXA1, and NOXO1 genes are transfected show weak basal ROS generation, with phorbol ester stimulus required for maximal activity 9, 14, 15, indicating the existence of a stimulus-dependent activation mechanism.
To address the question of whether the NOXO1β PX domain can target the plasma membrane in a specific phosphatidylinositol phosphate-dependent manner, we have determined the solution structure of the NOXO1β PX domain using NMR spectroscopy and re-evaluated lipid binding specificity using surface plasmon resonance. While protein:lipid overlay studies show that the NOXO1β PX domain binds to numerous phosphorylated PtdIns species, a sequence alignment of NOXO1β against other PX domains shows that the residue predicted to interact with the D3 position of the PtdIns ring is Ser, rather than Arg, suggesting that D3-phosphorylated PtdIns should not be a favorable ligand for NOXO1β. The structure of the NOXO1β PX domain described herein shows that the positions of potentially phosphate-coordinating Arg residues are most similar to those of the PI3K-C2α PX domain, which shows specificity for PtdIns(4,5)P2. In contrast to overlay experiments, SPR experiments identify selectivity of the NOXO1β PX domain for PtdIns(4,5)P2 and PtdIns(3,4,5)P3, and NMR-detected titration experiments show a structurally specific interaction of NOXO1β PX with PtdIns(4,5)P2. Localized increases and decreases in PtdIns(4,5)P2 concentration occur in a variety of cellular processes, including actin-mediated remodeling. Targeting by NOXO1β to localized pools of PtdIns(4,5)P2 could thus mediate Nox1 activity in a PtdIns phosphate-dependent manner.
Results
Insights into phospholipid specificity derived from the solution structure and fast timescale dynamics of the NOXO1β PX
We have solved the solution structure of the NOXO1β PX domain using NMR methods with the goal of using this structure to better understand the structural basis of PtdInsP specificity among PX domains. The resultant structures are shown in Figure 1A and B, while structural statistics are shown in Table 1. Backbone (heavy) atom RMSD over structured regions is 1.03 (2.05) Å. The comparatively high RMSD is a consequence of the need for perdeuteration during resonance assignment and the associated paucity of NOEs among non-exchangeable protons. Inclusion of NOEs from Ile(δ1), Leu, and Val methyl groups and Phe ring protons was critical to convergence. The structures satisfy NMR restraints, Ramachandran backbone and Whatcheck packing criteria.
Figure 1.

(A) Overlay of fourteen lowest energy structures of the NOXO1β PX domain. Enhanced τe values for the α1/α2 loop identify conformational diversity at the PtdInsP-binding pocket (arrow). Helices are colored light green, strands dark green, and extended/coil gray. (B) Low-energy structure, rotated 90° on the horizontal axis from A, with conserved secondary structural elements labeled. The fourth β-strand is common to NOXO1β and p47phox. (C) The R40 guanidino group interacts with the C-terminus of α3 and the D125 carboxylate. Interaction of the D43 carboxylate with R46 may mitigate electrostatic repulsion at the D3 position of the inositol ring, allowing binding to PtdIns(3,4,5)P2. Orientation of C matches that of A. Strands are colored dark green, helices light green, and coil gray.
Table I.
Refinement statistics for fourteen lowest-energy structures of NOXO1β-PX.
| Number of experimental restraints | |||||
|---|---|---|---|---|---|
| Distance restraints from NOEs | total | intra | sequential | medium | long |
| unambiguous | 559 | 85 | 155 | 134 | 185 |
| ambiguous | 67 | ||||
| Dihedral φ/ψ | 222 | ||||
| χ1 | 97 | ||||
| H-bond | 63 | ||||
| RDC | 153 | ||||
| 1H shift | 53 | ||||
| RMSD, energy, and average number of violations from experimental restraints | |||
|---|---|---|---|
| Restraint | RMSD | Energy (kcal/mol) | Avg. viols. |
| NOE distances, Å | 0.15 ± 0.02 | 162 | (> 0.5Å) 4 |
| Dihedral angles, °. | 1.9 ± 0.3 | 85 | (> 5°) 5 |
| 1H shifts, ppm | 0.09 ± 0.01 | 22 | (>0.2 ppm) 2 |
| RDC, Hz | 0.44 ± 0.04 | 148 | |
| Q-factora | 0.067 ± 0.007 | ||
| RMSD from idealized geometry | |
|---|---|
| Bonds, Å | 0.01 ± 0.00 |
| Angles, °. | 1.3 ± 0.04 |
| Improper, °. | 4.1 ± 0.2 |
| Total energy (kcal/mol)b | −6702 |
| Coordinate precision | backbone | heavy |
|---|---|---|
| Structuredc, Å | 1.03 | 2.05 |
| Ramachandram analysis | m.f. | a.a. | g.a. | dis. |
|---|---|---|---|---|
| All | 88.1 | 8.8 | 1.7 | 1.4 |
| Structuredc | 93.4 | 5.7 | 0.7 | 0.3 |
| What_check Z-score | |
|---|---|
| First generation packing | −1.96 |
| Second generation packing | −2.09 |
| χ1/χ2 normality | −1.23 |
Cornilescu et al. (1998). J. Am. Chem. Soc.120:6836–6837.
Includes all restraints, geometries, and electrostatic energies
Structured residues span 8–56, 84–120, and 135–138.
The growing body of PX domain structures yields a set of structure-based predictive rules for PtdIns phosphate-binding specificity. The crystal structure of the p40phox PX domain bound to PtdIns(3)P shows coordination of the D3 phosphate by R58 7. P47phox16 contains the structurally analogous R43, which presumably coordinates the D3 phosphate in the p47phox PX target PtdIns(3,4)P2. R50 in the CISK PX domain, which binds PtdIns(3,4,5)P3, PtdIns(3,5)P2, and to a lesser degree, PtdIns(4,5)P217, is likewise positioned to coordinate a D3 phosphate 18. [The CISK PX has also been described as PtdIns(3)P-specific 19.] Thus, a positively charged residue in this position is strongly conserved among PX domains that have high affinity for D3-phosphorylated PtdInsP’s. In contrast, a hydrogen bond acceptor/donor residue is typically found in PX domains that target non-D3-phosphorylated PtdIns head groups, e.g., T1462 in the PI3K-C2α PX domain and Y317 in the Bem1p PX domain, which target PtdIns(4,5)P2, and PtdIns(4)P, respectively.
Our structure shows that the NOXO1β PX domain has a Ser at the D3-interacting position, suggesting that PtdIns(3)P is not a primary target. Inspection of residues surrounding the PtdInsP binding pocket shows that the NOXO1β PX domain shows greatest similarity to the PI3K-C2α PX domain 20, 21, which specifically binds PtdIns(4,5)P2. Compared over 80 Cα atoms in secondary structure elements, NOXO1β PX exhibits a 1.49 Å RMSD vs. PI3K-C2α PX (2REA) and a 1.88 Å RMSD vs. p47phox PX (1KQ6). Figure 2 compares PtdInsP binding site residues of the PX domains of NOXO1β with p40phox [which binds to PtdIns(3)P], p47phox [PtdIns(3,4)P2], Bem1p [PtdIns(4)P], CISK [PtdIns(3,4,5)P3and PtdIns(3,5)P2] and PI3K-c2α [PtdIns(4,5)P2] and shows the presence of an Arg positioned to coordinate a D3 phosphate in p40phox, p47phox and CISK but the absence of a corresponding Arg in the PI3K-c2α, Bem1p, and NOXO1βPX domains. In all cases, the residue expected to interact with the D4 position is an Arg. An Arg at this position is highly conserved throughout PX domains whether they bind to D4-phosphorylated PtdIns and is not in itself diagnostic of PtdInsP specificity. Instead, factors that appear to contribute to selection for/against D4- and D5-phosphates are the Arg content and the size and conformational flexibility of the binding pocket and the loop immediately preceding helix α2.
