

Abstract
Neuroglial cells define brain homeostasis and mount defense against pathological insults. Astroglia regulate neurogenesis and development of brain circuits. In the adult brain, astrocytes enter into intimate dynamic relationship with neurons, especially at synaptic sites where they functionally form the tripartite synapse. At these sites astrocytes regulate ion and neurotransmitter homeostasis, metabolically support neurons and monitor synaptic activity; one of the readouts of the latter manifests in astrocytic intracellular Ca2+ signals. This form of astrocytic excitability can lead to release of chemical transmitters via Ca2+-dependent exocytosis. Once in the extracellular space, gliotransmitters can modulate synaptic plasticity and cause changes in behavior. Besides these physiological tasks, astrocytes are fundamental for progression and outcome of neurological diseases. In Alzheimer’s disease, for example, astrocytes may contribute to the etiology of this disorder. Highly lethal glial-derived tumors use signaling trickery to coerce normal brain cells to assist tumor invasiveness. This review sheds new light on the brain operation in health and disease, but also points to many unknowns.
Keywords: astrocytes, disease, health, metabolism, neurogenesis, signaling
Introduction
Neuroglia was initially described by Rudolf Virchow as a connective tissue that binds nervous elements together (Virchow 1858). Soon after, in 1870-ies, the cellular nature of glia was firmly established by Camillo Golgi (Golgi 1873; Golgi 1903). Astrocytes, a subset of glial cells that represent the main subject of this review, were named by Michael von Lenhossek (Lenhossek 1891). Subsequently, Rudolf Albert von Kölliker and William Lloyd Andriezen (Kölliker 1889; Andriezen 1893) sub-classified them into protoplasmic and fibrous astrocytes, located in the grey and white matter, respectively. Santiago Ramón y Cajal further studied astrocytes and their interactions with other neural elements (Ramón y Cajal 1913). As we know today, by using a gold chloride stain, Ramón y Cajal accidentally targeted intermediate filaments consisting mainly of glial fibrillary acidic protein (GFAP), a contemporary astrocytic marker (Kimelberg 2004), the gene promoter of which has proved as a pivotal tool used for molecular genetics approaches in studying astrocytes.
It is during the nascent period of (astro)glial research, i.e. in the second part of 19th century and the early 20th century that a variety of broad hypotheses were made in respect to function of glial cells in the brain. Hence, astroglia would: i) serve as a metabolic support for neurons (Camillo Golgi); ii) play a major role in uptake and/or degradation of neurotransmitters (Ernesto Lugaro); iii) act like an endocrine gland by providing for release of substances into the blood (Jean Nageotte); iv) regulate information processing and synaptic connectivity in the brain (Carl Ludwig Schleich); and v) control sleep and waking states (Santiago Ramón y Cajal) [for details see e.g., (Verkhratsky et al. 2011) and references therein]. However, the research testing these bold and fundamental hypotheses was realized at much later time, and mainly executed in the past three decades. Indeed, the newly attained evidence indicates that astrocytes are actively involved in many of the brain’s functions. Investigation of the roles of astrocytes in the functioning of nervous systems has recently emerged as one of the hottest fields in brain research. Professional societies are clamoring for symposia on the topic to be presented at their meetings. One such recent gathering occured at the International Society for Neurochemistry satellite meeting entitled “Glial cells in (patho)physiology” (http://lnmcp.mf.uni-lj.si/), held on August 24–27, 2011 in Ljubljana, Slovenia at the Slovenian Academy of Sciences and Arts. The new and exciting results presented at the meeting prompted the present review.
We start our journey by discussing the astroglial role in neurogenesis and proceed to structural relationships between astroglia and neurons. We believe that in order to understand a bidirectional metabolic and signaling dialogue between astrocyte and neurons, one has first to consider the hierarchy of structural associations, and to appreciate their plasticity. We then provide an update on novel findings in respect to an astrocytic homeostatic task, a metabolic support of the brain. What follows is a description of glial signaling components in neuronal-glial circuits: i) the astrocytic plasma membrane set of receptors and transporters, ii) astrocytic intracellular Ca2+ dynamics and consequential regulation of exocytotic/vesicular release of gliotransmitters, and iii) the role of gap junctions; in all these topics we emphasize the effects that glia may have on nearby neurons. Indeed, we next discuss the consequence of gliotransmitter release into the extracellular space (ECS), i.e., modulation of synaptic transmission and plasticity, and its contribution to sleep behavior. Finally, we use two examples of pathological states into which glial cells provide fundamental contribution: i) the Alzheimer’s disease in which astrocytes contribute to etiology; and ii) the glioma, a glial neoplasm which, in its invading quest, engages in active bidirectional signaling with surrounding normal cells in the brain.
Astroglia in neurogenesis
In the adult mammalian brain, neurogenesis, a process of generation of new neurons, is under normal conditions limited to two regions: lateral ventricles and the subgranular zone of dentate gyrus of the hippocampus. The newly formed immature neurons continuously generated in the walls of the lateral ventricles migrate through the rostral migratory stream to their final destination in the olfactory bulbs where they differentiate into periglomerular or granule neurons. New neurons generated in the subgranular zone of dentate gyrus of the hippocampus migrate only a short distance to the granular cell layer where they integrate into the existing neural circuits. Importantly, the survival of these cells depends on their activity and functional integration. Increasing evidence suggests that astrocytes are important players in the neurogenic niche. Astrocytes share some properties with neural stem cells (Laywell et al. 2000; Seri et al. 2001; Buffo et al. 2008) and create an environment conducive to neurogenesis (Song et al. 2002). Astrocytes regulate neurogenesis by the secretion of factors such as Wnt3 (Lie et al. 2005), interleukin-1β, interleukin-6, and insulin-like growth factor binding protein 6 (Barkho et al. 2006). These glial cells also control the neurogenic niche through membrane-associated factors (Song et al. 2002), albeit the identity of these factors and the mechanisms involved are largely unknown.