Figure 2.

Comparison of PtdInsP binding sites for PX domains with specificity for different PtdIns phosphates. View is into the PtdInsP-binding pocket. (A) p40phox (1H6H) bound to PtdIns(3)P; (B) p47phox (1KQ6), which shows preference for PtdIns(3,4)P2; (C) Bem1p (2CZO), which shows preference for PtdIns(4)P; (D) CISK (1XTE), which shows preference for PtdIns(3,4,5)P3 and PtdIns(3,5)P2; (E) PI3K-C2α (2REA), which shows preference for PtdIns(4,5)P2; (F) NOXO1β, which shows preference for PtdIns(4,5)P2 and PtdIns(3,4,5)P3. Helices α1 and α2 are labeled for reference. Circled residues are in position to coordinate the D3 phosphate/hydroxyl moiety of the inositol ring. Labeled Arg residues presumably coordinate phosphate/hydroxyl moieties at D4 and D5 (full electron density is missing for PI3K-C2α R1493). (G) Structure-based alignment of PX domains. The end of β3 and the start of α2 are marked. The blue box marks the residue that interacts with the D3 position of the PtdIns head group, the green box marks the residue that interacts with the D4 position (based on the p40phox and Grd19p PX structures). Residues potentially positioned to coordinate a D5 phosphate are shown in red(CISK N81 has been proposed to hydrogen bond to the D5 phosphate). Residues conserved in the α1/α2 loops of NOXO1β and PI3K-C2α are shown in bold. Residues in the loop immediately preceding α2 are conformationally divergent among solved PX domain structures (black box). NOXO1γ contains a five-residue insertion between NOXO1β L74/D75. Primary PtdInsP specificities for each PX domain are listed at right.
NOXO1β has a relatively open PtdInsP-binding pocket in that α2 loop-residues do not block access. Analysis of fast timescale dynamics based on 15N relaxation also show that that this loop is disordered. Figure 3 shows results of Modelfree v4.20 analysis 15N T1, T2, and NOE measurements and identify a clustering of residues exhibiting ns-scale flexibility (as manifested in elevated τe values) in the loop immediately preceding α2. This section spans the Arg residues (81, 84) potentially involved in coordination of a D5-phosphate, and the conformation of this loop as depicted in Figure 2F is one of a structurally diverse ensemble (Figure 1A, arrow). Notably, the corresponding loop is also disordered in the crystal structures of the PI3K-C2α PX domain, which binds PtdIns(4,5)P2. Whether disorder in this loop is a requisite for binding to PtdIns(4,5)P2 or simply a consequence of the longer loop found in these two PX domains will require further analysis, and preferably a structure of a PX domain bound to a lipid other than PtdIns(3)P. Most other regions in NOXO1β with elevated τe or Rex are sparsely distributed or of small magnitude, with the notable exception of the loop preceding strand β4.
Figure 3.

NOXO1β PX domain backbone dynamics as determined by 15N relaxation. Plotted are best-fit S2, τe and Rex as calculated by Modelfree 4.20 for motional models determined by FAST Model Free. Residues in the loop preceding helix α2 exhibit elevated values of τe.
Strand β4, unambiguously identified in our NOXO1β PX structure, runs antiparallel to β1 (Figure 1B) and is not a canonical component of PX domains. An analogous strand is found in the X-ray structure of the long construct (1KQ6) of the p47phox PX domain 22 but not in the X-ray (1O7K) or NMR (1GD5) structures of the short construct16, 23. Across known PX domains, this strand appears to be restricted to the NOXO1/p47phox family, although a C-terminal β-strand antiparallel to β3 is found the in the PX domain of Bem1p 24. The β4 strand in p47phox and NOXO1β redirects the C-terminus to point in the other direction, and emanate from the opposite side, relative to most known PX domain structures. This strand thus potentially alters positioning of the PX domain relative to the subsequent domains in NOXO1/p47phox as compared to other PX domain-containing proteins and shows that the α3-β4 loop and β4 strand are structurally conserved components of this family.
Structural impact of the R40Q mutation
The R40Q mutation to the NOXO1β PX domain disrupts lipid binding in vitro and membrane localization in vivo9. This mutation is the analog of the R42Q mutation in p47phoxwhich is found in patients with chronic granulomatous disease 25 and which also disrupts membrane localization in vivo13. This mutation is frequently used as a negative lipid-binding control for both p47phox and NOXO1β. However, our structure clearly shows that Ser41, and not Arg40, occupies the PtdIns D3 coordination position (Figure 1C). The side chain of R40/42 is largely buried in both NOXO1β and p47phox16, 22, 23, and in both domains, the Arg guanidino group mitigates the negative charges from D125 (NOXO1β)/D124 (p47phox) and the C-cap of helix α3 and seals the hydrophobic core from solvent. A Gln side chain lacks the positive charge and is too short to fulfill these interactions. Thus, the R40Q (NOXO1β)/R42Q (p47phox) mutation 9, 13 likely disrupts protein:lipid binding by destabilizing the PX domain, rather than abrogating a specific Arg:phosphate interaction, consistent with the findings that this mutation to p47phox in CGD patients results in absence, rather than mislocalization, of p47phox26.
Phospholipid binding specificity of the NOXO1β PX domain
SPR analysis
The notable structural similarity to a PtdIns(4,5)P2-specific PX domain prompted us to further examine lipid binding specificity through direct means. We performed SPR experiments in which we flowed NOXO1β PX across a phospholipid surface consisting of a PC:PE bilayer with/without 6 mol % PtdInsP. Figure 4A–C shows sample SPR traces for NOXO1β-PX binding to PtdIns(3)P and PtdIns(4,5)P2, and p40phox-PX binding to PtdIns(3)P. The high level of binding to the control surface is NOXO1β-specific, as p40phox-PX exhibits minimal (0–50 RU) binding to the reference lane and substantial (several hundred RU) binding to 3 mol % PtdIns(3)P under identical running conditions. We observed reproducible, statistically significant increased binding to PC:PE surfaces containing 6 mol % PtdIns(4,5)P2 and PtdIns(3,4,5)P3 at 5 and 10 μM protein concentrations (p < 0.05, Dunn’s post-hoc analysis). NOXO1β shows no significant binding vs. background to any of the other PtdInsP’s (Figure 4D–E). Binding to PtdIns(3,4)P2 approaches, but does not reach, statistical significance.
Figure 4.