Intermediate filament proteins, GFAP and vimentin, are expressed in astrocytes and appear to critically affect the processes by which astrocytes control neurogenesis and other aspects of neural plasticity and regeneration. The ablation of GFAP and vimentin in mice (Colucci-Guyon et al. 1994; Pekny et al. 1995; Eliasson et al. 1999; Pekny et al. 1999; Pekny and Pekna 2004; Pekny and Nilsson 2005; Pekny and Lane 2007) creates more permissive environment to transplantation of neural grafts or neural stem cells (Kinouchi et al. 2003; Widestrand et al. 2007) and increases axonal and synaptic regeneration (Menet et al. 2003; Pekny et al. 2004; Wilhelmsson et al. 2004; Cho et al. 2005). Neuronal differentiation of neural progenitor cells was increased when they were co-cultured with astrocytes lacking GFAP and vimentin (GFAP−/−Vim−/−) (Widestrand et al. 2007). Although the altered cellular distribution of Wnt3 in the GFAP−/−Vim−/− astrocytes could be associated with changed secretion of this pro-neurogenic factor and thus partially explain this finding, a direct cell-to-cell signaling from astrocytes to neural stem/progenitor cells and its involvement in neurogenesis remains an attractive alternative. It appears that GFAP and vimentin are important for the astrocyte-mediated inhibition of neural stem/progenitor cell differentiation and that this inhibition happens, at least partially, through the Notch signaling from astrocytes to neural stem/progenitor cells (Pekny et al., unpublished data).
Astrocytes are also an essential component of the neurovascular unit and directly regulate the properties of the blood-brain barrier. Thus, astrocytes can also regulate the neurogenic niche indirectly by determining the accessibility of blood-derived factors/molecules modulating neurogenesis (Barres 2008; Liebner et al. 2011).
Integration into the existing neuronal networks is essential for the survival of newly born neurons. It appears that astrocytes regulate this step by releasing signals that induce synapse formation as well as signals that initiate the removal of redundant synapses. Astrocyte-derived factors participate in synaptic development by inducing synapse formation and maturation (Nagler et al. 2001). Thrombospondins 1 and 2 are members of a family of extracellular glycoproteins with synaptogenic properties secreted by astrocytes (Christopherson et al. 2005). Their deficiency leads to reduced synaptic density during development (Christopherson et al. 2005). Expression of thromobospondins 1 and 2 is increased after experimental stroke and both thrombospondins co-localize mostly with astrocytes (Liauw et al, 2008). Compared to wild-type mice, mice deficient in thromobospondin 1 and 2 exhibited synaptic density and axonal sprouting deficit associated with impaired motor function recovery after stroke, despite no differences in infarct volume and blood vessel density (Liauw et al. 2008). Recently, Eroglu et al (Eroglu et al. 2009) identified the gabapentin receptor a2d-1 as a neuronal thrombospondin receptor that is responsible for excitatory synapse formation in the central nervous system (CNS). Astrocytes are also involved in maintaining synaptic contacts after injury. Hence, mice deficient in GFAP and vimentin have a more pronounced loss of synapses in the hippocampus in the acute phase (four days) after the entorhinal cortex lesion. Remarkably, synaptic recovery of these mice was also increased, reaching the levels on the uninjured side on day 14 after lesion (Wilhelmsson et al. 2004)
Recent data suggest that astrocytes also play a role in the elimination of redundant synapses during development. Immature astrocytes in the developing brain seem to be a source of a signal that triggers the expression of complement component C1q in developing neurons (Stevens et al. 2007). C1q localizes to synapses that are thus tagged for elimination through the activation of the complement cascade and deposition of C3b, an opsonin derived from the proteolytic activation of the complement component C3 (Stevens et al. 2007). Indeed, both mice deficient in C1q (C1q−/−) and mice deficient in C3 showed significant and sustained defects in CNS synapse elimination in the thalamus (Stevens et al. 2007). Also, C1q−/− mice show enhanced neocortical excitatory synaptic connectivity and epileptiform activity (Chu et al. 2010).
In summary, astrocytes both directly and indirectly control the neurogenic niche and integration and survival of newly formed neurons. Thus, astrocytes constitute an attractive target for therapeutic modulation of the generation and survival of newly formed neurons in pathological conditions such as stroke and/or neurodegenerative disease.
Structural associations between neurons and glia in developed brain
Glial cells in the brain tightly associate with their environment. Astrocytes form processes that contact other neural cells, be them glia and/or neurons, and extent onto non-cellular surroundings such a basal lamina, or cerebrospinal fluid (CSF) that fills ventricles (Reichenbach 1989). Virtually all vascular surfaces in the brain are covered by astroglial endfeet. Using these specialized contacts astrocytes can also reach the pia mater. Astrocytic processes in the parenchyma are in contact with and/or ensheath distinct neuronal elements, in particular synapses, with the coverage that varies in different regions of the brain (Ventura and Harris 1999; Hatton 2002). Due to strategic positioning at the cross-roads between the vasculature and neurons, astrocytes can mediate the exchange of molecules between these compartments. Moreover, it is in the neuropil that, so-called, peripheral astrocyte processes (PAPs), which form thin membrane out-foldings, engage with their neighboring neuronal counterparts to provide structural sites for bidirectional astrocyte-neuron communication. Due to their high density, PAPs may constitute half of the glial cell volume and about 80% of its surface (Chao et al. 2002). The perisynaptic processes, which ensheath synapses, display lamellar structure; their functions are multi-faceted, e.g., isolation of individual synapses from their environment, uptake of neurotransmitters and involvement in synaptic transmission and plasticity.
Interaction of neuronal, glial and vascular elements results in the formation of specialized functional territories which may range in size from a few nanometers up to centimeters. These territories may be referred to as domains, which more or less function autonomously. Coexisting glial domains can be hierarchically arranged starting with individual and very small glia-neuron contacts constituting a nanodomain (Fig. 1, left). For instance, glial “tongues” lining a synaptic cleft, or finger-like perinodal glial processes at axons, can be considered as nanodomains. At the next level, microdomains ensheath an individual synapse or a group of synapses (Fig. 1, left), as initially demonstrated for a specialized astrocyte, the Bergmann glial cell in the cerebellum (Grosche et al. 1999). A single astrocyte forms many microdomains, which can operate independently from each other, and have a degree of operational independence from the glial stem process from which they extend. Both nanodomains and microdomains have been shown to be dynamic and plastic structures (Hirrlinger et al. 2004).