SPR data for binding by the NOXO1β PX domain to phosphatidylinositols and acidic phospholipids. (A-C) Example SPR traces for 5 μM NOXO1β PX domain binding to 6 mol % (A) PtdIns(3)P and (B) PtdIns(4,5)P2. (C) shows 5 μM p40phox PX domain binding to 3 mol % PtdIns(3)P under the same experimental conditions for comparison. Solid line shows binding to PtdInsP lane, dashed line shows binding to the reference lane. (D-E) Analysis of NOXO1β PX binding to phospholipids in a PE/PC bilayer at (D) 5 μM and (E) 10 μM protein concentration. Labeled phospholipid was present at 6 mol %. Fold binding over reference is calculated as the ratio of baseline-subtracted target to baseline-subtracted reference lanes. Mean +/− std. dev. of binding shows significance (p < 0.05) vs. PtdIns for PtdIns(4,5)P2, and PtdIns(3,4,5)P3 using Dunn’s post-hoc method following ANOVA.
Because NOXO1β PX associated with the control PC:PE surface at a high level (hundreds of RUs for NOXO1β vs. tens of RUs for p40phox), we report our SPR results as ratios, rather than differences, between target and reference lipid lanes. This treatment is necessary because the absolute RUs for binding to the control surface is dependent on the amount of lipid deposited on the L1 chip surface, and this is difficult to control to the degree necessary to make analysis of RU differences consistent and meaningful over the ten lipid compositions we tested. ANOVA of the entire data set shows larger between than within group variance, indicating that (1) that the use of RU ratios is sufficiently robust to accommodate differences in initial lipid loading and (2) that the NOXO1β PX domain has quantitatively distinct binding preferences for specific PtdInsP’s. Post-hoc analysis using Dunn’s method identified binding to PtdIns(4,5)P2 and PtdIns(3,4,5)P3 as statistically significant. Although the use of RU ratios allows us to extract useful information from our SPR data, it does not allow fitting of association/dissociation curves or selection of a binding model. We also noted frequent clogging of the Biacore flowpath at protein concentration levels sufficient to saturate binding. Hence, we are unable to quantitatively determine KD, kon, or koff for NOXO1β PX.
NMR titration
Titration of the NOXO1β PX domain with diC4-PtdIns(4,5)P2 caused titrable, significant (greater than half of linewidth) chemical shift changes for numerous residues in helix α1 (W42, D43, Q47, K49-T51, E54) and residues in the α1α2 loop (D75) and in the α2 helix (R91, L94, D96 and L103). An expansion of the initial and terminal HSQC spectra in the titration series is shown in Figure 5A. The residues that exhibit chemical shift changes in the presence of PtdIns(4,5)P2 cluster on the membrane-binding surface proximal to the putative PtdIns(4,5)P2-binding pocket (Figure 5B). In contrast, titration with diC6-PtdIns(3,4)P2 did not cause concerted shift changes (Figure 5C), although some residues appear to have been broadened. These results are consistent with the SPR data in that they identify an interaction between the NOXO1β PX domain and PtdIns(4,5)P2. Importantly, they differentiate between binding to PtdIns(3,4)P2 and PtdIns(4,5)P2. Although we do observe an impact on intensity of several protein residues upon addition of PtdIns(3,4)P2, this interaction is much weaker and not structurally localized. Given that the NOXO1β PX domain is strongly basic (calculated pI ~11), we suggest that at least some of the previously observed in vitro associations of NOXO1β PX with anionic lipids is driven by nonspecific electrostatic interactions.
Figure 5.
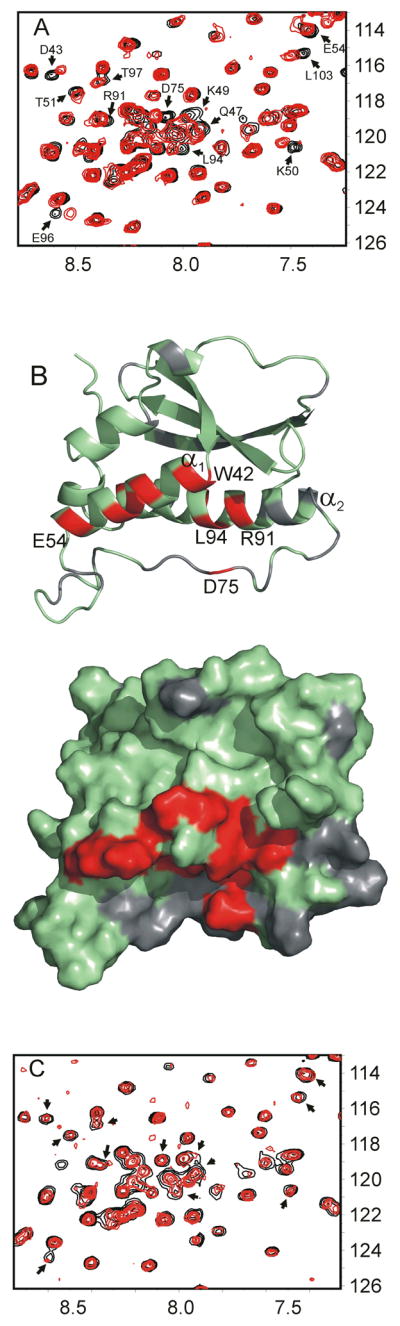
(A) Expansion of NOXO1β PX HSQC spectra in the absence (black) and presence (red) of 2-fold molar excess PtdIns(4,5)P2. Resonances exhibiting significant changes in chemical shift are identified by arrows. (B) Residues matching these resonances are colored red on the NOXO1β PX structure. These residues cluster along helix α1 and the putative PtdInsP-binding site residues in the α1/α2 loop and helix α2. The surface representation is in the same orientation as the ribbon diagram and shows clustering of PtdIns(4,5)P2-interacting residues at the phospholipid binding pocket. Residues exhibiting no shift changes are green, missing/overlapped residues are gray. (C) Expansion of HSQC spectra in the absence (black) and presence (red) of 2-fold molar excess PtdIns(3,4)P2. Arrows mark the same residues as in (A). Although several resonances broaden, no residues show titrable shift changes.
Comparison to other NOXO1 isoforms
NOXO1 has four splice variants 14, 15, with differences between proteins contained within the PX domain. NOXO1β is the major isoform. NOXO1α and NOXO1δ lack K50 (NOXO1β numbering), which lies in helix α1 and is completely surface exposed in our structure. Its absence would alter the helical register, resulting in L52 being surface-exposed and K53 being buried, likely disrupting proper folding of the PX domain. Both NOXO1α and δ localize to intracellular vesicles and/or form large aggregates when overexpressed 11, consistent with partial misfolding of the protein. NOXO1γ and δ differ from NOXO1β and α, respectively, in containing the five-residue insert GQASL between L74 and D75 (NOXO1β numbering). Because this insertion occurs in the long α1-α2 loop, it is less likely than the K50 deletion to disrupt the PX domain structure. Indeed, while attempts to purify the NOXO1α PX domain were unsuccessful, we were able to express and purify the NOXO1γPX domain. Figure 6 shows an overlay of HSQC spectra of the NOXO1β and NOXO1γ PX domains. The similarity of the spectra indicate that both PX domains are folded and that they adopt the same overall fold. The most prominent shift changes occur at the insertion point, spanning L73-A76 and at residues proximal to the insertion point, including L94 in the α2 helix and the W42 indole. Residues at the position of the Trp often form the back of the PtdIns-binding pocket in other PX domain structures. Smaller but concerted shift changes are observed in neighboring residues spanning L70-V82. NOXO1γ exhibits decreased localization to the plasma membrane and is largely localized in the nucleus 11 or cytoplasm 10. This change in localization could be a direct result of differing PtdInsP-specificity or -affinity between the isoforms, or an indirect result of differing intra- or intermolecular interactions between the PX domain and other NOXO1 domains or distinct proteins. Notably, the residues that show shift changes between isoforms are included among those that show shift changes upon addition of PtdIns(4,5)P2. A more detailed analysis of NOXO1γ will be required to determine the impact of the five-residue insertion on protein structure and lipid binding.
Figure 6.

HSQC spectra of NOXO1β (black) and NOXO1γ (red) PX domains. Overall similarity indicates that both proteins adopt the same fold. Residues that exhibit large shift changes cluster around the insertion point (L74/D75) either in sequence (L73-L79) or space (L94, W42).
Discussion
We have determined the solution structure of the NOXO1β PX domain and investigated its preferential PtdInsP target using lipid overlay- (Supplemental Figure 1), SPR-, and NMR-based experiments. Our structural data show that both the PtdInsP-binding pocket size and the presence of localized disorder are similar to that observed for the PtdIns(4,5)P2-binding PI3K-c2α, but distinct from the PtdIns(4)P-binding Bem1P PX domains, which has a smaller and more rigid pocket. Stahelinet al. have proposed that the loop immediately preceding helix α2 establishes a steric barrier against D5-phosphorylated PtdInsP’s in Bem1p 24. This loop is several residues longer in NOXO1β, CISK and PI3K-C2α than in p40phox, p47phox or Bem1p (Figure 2G). The crystal structures of the PI3K-C2α PX domain are missing electron density for residues in this loop (e.g., no density for residues 1488–1497 in 2IWL, weak side chain density for residues 1490–1496 in 2REA), suggesting that the PtdIns(4,5)P2-binding loop is disordered in the unbound state. In contrast, the analogous loop residues in crystal structures of p40phox, p47phox, and Bem1p are well defined. Additional selectivity for a D5-phosphate presumably arises from the Arg residues present in this loop in PI3K-C2α, CISK and NOXO1β but absent in Bem1p, p40phox and p47phox. Mutation of R1493A in PI3K-C2α causes a 23-fold increase in Kd for PtdIns(4,5)P221. Notably, this residue is conserved in NOXO1β (R81, Figure 2G), consistent with coordination of a D5 phosphate. While the PI3K-C2α PX domain is specific for PtdIns(4,5)P227, our SPR experiments show that NOXO1β is also able to interact with PtdIns(3,4,5)P3. Both NOXO1β and PI3K-C2α PX domains contain an Asp at the beginning of α1 (D43 and D1464 in NOXO1β and PI3K-C2α, respectively) which would provide unfavorable electrostatic interactions with the D3 phosphate. However, the negative change could be mitigated in NOXO1β by R46, which is on the same side of α1 and forms a salt bridge to D43 in our structures (Figure 1C). The Gln at residue 1467 in PI3K-C2α cannot form a similar interaction.
The differences in PtdInsP-binding specificity observed for the NOXO1β PX domain may result from the different physical presentations of target phospholipids in each of the assays used. Lipid overlays present long-chain PtdInsP’s adhered to nitrocellulose membranes, SPR presents long-chain PtdInsP’s embedded in a PE:PC bilayer, and NMR presents short-chain PtdInsP in solution. Lipid overlay assays, while convenient, have the potential to generate misleading data, especially with regards to phospholipid specificity of PX domains 28. Collectively, in our (Supplemental Data) and others’ hands 9–11, the NOXO1β PX domain exhibits binding to PtdIns’s (3)P, (4)P, (5)P, and (3,5)P2, as well as PA and PS. Notably, each group has obtained a different subset of binding specificities, highlighting a difficulty in interpreting NOXO1β binding data. Surface plasmon resonance analysis, while substantially more time, labor, and equipment intensive than lipid overlay experiments, has been identified as a preferable method of testing phospholipid-binding specificity of proteins 29. Our SPR analysis reveals a statistically significant increase in binding by the NOXO1β PX domain to PtdIns(4,5)P2 and PtdIns(3,4,5)P3 vs. control (PE/PC or PE/PC/PtdIns). Binding to PtdIns(3,4)P2 approaches significance, while binding to PtdIns monophosphates is not significant. In comparison, only minimal binding to either PtdIns(4,5)P2 or PtdIns(3,4,5)P3 is observed in lipid overlay experiments. Our NMR titration experiments extend our SPR results and further differentiate between site-specific binding to PtdIns(4,5)P2 vs. weaker, nonspecific binding to PtdIns(3,4)P2.
In quiescent cells, PtdIns(4)P and PtdIns(4,5)P2 each constitute nearly 50 % of the phosphorylated PtdIns content, with PtdIns(3)P and PtdIns(5)P constituting ~2 % each and PtdIns(3,4)P2, PtdIns(3,5)P2, and PtdIns(3,4,5)P3 constituting ~0.1 % each 30. PtdIns(4,5)P2 is concentrated in the plasma membrane, PtdIns(4)P is distributed between the plasma membrane and the Golgi, and PtdIns(3)P is largely found in early endosomes 31, 32. Our structure and NMR- and SPR-derived binding data suggest that the previously observed constitutive localization to the membrane by NOXO1β is due to its interaction with PtdIns(4,5)P2, rather than PtdIns(3)P or PtdIns(4)P as suggested by lipid-overlay experiments. As noted by Stahelin et al. 33, PX domain-mediated targeting in vivo depends both on the affinity for a particular PtdInsP of the PX domain and on the availability of each PtdInsP species. Given that the NOXO1β PX domain shows particular affinity to PtdIns(4,5)P2 as measured by SPR and NMR titration experiments, and that this phospholipid is by far the predominant PtdInsP in resting cell plasma membranes, we suggest that PtdIns(4,5)P2 is the primary lipid target of NOXO1β.
Nox1-generated ROS are involved in numerous cellular processes involving cytoskeletal remodeling and cell migration 34–36. Notably, localized accumulation of PtdIns(4,5)P2 is associated with recruitment of actin-modifying proteins and actin polymerization, a hallmark of cell migration and cytokinesis 37. Thus, while PtdIns(4,5)P2 is distributed throughout the plasma membrane in quiescent cells, variation in localized concentration is observed in multiple cellular processes. Our finding that NOXO1β targets PtdIns(4,5)P2 thus provides a possible link between accumulation at sites of cytoskeletal remodeling of PtdIns(4,5)P2 and the spatially and temporally localized activation of Nox1. The ability of NOXO1β to bind to PtdIns(3,4,5)P3 also suggests that phosphorylation at D3 of PtdIns(4,5)P2 will not abrogate targeting by NOXO1β and thus not inhibit ROS production by Nox1. Overall, our structure, SPR, NMR-titration analyses of the NOXO1β PX domain suggest a primary in vivo target of PtdIns(4,5)P2. However, prediction of PtdIns specificity based on PX domain structure remains challenging, likely due to observed flexibility at the ligand-binding pocket and conformational changes at the pocket upon ligand binding.