Figure 1.

Schematic representation of the various types of co-existing glial domains, illustrated layered over two background drawings: i) a pyramidal cell (left) from Ramón y Cajal (Ramón y Cajal 1911); and ii) a figure from Vogt and Vogt (Vogt and Vogt 1937), showing the transition (vertical arrows) between the striate (center) and the occipital (right) cortex in a human brain. Glial cells form a specific functional organization at different levels from nanodomains up to superdomains for their interaction with neurons. Individual synapses are associated with their ensheathing glial microdomains, but parallel stimulation of related inputs may integrate (oligo-)cellular domains involving the whole glial cell (or a few of them) and their neuronal partners. Such cellular domains can be of columnar or spherical (center, n1) shape. Appropriate stimulation may then activate, via gap junctional coupling, networks (center, ni and nii), which are likely dynamic and consist of many astrocytes. Macrodomains of variable size may form depending on neuronal activity. A further progression of integration will result in the generation of very large functional units, so-called superdomains, corresponding to entire cortical areas or gyri. Eventually, even a whole hemisphere associated with huge astrocytic populations may transiently be involved, putatively mediating events such as spreading depression and seizures.
In certain areas of the brain, for example in olfactory cortex, larger synaptic aggregates or glomerula serve as dominant functional units. Such cellular domains (Fig. 1, left) are enwrapped by a glial envelope which probably has similar functions as a microdomain. Depending on the size of this cellular domain, a whole astrocyte or even several astrocytes take part in its formation. It depends on the functional requirements whether a given glial element forms a microdomain or a cellular domain (Fig. 1; center, n1). For example, individual microdomains of a Bergmann glial cell can be activated when individual parallel fibers are weakly stimulated, while many microdomains or even a whole Bergmann glial cell (and, thus, a cellular domain) can be activated by strong stimulation (Grosche et al. 1999). In the cerebellum one Purkinje cell constitutes the smallest functional neuronal unit. However, a single Purkinje neuron is surrounded by about eight Bergmann glial cells. Such astrocytic clusters supporting a functional neuronal unit are defined here as oligocellular domains; they constitute a transition to larger arrays which can be called mesodomains (Fig. 1, center).
At the next level in the hierarchy, assemblies of glial oligocellular domains and mesodomains form large (“macroscopic”) functional units, such as columnar units of the cerebral cortex, for example ocular dominance columns of the visual cortex or “barrels” of the somatosensory cortex. Astrocytes may contribute to such larger units based on the formation of extensive cellular networks (Fig. 1, center). A network or an overlapping conglomerate of glial networks (Fig. 1; center, n i-n ii) are generated by the gap junctional coupling between the astrocytes. Mechanisms of extracellular homeostasis like spatial buffering of K+ ions, as well as the exchange of signals (e.g., Ca2+ waves) partially rely on this intercellular coupling. The astrocytic networks are experimentally visualized using an injection of a dye into one cell with subsequent dye spread to adjacent cells. An assembly of glial networks may be considered as glial macrodomains (Fig. 1, right). It has been shown that physiological stimulation of neurons in the barrel cortex of mice causes astrocytic Ca2+ waves which remained restricted to the very barrel (i.e., macrodomain) that was stimulated (Schipke et al. 2008). Groups of macrodomains form superdomains which may be related to cortical gyri or fields. Depending on the degree and distribution of neuronal excitation, the whole repertoire of glial domains may be switched on in series within the very same part of the brain. At the pathological end of this scale, phenomena such as spreading depression can expand over the entire cortex, thus involving the entire glial population. The various glial domains can be transient and variable and appear to be important for brain function.
Astroglia in brain metabolism
Astrocytes account for a large fraction of the brain volume and their ultrastructure is, particularly in the human brain, enormously elaborate with an immense network of fine processes (Fig. 2) (Oberheim et al. 2006). This enables astrocytes to monitor an extremely large number of synapses thereby being actively involved in neurotransmitter homeostasis (Newman 2003; Ransom et al. 2003; Hertz and Zielke 2004; Waagepetersen et al. 2009). The finding that astrocytes possess the machinery to release glutamate and other transmitters in a Ca2+-dependent manner has led to the notion that they actively participate in the signaling processes in a concerted action with neurons (Parpura et al. 1994; Parpura and Zorec 2010). It should be noted that in the brain, astrocytes, rather than neurons, are capable of de novo synthesis of glutamate and of storing glucose in the form of glycogen (Hertz and Zielke 2004).
Figure 2.

Brain tissue is largely divided between domains of astrocytes. Mouse astrocytes, whose gap juncrions were closed, were filled with two different dyes to visualize their respective domains. In a human brain, a single astrocyte domain can encompass over million neuronal synapses. Reprinted from (Pekny and Wilhelmsson 2006).
The contribution of astrocytes to the overall brain metabolism has been somewhat controversial but as discussed in detail by Hertz et al. (Hertz et al. 2007) the astrocytes have a high oxidative metabolism which is compatible with the prominent expression of enzymes involved in oxidative metabolism as well as a high content of mitochondria even in the fine processes as shown by a transcriptomic analysis of freshly isolated astrocytes and electron microscopic images of astrocytic fine processes in brain cortical sections (Lovatt et al. 2007).