Materials and Methods
Protein expression and purification
The human NOXO1β PX domain (residues 1–144) was isolated by PCR from the full-length clone (gift of Tom Leto), inserted into pGEX-6P1, and expressed as a GST-fusion protein in BL21(DE3)CodonPlus RIPL cells (Stratagene) and purified as previously described38. Optimal expression of soluble protein was achieved at 32 °C. Later experiments used a pET47b-based plasmid into which maltose-binding protein had been inserted and the protease cleavage site modified to match that of pGEX-6P1. This plasmid gave substantially higher expression levels (10 mg final product/L culture). Purification followed that of the GST-fusion with the addition of a Ni-NTA (Qiagen) column step for initial capture and elimination of the GSH column step. The plasmid coding for the NOXO1γ PX domain was constructed by mutagenesis of the NOXO1β/pGEX-6P1 construct using the Quick change protocol (Stratagene). NOXO1γ PX expression (15N) and purification followed that of NOXO1β PX.
All proteins were expressed in M9 minimal media, and all proteins were cleaved from their fusion partners using Prescission Protease (GE Biosciences). Protein samples for SPR were dialyzed into 20 mM HEPES, pH 7.1, 137 mM KCl, 1 mM MgCl2, 1 mM EGTA (SPR buffer). Perdeuterated samples were grown in >99 % D2O (Spectra Stable Isotopes) with 15NH4Cl (Sigma-Isotec, CIL) and 2H713C6-glucose (CIL) as sole nitrogen and carbon sources. I(δ1)LV-methyl protonated samples were produced by addition of 100 mg 2-keto-3-(methyl-d3)-butyric acid, 50 mg 2-ketobutyric acid (13C4, 3,3-d2) (CIL and Sigma-Isotec) prior to induction. I(δ1)LV-methyl, 1H/12C/15N-F samples were produced by addition of 100 mg 2-keto-3-(methyl-d3)-butyric acid, 50 mg 2-ketobutyric acid (13C4, 3,3-d2), and 40 mg 15N phenylalanine (CIL) prior to induction.
The p40phox PX domain (residues 2–149) was cloned into pET49b (EMD) as a GST-His6-fusion and expressed in BL21(DE3) CodonPlus RIPL cells (Stratagene) overnight at 15 °C. Fusion protein was captured by application to GSH-Sepharose FF resin (GE Biosciences). The fusion protein was eluted from the column, cleaved from the GST tag overnight using Prescission Protease, purified by sequential application to GSH-Sepharose FF and Source Q columns (GE Biosciences), and dialyzed into SPR buffer.
NMR Data Acquisition
NMR spectra were acquired at 293K on a Bruker Avance DRX-600 spectrometer equipped with a triple resonance Z-gradient Cryoprobe. Samples were dialyzed into 100 mM NaPi, 100 mM NaCl, 0.1 mM EDTA, pH 6.5. Five percent (v/v) 2H5 glycerol was added after samples were concentrated. NOXO1β and γ PX domains were concentrated to ~500 μM for all NMR experiments except as otherwise noted. Use of a 4 mm Shigemi sample tube inserted into a 5 mm flat bottom tube containing D2O provided 30% shorter 1H pulses (vs. a 5 mm sample tube), allowed use of 100 % H2O for the sample buffer, and afforded equivalent signal/noise as a same-concentration sample in a 5 mm NMR tube. As previously noted 38, perdeuteration of the protein was necessary to observe magnetization transfer among side-chain carbons. Analysis of the 15N relaxation experiments yielded a rotational correlation time of 14.6 ns. Size exclusion chromatography was consistent with a monomeric protein, and we noticed no concentration-dependent changes in chemical shifts between 50 μM and 1 mM. The long correlation time is a consequence of temperature (293 K) and inclusion of glycerol, both of which extended protein stability. For PtdInsP titration experiments, 2H/15N-labeled protein (80 μM) was dialyzed into 20 mM HEPES, pH 7.2, 137 mM KCl, 0.1 mM EGTA. DiC6-PtdIns(3,4)P2 (Cayman Chemical, Ann Arbor, MI) or diC4-PtdIns(4,5)P2 (Echelon Biosciences, Inc., Salt Lake City, UT) was dissolved in titration buffer at 160 μM and added to the protein solution to give protein:lipid ratios of 1:0, 0.25, 0.5, 0.75, 1, and 2.
Resonance Assignment
HN, N, C, Cα, and Cβ shifts were assigned as described 38. Methyl groups were assigned using the HmeCmeCGCBCA sequence 39, an HmeCmeCBCA sequence which provided higher sensitivity for Val residues and was derived from the previous sequence by deleting one carbon transfer step, and the H(C)C(CO)NH/(H)CC(CO)NH TOCSY sequences 40. We were able to assign 3/3 Ile, 10/10 Val and 42/44 Leu methyl groups. Data were collected on both fully 2H/13C/15N/1H(Iδ1/L/V)-labeled samples and on samples containing Leu and Val residues that were labeled 1H/13C on one methyl group and 2H/12C on the other methyl group 39. All Gln NH2 resonances were assigned using a 2D version of the H2N(CO)CGCB sequence 41. Phenylalanine resonances were assigned through analysis of NOESY, COSY, and TOCSY spectra. NMR data were processed using NMRPipe 42 and analyzed using NMRView 43.
Restraint Collection
Three-dimensional 15N- and/or 13C-edited NOESY-HSQC spectra 44 were collected in H2O at 20 °C on 2H/15N, 2H/13C/15N, 2H/13C/15N/1H(Iδ1/L/V) and 2H/13C/15N/1H(Iδ1/L/V)+(15N- F) samples in H2O and D2O at 20 °C with 200 ms and 300 ms mixing times. A three-dimensional, CT-NOESY-HSQC was collected in D2O with a 300 ms mixing time. Isotopic half-filtered NOESY spectra 45 were also collected on the 2H/13C/15N/1H(Iδ1/L/V)+(15N-F) labeled sample in H2O. Backbone 15N relaxation data were collected using standard 2D sensitivity-enhanced experiments. R1 data were collected with 0.12, 0.24 (x2), 0.64, 1.28 and 2.56 s relaxation delays; R2 data were collected with 0.017, 0.025 (x2), 0.034, 0.051, and 0.076 s relaxation delays; heteronuclear NOE data were collected using a 6 s recycle delay with 3 s proton saturation applied by a train of 120 ° pulses separated by 5 ms delays. Data were analyzed using Modelfree 4.20 46, 47 with the FAST Model Free scripts 48.
1DHN were obtained using in-phase/anti-phase HSQC or TROSY-HNCO sequences 49, 50 applied to isotropic protein samples or those embedded in acrylamide gels. 51–54. Uncharged polyacrylamide gels (7 % acrylamide, 19:1 acrylamide:bis-acrylamide, Sigma) were polymerized overnight in a 6 mm diameter gel funnel (New Era Enterprises, Vineland, NJ), extensively dialyzed, and dried at room temperature. The dried gel was reconstituted in the 6 mm funnel by addition of 400 μl of NOXO1β PX (0.5 mM concentration in NMR buffer) and overnight hydration. Fully hydrated gels were inserted into a 5 mm NMR tube using the 6.0–4.2 mm funnel. Charged gels were prepared at 7% acrylamide with 25% 3-(acrylamidopropyl)-trimethylammonium chloride (APTMAC). Gels were polymerized inside of a piece of 1/8” (i.d.) Tygon tubing, dialyzed extensively against water, and dried at room temperature for ~2 days on a sheet of Teflon. The dried gel was placed into a 5 mm Shigemi tube and hydrated overnight with 300 μl of 0.5 mM NOXO1β PX in NMR buffer. The gel was longitudinally compressed to 90 % of its original length using the Shigemi plunger. Initial estimates of Da and Rh were estimated from the distribution of 1DHN55 and from calculations using homology models of NOXO1β based on p47phox56. These values were refined at the Xplor-NIH water refinement stage and used iteratively in Aria calculations.
Structure calculation
Initial structures were calculated using Aria2.2/CNS 1.2 57, 58, which produced unambiguous and ambiguous assignment tables. These restraints were used for de novo structure calculation using Xplor-NIH 2.26. Xplor was used for final structure calculations to facilitate incorporation of 1H chemical shift and RDC restraints and to simplify execution of the explicit water refinement. Thirty-two structures were calculated in vacuo without electrostatics and the fourteen low-energy structures then refined in explicit water with the electrostatic potential turned on. Restraints involving Phe side chains were set at 1.8 Å for the lower limit and either 4.0 or 6.0 Å for the upper limit, depending on cross peak intensity. All other restraints were automatically calibrated by Aria. For water refinement, the restraints were rescaled and binned based on internal distance standards using a total range of 1.8 to 5, 6, or 7Å depending on type of experiment and mixing time (e.g., the upper bounds for distances between methyl protons measured by the 300 ms NOESY was set to 7Å). Xplor calculations used the initial unambiguous and ambiguous distance restraints, 55 χ1 (confidence >= 0.7) 59 and 222 backbone φ+ψ60, 61 dihedral angle restraints, 53 1H methyl chemical shifts, 92 1HN-15N residual dipolar coupling restraints obtained from axially compressed acrylamide gels 51, 52 and 64 1H-15N residual dipolar couplings obtained from charged acrylamide gels 53, 54. The hydrogen bond restraint list was built iteratively based on the presence of secondary structural elements identified from backbone dihedral angles, cross-strand HN-HN NOEs, and analysis of structures using HB-PLUS 62. Ultimately, we incorporated 63 hydrogen bond restraints. Water refinement was carried out using Xplor-NIH 2.26with protocols developed by Spronk and Nilges63, 64. Structural statistics for fourteen low-energy structures are shown in Table I. Structure Figures were prepared using Pymol65 and CorelDraw.
SPR experiments
Liposomes were prepared as previously described 66. Briefly, target lipids including 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidic acid (POPA), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (POPS), or dipalmitoyl PtdIns phosphates were dissolved in CHCl3/CH3OH/H2O (1:2:0.8) and added to 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) or -phosphoethanolamine (POPE) in CHCl3 at a ratio of 74:20:6 POPC:POPE:target lipid. The mixture was dried first under N2 and then under vacuum to remove residual CHCl3, hydrated in SPR buffer, and extruded 19 times with a LiposoFast extruder (Avestin, Ottawa, ON) through two stacked polycarbonate membranes (0.1 μm pore size). Dipalmitoyl PtdIns phosphates were purchased from Cell Signals, Kinnear, OH; all other lipids were purchased from Avanti Polar Lipids, Alabaster, AL.
SPR data were collected at 25 °C on a Biacore T100. Liposomes (0.1 mM total lipid concentration) were immobilized on an L1 chip by flowing over the surface at 5 μl/min for 600 sec. Typical lipid loading was ~6,000 RUs per lane. This value suggests that the lipids are forming a contiguous bilayer on the chip surface, rather than remaining as intact liposomes 67, and minimizes nonspecific protein binding to the L1 chip surface without introducing mass transfer effects 24, 29. The bilayer was washed with three 6 μl injections of 50 mM NaOH at 100 μl/min interspersed with SPR buffer and blocked with a 25 μl injection of 0.1 mg/ml fatty acid-free BSA at 5 μl/min. Following the wash steps, SPR buffer was flowed across reference and target lanes at 5 μl/min for 48 sec. Protein solutions at 0, 0.05, 0.1, 0.5, 1.0, 5.0 and 10.0 μM NOXO1β PX or p40phox PX (control) were injected at 5 μl/min and data collected for 1,200 sec, followed by regeneration of the chip surface by washing with 40 mM CHAPS (Anatrace, Maumee, OH) and 40 mM n-octylβ-D-glucopyranoside (Anatrace). Three independent sensorgrams were collected for each lipid and protein concentration tested.
SPR Analysis
Because we observed high levels of binding to the background PE/PC lipids in the reference lane (Dashed line, Figure 4A & B), we have used the ratios, rather than the differences, between baseline-subtracted target lipid vs. reference lane responses to assess relative binding by the NOXO1β PX domain to different phospholipids (as suggested by Eric Roush, Biacore Application Scientist). This minimizes errors derived from between-run differences in initial lipid loading. Specifically, we use the mean RU value of the 40 sec prior to protein injection as a baseline and subtract this from the response curve after protein injection. The target/reference ratio is calculated as the mean of the ratios of the baseline-subtracted target and reference RUs between 160 and 560 sec after protein injection. The 160 sec delay allows for stabilization of protein binding to the target and reference lanes. The standard deviation for a given target:reference ratio is ~0.01 over the 400 sec averaging period. SPR binding data at 5 μM and 10 μM protein concentration were analyzed in SigmaPlot v11 (Systat) using the Kruskal-Wallis one way ANOVA on ranks (p=0.005 at 5 μM; p=0.003 at 10 μM) with subsequent application of Dunn’s post-hoc test for unequal variances with p=0.05 as the criteria for significance.
Supplementary Material
Acknowledgments
This work is funded by NIH R01AI064609 to D.A.H and R01AI022564 to L.C.M. D.A.H thanks Pascal Mercier (Chenomx) and Charles Schwieters (NIH) for Xplor/water refinement scripts. D.A.H. and N.Y.D. also thank Roy Hantgan (WFU Sch. Med.) and Eric Roush (Biacore) for advice and comments regarding SPR experiments.