Glutamatergic neurotransmission is likely to account for a considerable part of the total energy requirement related to neural signaling processes (Attwell and Laughlin 2001). While there is little doubt that glucose is the main energy substrate for the brain (McKenna et al. 2012), it may be less clear what may be the direct roles of glycogen and lactate both of which are derived from glucose (McKenna et al. 2012). In case of lactate the discussion has been focused on its role relative to that of glucose as the energy substrate for neurons [e.g. (Bouzier-Sore et al. 2003; Bak et al. 2006)]. Regarding glycogen, it has been debated whether it should be viewed as an energy store which can be mobilized during an episode of aglycemia or it may be actively involved in the maintenance of neural functions during physiological activity (Swanson 1992; Swanson and Choi 1993; Ransom and Fern 1997; Wender et al. 2000; Brown et al. 2005; Dienel et al. 2007). In this context it is of importance that Shulman et al (Shulman et al. 2001), based on functional imaging studies in the brain in vivo proposed a considerable activity of the glycogen-shunt, i.e. the concerted action of glycogen synthesis and glycogen degradation involving glycogen synthase and glycogen phosphorylase as illustrated in Schousboe et al. (Schousboe et al. 2010). The availability of specific pharmacological tools to inhibit glycogen phosphorylase has enabled detailed studies of the functional role of the glycogen-shunt to be performed showing that it clearly plays an important role in memory formation as well as in the maintenance of neurotransmission (Waagepetersen et al. 2000; Gibbs et al. 2006; Dienel et al. 2007; Suh et al. 2007; Walls et al. 2008; Sickmann et al. 2009; Walls et al. 2009; Suzuki et al. 2011).
The location of the astrocytic endfeet at the capillaries combined with the abundant expression of the glucose transporter GLUT1 (McKenna et al. 2012) provides these cells with an easy access to glucose supply. Subsequent to cellular uptake, glucose is phosphorylated by hexokinase type I which in the astrocytes is associated with the mitochondria (Lynch et al. 1991; Griffin et al. 1992). It is important to note that even though the phosphorylation process is highly efficient it has been convincingly demonstrated that the astrocytes contain a nonnegligible concentration of free, non-phosphorylated glucose (Prebil et al. 2011). The product of glucose phosphorylation, glucose-6-phosphate can enter the glycolytic pathway to form pyruvate and lactate or the pentose phosphate pathway which accounts for a small percentage of the glucose metabolism (McKenna et al. 2012). Glucose-6-phosphate may alternatively act as substrate for glycogen synthesis and glycogen may subsequently be degraded by the action of glycogen phosphorylase ultimately leading to regeneration of glucose-6-phosphate. i.e. the processes constituting the glycogen-shunt (Walls et al. 2009; Schousboe et al. 2010). The activities of glycolysis alone and that of the concerted action of glycogenolysis and glycolysis involving the glycogen-shunt, respectively appear to be regulated interdependently as inhibition of glycogen phosphorylase leads to a super-compensation of the rate of glycolysis both in the brain in vivo and in cultured astrocytes (Dienel et al. 2007; Walls et al. 2009; Schousboe et al. 2010; Schousboe et al. 2011). It appears that this interdependence of glycogen-shunt activity and glycolysis plays an important role in the supply of energy for the maintenance of glutamatergic activity (Walls et al. 2009; Schousboe et al. 2011). It should also be noted that inhibition of the glycogen-shunt has a significant effect on both vesicular glutamate release and its trans-membrane transport (Sickmann et al. 2009; Schousboe et al. 2010; Schousboe et al. 2011). This together with the demonstration that re-uptake of glutamate in glutamatergic neurons can only be supported by glucose metabolism and not by that of lactate (Bak et al. 2006) strongly argues that the glycogen-shunt as well as the glycolytic process are of fundamental significance for maintenance of glutamatergic activity. In this context it should also be mentioned that the astrocytic glutamate uptake mediated by the high affinity glutamate transporters abundantly expressed in the plasma membrane appears to be coupled to adenosine 5′-triphosphate (ATP) generated in the glycolytic pathway rather than by ATP generated by mitochondrial oxidative phosphorylation (Schousboe et al. 2010; Schousboe et al. 2011). This is likely explained by the association of the ATP producing glycolytic enzymes glyceraldehyde-3-phospate dehydrogenase and phosphoglycerate kinase with the astrocytic plasma membrane (Schousboe et al. 2011). This enables formation of a metabolomic complex between these enzymes, the Na+/K+-ATPase and the glutamate transporters of which the latter two also form complexes in the plasma membrane [see (Schousboe et al. 2011)]. Such a metabolomic structure would highly increase the efficiency of usage of the ATP produced in the glycolytic pathway, as discussed in Schousboe et al. (Schousboe et al. 2011).
Ionotropic receptors and transporters provide for fast neuronal-glial signaling at the synaptic level
In the CNS the majority of synaptic contacts are tightly covered by astroglial perisynaptic processes (Araque et al. 1999), which provide for structural separation between synapses and are likely to participate in the maintenance and regulation of synaptic transmission. In the grey matter of mammals single astrocyte can cover 20 – 100 thousand synapses in rodents and up to 2 millions synapses in primates and humans (Oberheim et al. 2006; Verkhratsky et al. 2011). This morphological organization obviously calls for specific mechanisms of local signaling that allow for neuronal-glial integration at the level of individual synapses. The local neuronal-glial signaling at the synaptic level involves activation of multiple neurotransmitter receptors localized at the astroglial plasma membrane. Activation of astroglial metabotropic receptors is mainly associated with intracellular Ca2+ signaling originating from the endoplasmic reticulum (ER) (Verkhratsky et al. 1998; Agulhon et al. 2008), which regulates various functions such as metabolic activity, gene expression and release of gliotransmitters. Astroglial Ca2+ signals, however, tend to be slow and their relevance for rapid regulation of synaptic transmission and plasticity has been recently questioned (Fiacco et al. 2007; Agulhon et al. 2010). Astroglial processes are endowed with several types of ionotropic receptors and transporters that may be responsible for fast signaling activated during the course of synaptic transmission. Highly localized ion signals produced by these molecules can potentially regulate synaptic transmission through several pathways.