Abbreviations
- CISK
cytokine-independent survival kinase
- NMR
nuclear magnetic resonance
- PI3K
phosphatidylinositol 3-kinase
- POPA
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidic acid
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine
- POPE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine
- POPS
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced)
- Nox
NADPH oxidase
- NOXO1
Nox organizer 1
- NOXA1
Nox activator 1
- PX
phox homology
- RMSD
root-mean squared deviation
- ROS
reactive oxygen species
- RU
resonance units
- SPR
surface plasmon resonance
- 2D
two dimensional
- 3D
three dimensional
Footnotes
Accession numbers
Resonance assignments have been deposited at BMRB under accession code 16749. Structures and related data have been deposited at RCSB under accession code 2L73.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kim YS, Morgan MJ, Choksi S, Liu ZG. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol Cell. 2007;26:675–687. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 2.Komatsu D, Kato M, Nakayama J, Miyagawa S, Kamata T. NADPH oxidase 1 plays a critical mediating role in oncogenic Ras-induced vascular endothelial growth factor expression. Oncogene. 2008;27:4724–4732. doi: 10.1038/onc.2008.102. [DOI] [PubMed] [Google Scholar]
- 3.Shinohara M, Adachi Y, Mitsushita J, Kuwabara M, Nagasawa A, Harada S, Furuta S, Zhang Y, Seheli K, Miyazaki H, Kamata T. Reactive oxygen generated by NADPH oxidase 1 (Nox1) contributes to cell invasion by regulating matrix metalloprotease-9 production and cell migration. J Biol Chem. 2010;285:4481–4488. doi: 10.1074/jbc.M109.071779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Dutta S, Rittinger K. Regulation of NOXO1 Activity through Reversible Interactions with p22 and NOXA1. PLoS One. 2010;5:e10478. doi: 10.1371/journal.pone.0010478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng G, Lambeth JD. Alternative mRNA splice forms of NOXO1: Differential tissue expression and regulation of Nox1 and Nox3. Gene. 2005;356:118–126. doi: 10.1016/j.gene.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 7.Bravo J, Karathanassis D, Pacold CM, Pacold ME, Ellson CD, Anderson KE, Butler PJG, Lavenir I, Perisic O, Hawkins PT, Stephens L, Williams RL. The crystal structure of the PX domain from p40phox bound to phosphatidylinositol 3-phosphate. Mol Cell. 2001;8:829–839. doi: 10.1016/s1097-2765(01)00372-0. [DOI] [PubMed] [Google Scholar]
- 8.Zhou CZ, de La Sierra-Gallay IL, Quevillon-Cheruel S, Collinet B, Minard P, Blondeau K, Henckes G, Aufrère R, Leulliot N, Graille M, Sorel I, Savarin P, de la Torre F, Poupon A, Janin J, van Tilbeurgh H. Crystal structure of the yeast Phox homology (PX) domain protein Grd19p complexed to phosphatidylinositol-3-phosphate. J Biol Chem. 2003;278:50371–50376. doi: 10.1074/jbc.M304392200. [DOI] [PubMed] [Google Scholar]
- 9.Cheng G, Lambeth JD. NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the phox homology (PX) domain. J Biol Chem. 2004;279:4737–4742. doi: 10.1074/jbc.M305968200. [DOI] [PubMed] [Google Scholar]
- 10.Takeya R, Taura M, Yamasaki T, Naito S, Sumimoto H. Expression and function of Noxo1gamma, an alternative splicing form of the NADPH oxidase organizer 1. FEBS J. 2006;273:3663–3677. doi: 10.1111/j.1742-4658.2006.05371.x. [DOI] [PubMed] [Google Scholar]
- 11.Ueyama T, Lekstrom K, Tsujibe S, Saito N, Leto TL. Subcellular localization and function of alternatively spliced Noxo1 isoforms. Free Radic Biol Med. 2007;42:180–190. doi: 10.1016/j.freeradbiomed.2006.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banfi B, Clark RA, Steger K, Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem. 2003;278:3510–3513. doi: 10.1074/jbc.C200613200. [DOI] [PubMed] [Google Scholar]
- 13.Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nature Cell Biol. 2001;3:675–678. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- 14.Geiszt M, Lekstrom K, Witta J, Leto TL. Proteins homologous to p47phox and p67phox support superoxide production by NAD(P)H oxidase 1 in colon epithelial cells. J Biol Chem. 2003;278:20006–20012. doi: 10.1074/jbc.M301289200. [DOI] [PubMed] [Google Scholar]
- 15.Takeya R, Ueno N, Kami K, Taura M, Kohjima M, Izaki T, Nunoi H, Sumimoto H. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J Biol Chem. 2003;278:25234–25246. doi: 10.1074/jbc.M212856200. [DOI] [PubMed] [Google Scholar]
- 16.Hiroaki H, Ago T, Ito T, Sumimoto H, Kohda D. Solution structure of the PX domain, a target of the SH3 domain. Nature Struct Biol. 2001;8:526–530. doi: 10.1038/88591. [DOI] [PubMed] [Google Scholar]
- 17.Xu J, Liu D, Gill G, Songyang Z. Regulation of cytokine-independent survival kinase (CISK) by the Phox homology domain and phosphoinositides. J Cell Biol. 2001;154:699–705. doi: 10.1083/jcb.200105089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xing Y, Liu D, Zhang R, Joachimiak A, Songyang Z, Xu W. Structural basis of membrane targeting by the phox homology domain of cytokine-independent survival kinase (CISK-PX) J Biol Chem. 2004;279:30662–30669. doi: 10.1074/jbc.M404107200. [DOI] [PubMed] [Google Scholar]
- 19.Virbasius JV, Song X, Pomerleau DP, Zhan Y, Zhou GW, Czech MP. Activation of the Akt-related cytokine-independent survival kinase requires interaction of its phox domain with endosomal phosphatidylinositol 3-phosphate. Proc Natl Acad Sci U S A. 2001;98:12908–12913. doi: 10.1073/pnas.221352898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parkinson GN, Vines D, Driscoll PC, Djordjevic S. Crystal structures of PI3K-C2α PX domain indicate conformational change associated with ligand binding. BMC Structural Biology. 2008;8:13. doi: 10.1186/1472-6807-8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stahelin RV, Karathanassis D, Bruzik KS, Waterfield MD, Bravo J, Williams RL, Cho W. Structural and membrane binding analysis of the phox homology domain of phosphoinositide 3-kinase-C2α. J Biol Chem. 2006;281:39396–39406. doi: 10.1074/jbc.M607079200. [DOI] [PubMed] [Google Scholar]
- 22.Wahl M, Delbrueck H, Oschkinat H, Heinemann U. PDB entry 1KQ6; p47phox PX domain 2003 [Google Scholar]
- 23.Karathanassis D, Stahelin RV, Bravo J, Perisic O, Pacold CM, Cho W, Williams RL. Binding of the PX domain of p47phox to phosphatidylinositol 3,4-biphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J. 2002;21:5057–5068. doi: 10.1093/emboj/cdf519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stahelin RV, Karathanassis D, Murray D, Williams RL, Cho W. Structural and membrane binding analysis of the phox homology domain of Bem1p. J Biol Chem. 2007;282:25737–25747. doi: 10.1074/jbc.M702861200. [DOI] [PubMed] [Google Scholar]
- 25.Noack D, Rae J, Cross AR, Ellis BA, Newburger PE, Curnutte JT, Heyworth PG. Autosomal recessive chronic granulomatous disease caused by defects in NCF-1, the gene encoding the phagocyte p47-phox: Mutations not arising in the NCI-1pseudogenes. Blood. 2001;97:305–311. doi: 10.1182/blood.v97.1.305. [DOI] [PubMed] [Google Scholar]
- 26.Heyworth PG, Cross AR. Chronic granulomatous disease mutations and the PX domain. Nature Cell Biol. 2002;4:E110. doi: 10.1038/ncb0502-e110a. [DOI] [PubMed] [Google Scholar]