Ionotropic receptors in perisynaptic processes
Expression of neurotransmitter receptors in astrocytes in different brain regions is highly heterogeneous and is likely to be regulated by local neurotransmitter environment and often the assortment of receptors expressed by astroglia matches that present in the their neuronal neighbors (Verkhratsky et al. 1998; Verkhratsky 2011). In the cerebellum, for example Purkinje neurons and their close associates Bergmann glial cells express the almost identical receptor assortment that includes receptors for adrenalin/epinephrine, histamine, glutamate, γ-amino butyric acid (GABA) and ATP, these receptors being congruent to the neurotransmitters released in this anatomical region (Kirischuk et al. 1995; Kirischuk et al. 1996b; Kirischuk et al. 1996a; Verkhratsky et al. 1998). In basal ganglia rich in dopaminergic transmissions astrocytes specifically express dopamine receptors (Miyazaki et al. 2004); similarly in the spinal cord where glycine is utilized as a primary inhibitory transmitter astrocytes demonstrate specific expression of glycine receptors (Kirchhoff et al. 1996).
There are at least three types of ionotropic receptors expressed in astrocytes in the mammalian brain which are represented by α-amino-3-hydroxy-5-methyl-isoxazole propionate (AMPA) and N-methyl-D-aspartate (NMDA) types of tetrameric glutamate receptors (GluRs) and P2X trimeric purinoceptors (Lalo et al. 2011a). Ionotropic GluRs of AMPA type are the most abundant in astroglia being detected in cortex, hippocampus and cerebellum (Condorelli et al. 1999; Gallo and Ghiani 2000; Seifert and Steinhauser 2001). Depending on the expression of the GluR2 subunit, some of astroglial AMPA receptors show moderate (PCa/Pmonovalent ~ 1 –1.5) Ca2+ permeability (Seifert and Steinhauser 2001), although fast desensitization of these receptors when activated by glutamate substantially limits Ca2+ entry (Kirischuk et al. 1999). Astroglial NMDA receptors were detected and characterized in detail both in vitro and in situ (Verkhratsky and Kirchhoff 2007; Lalo et al. 2008; Oliveira et al. 2011) and their expression seems to be confined to cortex and the spinal cord. Astroglial NMDA receptors are characterized by weak Mg2+ block at −80 mV [the block develops at more negative membrane potentials (Palygin et al. 2011)] and moderate Ca2+ permeability [PCa/Pmonovalent ~ 3; (Palygin et al. 2010)]. Cortical astrocytes also express heteromeric P2X1/5 receptors that are exceptionally sensitive to ATP 2+ (EC50 ~ 40 – 50 nM), are moderately permeable to Ca (PCa/Pmonovalent ~ 2) and show little desensitization in the presence of the agonist (Lalo et al. 2008; Palygin et al. 2010). Cortical astrocytes and cerebellar Bergmann glial cells also express P2X7 purinoceptors (Habbas et al. 2011; Oliveira et al. 2011), which, most likely, are associated with pathological purinergic signaling. Ionotropic receptors present in astroglia are activated during synaptic transmission forming the basis for glial synaptic currents (GSCs) that directly depend on presynaptic neurotransmitter release (Lalo et al. 2011b). Furthermore spontaneous (“miniature”) AMPA, NMDA and P2X1/5 receptors-mediated GSCs were readily detected in cortical astrocytes indicating close apposition of astroglial membranes containing ionotropic receptors to the sites of neurotransmitter release from presynaptic terminals (Lalo et al. 2006; Lalo et al. 2011b; Lalo et al. 2011a).
Astroglial Na+-dependent transporters and exchangers
Perisynaptic processes of astrocytes contain several transporters and exchangers that provide substantial Na+ fluxes. First, astrocytes are rich in Na+-dependent glutamate transporters (of EAAT1/GLAST and EAAT2/GLT-1 types), which represent the major element of glutamate homeostasis in the brain (Danbolt 2001; Gadea and Lopez-Colome 2001). Activation of glutamate transporters by synaptically released glutamate triggers inward currents mainly carried by Na+ that result in substantial (10 – 30 mM) increase in cytosolic Na+ concentration ([Na+]i) in astrocytes (Kirischuk et al. 2007; Bennay et al. 2008). The second Na+-dependent neurotransmitter transporter is represented by GABA transporters of GAT family, which operate with a stoichiometry of 2Na+/1GABA (Heja et al. 2009). Transmembrane Na+ gradients also govern several Na+ dependent exchangers involved in Ca2+ homeostasis (Na+/Ca2+ exchanger or NCX) and in H+/OH-/HCO3− transport critically important for acid/base homeostasis (Deitmer and Rose 2010). Astroglial NCX may operate in both forward (Ca2+ extrusion coupled with Na+ influx) and reverse (Ca2+ entry coupled with Na+ extrusion) modes (Goldman et al. 1994; Matsuda et al. 1996; Kirischuk et al. 1997; Rojas et al. 2007); the switch between NCX operational modalities depends on membrane potential and transmembrane Na+/Ca2+ gradients (Kirischuk et al. 1997). As a result NCX represents a powerful and capacious Ca2+/Na+ buffer that may rapidly and substantially affect local cytosolic concentrations of both ions.
Localized Ca2+ and Na+ signals integrate neuronal-glial networks at the synaptic level
Synaptic transmission activates several ion flux pathways present in perisynaptic astroglial processes. Importantly, molecules responsible for these pathways are strategically positioned. Morphological and functional studies indicate that ionotropic receptors, glutamate transporters and NCXs are all concentrated in the perisynaptic astroglial processes (Conti et al. 1994; Conti et al. 1996; Minelli et al. 2007; Lalo et al. 2011b). This results in local microdomains of high [Ca2+]i and [Na+]i that mediate local signaling cascades (Fig. 3). In particular, changes in [Na+]i can control several pathways which may effectively regulate synaptic transmission and plasticity. First, changes in [Na+]i directly control the efficacy of glutamate uptake; increases in [Na+]i inhibit glutamate transporters. It is feasible to speculate therefore that at the very peak of synaptic transmission event, which rapidly increases astroglial [Na+]i through activation of both ionotropic receptors and glutamate transporters, glutamate uptake is transiently inhibited therefore assuring high concentration of transmitter in the synaptic cleft securing transmission event. Increased [Na+]i turns the NCX into the reversed mode thus providing for additional local Ca2+ influx and for lowering [Na+]i which restores the efficacy of glutamate uptake. Incidentally, increase in [Na+]i may also facilitate the reverse mode of GABA transporter (which due to its stoichiometry is exceptionally sensitive to [Na+]i gradients) that can release GABA from astrocyte with yet uncharacterized inhibitory effects on synaptic transmission (Heja et al. 2009). Finally, increase in [Na+]i stimulates Na+/K+ pump that (i) facilitates astroglial K+ uptake thus participation in the ion homeostasis in the synaptic cleft and (ii) activates lactate shuttle (Magistretti 2006, 2009) that can specifically and focally supply active synapses with energy substrate.