- 27.Song X, Xu W, Zhang A, Huang G, Liang X, Virbasius JV, Czech MP, Zhou GW. Phox homology domains specifically bind phosphatidylinositol phosphates. Biochemistry. 2001;40:8940–8944. doi: 10.1021/bi0155100. [DOI] [PubMed] [Google Scholar]
- 28.Zhan Y, Virbasius JV, Song X, Pomerleau DP, Zhou GW. The p40phox and p47phox PX domains of NADPH oxidase target cell membranes via direct and indirect recruitment by phosphoinositides. J Biol Chem. 2002;277:4512–4518. doi: 10.1074/jbc.M109520200. [DOI] [PubMed] [Google Scholar]
- 29.Narayan K, Lemmon MA. Determining selectivity of phosphoinositide-binding domains. Methods. 2006;39:122–133. doi: 10.1016/j.ymeth.2006.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 31.Behnia R, Munro S. Organelle identity and the signposts for membrane traffic. Nature. 2005;438:597–604. doi: 10.1038/nature04397. [DOI] [PubMed] [Google Scholar]
- 32.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 33.Stahelin RV, Ananthanarayanan B, Blatner NR, Singh S, Bruzik KS, Murray D, Cho W. Mechanism of membrane binding of the phospholipase D1 PX domain. J Biol Chem. 2004;279:54918–54926. doi: 10.1074/jbc.M407798200. [DOI] [PubMed] [Google Scholar]
- 34.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sadok A, Pierres A, Dahan L, Prevot C, Lehmann M, Kovacic H. NADPH oxidase 1 controls the persistence of directed cell migration by a Rho-dependent switch of alpha2/alpha3 integrins. Mol Cell Biol. 2009;29:3915–3928. doi: 10.1128/MCB.01199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shinohara M, Shang WH, Kubodera M, Harada S, Mitsushita J, Kato M, Miyazaki H, Sumimoto H, Kamata T. Nox1 redox signaling mediates oncogenic Ras-induced disruption of stress fibers and focal adhesions by down-regulating Rho. J Biol Chem. 2007;282:17640–17648. doi: 10.1074/jbc.M609450200. [DOI] [PubMed] [Google Scholar]
- 37.Saarikangas J, Zhao H, Lappalainen P. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol Rev. 2010;90:259–289. doi: 10.1152/physrev.00036.2009. [DOI] [PubMed] [Google Scholar]
- 38.Davis NY, McPhail LC, Horita DA. Backbone (1)H, (15)N, and (13)C resonance assignments for the NOXO1β PX domain. Biomol NMR Assign. 2011;5:139–141. doi: 10.1007/s12104-010-9286-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tugarinov V, Kay LE. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J Am Chem Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- 40.Link Y, Wagner G. Efficient side-chain and backbone assignment in large proteins: Application to tGCN5. J Biomol NMR. 1999;15:227–239. doi: 10.1023/a:1008343915382. [DOI] [PubMed] [Google Scholar]
- 41.Farmer BT, II, Venters RA. Assignment of aliphatic side-chain 1HN/15N resonance in perdeuterated proteins. J Biomol NMR. 1996;7:59–71. doi: 10.1007/BF00190457. [DOI] [PubMed] [Google Scholar]
- 42.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 43.Johnson BA, Blevins RA. NMRView: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 44.Xia Y, Yee A, Arrowsmith CH, Gao X. 1HC and 1HN total NOE correlations in a single 3D NMR experiment. 15N and 13C time-sharing in t1 and t2 dimensions for simultaneous data acquisition. J Biomol NMR. 2003;27:193–203. doi: 10.1023/a:1025407905478. [DOI] [PubMed] [Google Scholar]
- 45.Otting G, Wüthrich K. Extended heteronuclear editing of 2D 1H NMR spectra of isotope-labeled proteins, using the X(ω1, ω2) double half filter. J Magn Reson. 1989;85:586–594. [Google Scholar]
- 46.Mandel AM, Akke M, Palmer AG., III Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 47.Palmer AG, III, Rance M, Wright PE. Intramolecular motions of a zinc finger DNA-binding domain from Xfin characterized by proton-detected natural abundance 13C heteronuclear NMR spectroscopy. J Am Chem Soc. 1991;113:4371–4380. [Google Scholar]
- 48.Cole R, Loria JP. FAST-Modelfree: a program for rapid automated analysis of solution NMR spin-relaxation data. J Biomol NMR. 2003;26:203–213. doi: 10.1023/a:1023808801134. [DOI] [PubMed] [Google Scholar]
- 49.Yang D, Venters RA, Mueller GA, Choy WY, Kay LE. TROSY-based HNCO pulse sequences for the measurement of 1HN-15N, 15N-13CO, 1HN-13CO, 13CO-13Cα and 1HN-13Cα dipolar couplings in 15N, 13C, 2H-labeled proteins. J Biomol NMR. 1999;14:333–343. [Google Scholar]
- 50.Ottiger M, Delaglio F, Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J Magn Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- 51.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J Biomol NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 52.Tycko R, Blanco FJ, Ishii Y. Alignment of biopolymers in strained gels: A new way to create detectable dipole-dipole coulpings in high-resolution biomolecular NMR. J Am Chem Soc. 2000;122:9340–9341. [Google Scholar]
- 53.Cierpicki T, Bushweller JH. Charged gels as orienting media for measurement of residual dipolar couplings in soluble and integral membrane proteins. J Am Chem Soc. 2004;126:16259–16266. doi: 10.1021/ja046054g. [DOI] [PubMed] [Google Scholar]
- 54.Meier S, Häussinger D, Grzesiek S. Charged acrylamide copolymer gels as media for weak alignment. J Biomol NMR. 2002;24:351–356. doi: 10.1023/a:1021609207024. [DOI] [PubMed] [Google Scholar]
- 55.Clore GM, Gronenborn AM, Tjandra N. Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J Magn Reson. 1998;131:159–162. doi: 10.1006/jmre.1997.1345. [DOI] [PubMed] [Google Scholar]
- 56.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 57.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR System: A new software suite for macromolecular structure determination. ActaCryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 58.Rieping W, Habeck M, Bardiaux B, Bernard A, Malliavin TE, Nilges M. ARIA2: Automated NOE assignment and data integration in NMR structure calculation. Bioinformatics. 2007;23:381–382. doi: 10.1093/bioinformatics/btl589. [DOI] [PubMed] [Google Scholar]
- 59.Berjanskii MV, Neal S, Wishart DS. PREDITOR: a web server for predicting protein torsion angle restraints. Nucleic Acids Res. 2006;34:W63–W69. doi: 10.1093/nar/gkl341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 62.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J Mol Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 63.Nabuurs SB, Nederveen AJ, Vranken W, Doreleijers JF, Bonvin AM, Vuister GW, Vriend G, Spronk CA. DRESS: a database of REfined solution NMR structures. Proteins. 2004;55:483–486. doi: 10.1002/prot.20118. [DOI] [PubMed] [Google Scholar]
- 64.Linge JP, Williams MA, Spronk CA, Bonvin AM, Nilges M. Refinement of protein structures in explicit solvent. Proteins. 2003;50:496–506. doi: 10.1002/prot.10299. [DOI] [PubMed] [Google Scholar]
- 65.DeLano WL. The PyMOL molecular graphics system. DeLano Scientific, LLC; Palo Alto, CA: 2008. [Google Scholar]
- 66.Shen K, Sergeant S, Hantgan RR, McPhail LC, Horita DA. Mutations in the PX-SH3A linker of p47phox decouple PI(3,4)P2 binding from NADPH oxidase activation. Biochemistry. 2008;47:8855–8865. doi: 10.1021/bi8005847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Erb EM, Chen X, Allen S, Roberts CJ, Tendler SJ, Davies MC, Forsen S. Characterization of the surfaces generated by liposome binding to the modified dextran matrix of a surface plasmon resonance sensor chip. Anal Biochem. 2000;280:29–35. doi: 10.1006/abio.1999.4469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.