Figure 3.

Local signaling mediated by ionotropic receptors and transporters in astroglial perisynaptic processes. Synaptic release of neurotransmitters (glutamate and/or ATP) activates ionotropic receptors and glutamate transporter, which generate Na+ and Ca2+ influx. Increase in [Na+]i can assume a signaling role through modulating neurotransmitter transporters, switching the reverse mode of NCX and stimulating Na+/K+ pump. This in turn can influence synaptic transmission and plasticity by affecting the time kinetics of glutamate removal from the cleft and through stimulating local metabolic support via the lactate shuttle.
To conclude, concerted activation of ionotropic receptors and ion transporters residing in astroglial perisynaptic processes results in fast and highly localized ion signaling; this in turn affects several pathways modulating synaptic transmission and plasticity.
Integration in neuronal-glial circuits: Gliotransmitters
Astrocytes are multifunctional housekeeping cells, involved in bi-directional communication with neurons; in doing so, these glial cells modulate synaptic transmission and plasticity (Araque et al. 1999; Newman 2003; Ni et al. 2007; Halassa et al. 2009). In this heterocellular signaling, astrocytes often utilize regulated exocytosis (Parpura et al. 2010; Parpura and Zorec 2010). This process requires vesicles containing a chemical transmitter, or a blend of them. Upon a merger of vesicular and plasma membranes, transmitter(s) stored within the vesicular lumen is(are) secreted into the ECS. Once in the ECS, gliotransmitters can act upon adjacent cells (paracrine function), or on the very cell which secreted them (autocrine function). Regulated exocytosis is set-off by an increase in the cytosolic [Ca2+]. Once triggered, this process can be quick-tempered as is the case in neurons, where a sub millisecond delay can be achieved when participating vesicles are organized in active zones, already pre-filled with transmitter, primed and docked to the plasma membrane. Regulated exocytosis in astrocyte, however, appears to be slower than in neurons (Kreft et al., 2004), in part due to slow vesicle delivery to the plasma membrane, albeit other undisclosed mechanisms may play a role as well. Astrocytes have vesicles which contain amino acids (e.g., glutamate and D-serine) and/or nucleotides (e.g., ATP) serving as gliotransmitters. Undeniably, astrocytes can also release vesicle-loaded peptides, as discussed elsewhere (Parpura et al. 2010).
Amino acid glutamate is synthesized de novo within astrocytes as a by-product of the tricarboxylic acid cycle (Westergaard et al. 1996; Hertz et al. 1999). D-serine is generated from L-serine by serine racemase, an enzyme found in astrocytes (Wolosker et al. 1999). ATP is produced via glycolysis and oxidative phosphorylation. Once released into the ECS, ATP can mediate intercellular signaling by acting directly onto purinergic receptors. Alternatively, but more frequently additionally, upon its hydrolysis by membrane-bound ecto-nucleotidases, the extracellular degradation products, adenosine diphospate and adenosine, can activate diverse plasma membrane receptors [reviewed in (Fields and Burnstock 2006)].
Evidence for Ca2+-dependent gliotransmitter release from astrocytes was initially demonstrated for glutamate, which release from cultured astrocytes was stimulated upon elevation of their cytosolic Ca2+ caused by a Ca2+ ionophore used as a secretagogue (Parpura et al. 1994). Similarly, Ca2+ ionophore-stimulated astrocytes can also release D-serine, a co-agonist of the glycine binding site of the NMDA receptor (Mothet et al. 2005). In support of ATP being released from astrocytes by Ca2+-dependent exocytosis, astrocytes exposed to nitric oxide (NO) showed an increase in cytosolic Ca2+ with consequential release of ATP into the ECS (Bal-Price et al. 2002). The release of peptides, e.g., atrial natriuretic peptide, from cultured astrocytes was demonstrated by monitoring optically fluorescently labeled vesicles in cells exposed to Ca2+ ionophore (Krzan et al., 2003). These initial findings have been corroborated by many laboratories using a variety of techniques to detect release of these gliotransmitters from astrocytes [e.g., (Bezzi et al. 2004; Kreft et al. 2004; Montana et al. 2004; Bowser and Khakh 2007; Jourdain et al. 2007; Pangrsic et al. 2007; Marchaland et al. 2008; Malarkey and Parpura 2011); reviewed in (Parpura and Zorec 2010)].
There are multiple sources of Ca2+ for Ca2+-dependent gliotransmitter release from astrocytes. We illustrate the complexity of Ca2+ sources using an example of exocytotic glutamate release, since it has been studied in most details. Hence, the majority of cytosolic Ca2+ necessary for astrocytic glutamate release originates from the ER store as determined using a blocker of ER-specific Ca2+-ATPases (Araque et al. 1998; Bezzi et al. 1998; Innocenti et al. 2000; Hua et al. 2004; Montana et al. 2004); based on further pharmacological evidence both inositol 1,4,5-trisphosphate (InsP3)- and ryanodine-sensitive receptors serve as channels for delivery of Ca2+ from the ER lumen to the cytosol (Hua et al. 2004) (Fig. 4). It should be noted, however, that the functionality of ryanodine receptors in astrocytes in situ is still debated (Beck et al. 2004).
Figure 4.

Ca2+- dependent vesicular release of gliotransmitters from astrocytes. The sources of Ca2+ for cytosolic Ca2+ increase are: the endoplasmic reticulum (ER) and the extracellular space (ECS). Cytosolic Ca2+ accumulation could be caused by the entry of Ca2+ from the ER store that possess inositol 1,4,5 trisphospate and ryanodine receptors. Store specific Ca2+-ATPase fills these stores with Ca2+. Ultimately, this (re)filling requires Ca2+ entry from the ECS through store-operated Ca2+ entry (SOCE). Additional Ca2+ entry from the ECS to the cytosol can be mediated by plasma membrane voltage -gated Ca2+ channels (VGCCs) and Na+/Ca2+ exchangers (NCXs), the latter operating in the reverse mode. Mitochondria represent a source/sink of cytosolic Ca2+. Mitochondrial Ca2+ uptake from the cytosol to its matrix is mediated by the Ca2+uniporter. Free Ca2+ exits the mitochondrial matrix through the Na+/Ca2+ exchanger and also by brief openings of the mitochondrial permeability transition pore. Increase of cytosolic Ca2+ is sufficient and necessary to cause vesicular fusions and release of gliotransmitters (GT). This process requires the activity of the ternary SNARE complex (SNAREs) consisting of: i) synaptobrevin 2 and/or cellubrevin located at vesicular membrane and ii) the binary cis complex pre-formed at the plasma membrane and composed of syntaxin (shown in an open form) and SNAP-23. Astrocytic vesicles are filled by various vesicular GT transporters, which for this action utilize the proton gradient generated by the vacuolar type H+-ATPase. Drawing is not to scale.
The Ca2+ entry across the astrocytic plasma membrane into to the cytosol eventually represents the source of this ion for (re)filling of the ER Ca2+ store. This can occur via store-operated Ca2+ entry (SOCE). Plasmallemal channels mediating this event become activated when ER Ca2+ is depleted (Takemura and Putney 1989; Golovina 2005). In particular, the canonical transient receptor potential (TRPC) 1 protein containing channels contribute to Ca2+-dependent glutamate release from astrocytes (Malarkey et al. 2008) (Fig. 4).
In addition to SOCE, Ca2+ entry from the ECS to the cytosol can be mediated by plasma membrane voltage- and ligand-gated Ca2+ channels. Astrocytes in acute slices from ventrobasal thalamus showed intrinsic cytosolic Ca2+ oscillations which lead to glutamate release (Parri et al. 2001); these oscillations dually draw Ca2+ from the ER store and the ECS by the Ca2+ entry via voltage-gated Ca2+ channels (VGCCs). Whether the activation of astrocytic ionotropic transmitter receptors, which leads to cytosolic Ca2+ influx [reviewed in (Lalo et al. 2011a)], plays a role in exocytotic gliotransmission remains to be determined. Another pathway for Ca2+ entry across the plasma membrane into the astrocytic cytosol is offered by NCXs operating in the reverse mode (Kirischuk et al. 1997; Rojas et al. 2008). Resulting cytosolic Ca2+ increases can cause exocytotic glutamate release from astrocytes (Paluzzi et al. 2007; Reyes and Parpura 2009).
Mitochondria represent a source/sink of intracellular Ca2+ which can modulate the magnitude of glutamatergic exocytosis in astrocytes (Reyes and Parpura 2008; Reyes et al. 2011). At elevated cytosolic Ca2+ levels, these organelles take up Ca2+ into the matrix via Ca2+ uniporter. As the cytosolic Ca2+ declines, free Ca2+ exits the matrix through the mitochondrial Na+/Ca2+ exchanger as well as via transient openings of the mitochondrial permeability transition pore. Indeed, the resulting buffering of cytosolic Ca2+ levels, caused by activity of these mitochondrial proteins, reflects in modulation of the magnitude of exocytotic glutamate release from astrocytes (Reyes and Parpura 2008). The contribution of cytosolic proteins that can buffer Ca2+ and buffering capacities of the nucleus in such modulation remains elusive at the moment.
Taken together, a concerted effort of various molecular entities located at the plasma membrane, the ER and mitochondria regulate cytosolic Ca2+ levels which in turn drive Ca2+-dependent regulated exocytosis. Interestingly, there could be additional levels of modulation of this process. It has been demonstrated that cytosolic glutamate concentrations as well as the availably of vesicular glutamate transporter (VGLUT) 3, that mediates packing of this gliotransmitter into vesicles (see below), can affect the magnitude of exocytotic release (Ni and Parpura 2009). Furthermore, there has been demonstration of the immunophillin-mediated enhancement of the glutamatergic output from astrocytes which had been attributed to a downstream of Ca2+ modulation, likely at the level of secretory machinery (Reyes et al. 2011).
Indeed, astrocytes express proteins of the exocytotic secretory machinery, in particular the soluble N-ethyl maleimide-sensitive fusion protein attachment protein receptor (SNARE) complex composed of: synaptobrevin 2, and its homologue cellubrevin; syntaxins 1, 2 and 4; and synaptosome-associated protein of 23 kDa (SNAP-23) [(Paco et al. 2009) and reviewed in (Montana et al. 2006)]. This complex is associated with various proteins, most notably synaptotagmin 4 (Zhang et al. 2004a). Although, the expression of mutated synaptotagmin 4 showed a dominant-negative effect by reducing Ca2+-dependent glutamate release from astrocytes (Zhang et al. 2004a), the lack of Ca2+-binding by this protein (Dai et al. 2004) leaves the identity of a Ca2+ sensor for regulated exocytosis in astrocytes unknown. The uptake of glutamate and ATP into vesicles is carried out by vesicle membrane bound proteins, VGLUTs and the vesicular nucleotide transporter (VNUT) (Sawada et al. 2008), respectively. These transporters uptake glutamate/ATP into vesicles using the proton concentration gradient established across the vesicular membrane by another vesicular membrane protein, vacuolar type H+-ATPase (V-ATPase). While it is widely accepted that neurons express these proteins (Takamori 2006), only recently it has been demonstrated that astrocytes express all the three isoforms of VGLUTs (1, 2, 3), VNUT as well as V-ATPase (Fremeau et al. 2002; Bezzi et al. 2004; Montana et al. 2004; Wilhelm et al. 2004; Zhang et al. 2004b; Anlauf and Derouiche 2005; Sawada et al. 2008). The release of glutamate, D-serine and/or ATP has consequences on synaptic physiology and animal behavior, as discussed below.
Integration in neuronal-glial circuits: Gap junctions
Most gap junctions in the brain occur between glial cells, where coupling strength can be very high. In particular, astrocytes are widely interconnected via gap junctions, which mainly consist of connexin (Cx) 43 and, to a lesser extent, Cx30. The intensity of astroglial coupling led to the concept of astrocytes providing a functional syncytium in which cell responses are highly coordinated and effective buffer volume is quite high (Fig. 5).
Figure 5.

Astrocyte gap junction mediated pathways for modulation of synaptic transmission. A) Gap junctions between astrocytes (1), or reflexive gap junctions between processes of a cell (2) facilitate movement of ions, second messengers, and nutrients within and between astrocytes. Synaptic activity (3) stimulates a Ca2+ wave (4) that is transmitted through gap junctions by Ca2+ (green), InsP3 (gray), and primarily through pannexin 1 channels (yellow channel) and, under some conditions, unpaired connexon channel opening that allows release of ATP (red arrow). B) The Ca2+ wave (4) spreads to a third astrocyte to the right. Na+ spikes are buffered and spread to adjacent cells (5). C) Increased K+ is taken up from areas of neuronal activity and spatially buffered by gap junctions (6). Metabolic fuel (7) is delivered through gap junctions in a directional manner, from blood vessels to areas of synaptic activity. Gliotransmitter release and other changes in astrocyte activity lead to modulation of the synaptic activity due to gap junction communication between astrocytes (8). Note at the top of each panel of this figure the presence of the blood vessel (illustrated as a horizontal red rod) which is enwrapped by the astroglial endfeet. Astroglia play active roles in so-called “neurovascular coupling” both by inducing the blood-brain barrier and through release of vasoactive agents.
One way in which such astrocyte signaling occurs is through generation of intracellular Ca2+ waves. Astroglial Ca2+ waves can be generated spontaneously and/or in response to neuronal activity. They are believed to provide a pathway of modulation between domains of separate neurons. Gap junctions, by direct intercellular exchange of Ca2+ or another second messenger molecule (e.g., InsP3) seem to determine speed and directionality of the waves (Fig. 5A). However, a considerable fraction of the waves is now known to be mediated by extracellular routes involving the release of ATP (via plasmalemmal channels and/or exocytosis) from the intracellular compartment into the extracellular environment and subsequent paracellular diffusion and action on neighboring cells through purinergic receptors. The channels responsible for ATP release were originally believed to be gap junction “hemichannels” on the cell surface, although pannexin 1 channels seem more likely candidates (Scemes et al. 2007) (Fig. 5A). These channels, which show minor sequence homology with invertebrate gap junction proteins, innexins, but none at all to the vertebrate connexins, open in response to elevated K+, activation of P2X7 receptors, stretch and membrane depolarization.
Astrocyte gap junctions also facilitate the spread of Na+ elevations from sites of local increases to adjacent astrocytes (Fig. 5B). Intense neuronal excitatory synaptic activity leads to Na+ spikes in astrocytes due to cotransport of Na+ with glutamate (Kimelberg et al. 1989; Langer and Rose 2009). In hippocampal slice preparations these intracellular Na+ increases spread to neighboring astrocytes through gap junctions (Langer et al. 2011). Gap junction mediated Na+ waves would alter the glutamate uptake from synapses thereby modulating their activity. Because of the effect of Na+ concentration on glycolysis and glycogen breakdown this form of communication changes metabolic activity of the astrocytes (Pellerin and Magistretti 1994). This may be one way that activity dependent, directional movement of metabolites is orchestrated as discussed below. The astrocyte syncytium forms a metabolic network that can provide energy supply to active neurons. In the grey matter, all or almost all astrocytes are highly polarized cells, with one process encircling a blood vessel as astrocytic endfeet and a second neuronal pole present in the proximity of neuronal synapses. By gap junction coupling they are assumed to provide a network that delivers metabolites from the perivascular space to the neuronal site. Since glucose and its metabolite lactate are the main energy carriers for neurons, it has long been thought that the astroglial network may function as a supportive conduit to serve for the transport of these energy donors from the vasculature to neurons. In fact, as has been shown recently, gap junction mediated cell-to-cell diffusion of these energetic metabolites seems to be one main function of the astroglial network, delivering energy to sites of high neuronal demand (Rouach et al. 2008; Gandhi et al. 2009). It is noteworthy that this metabolic network exhibits activity dependent traits, since it is responsive to increased synaptic activity at AMPA receptor sites.
In addition to these astroglial properties for intercellular transmission of Ca2+ waves and metabolites, astrocytes have also been regarded to function as a buffer volume to cope with local increases of external ions such as K+, which may rise to high levels upon action potential generation (Fig. 5C). Coupling of astrocytes through gap junctions offers the possibility to dissipate local peaks of K+ concentrations within the syncytium, by substantially increasing the volume of the buffer (Seifert and Steinhauser 2011). In this context it is noteworthy to mention that gap junctions can be formed between processes originating from a single cell. This type of gap junction is termed “reflexive”, where the contacts of a cell upon itself allow creation of subcellular microdomains acting in a cooperative way, for instance at perisynaptic sites (Wolff et al. 1998; Schipke and Kettenmann 2004).
The second class of macroglial cells, oligodendrocytes, is interconnected by connexin types that differ from the astroglial pattern (Nagy et al. 2004; Theis et al. 2005). The most abundant connexin expressed in oligodendrocytes, and also in its peripheral relative, the Schwann cell, is Cx32. An additional connexin, Cx29, which is likely unable to form gap junctions, but apposes with axonal Shaker type K+ channels is also expressed in Schwann cells and in oligodendrocytes, while oligodendrocytes also express Cx47. Both Schwann cells and oligodendrocytes are responsible mainly for the formation and maintenance of the myelin sheath. Most work regarding myelin connexins has been done on Schwann cells, due to the discovery that the X-linked form of a peripheral neuropathy, the type X Charcot Marie Tooth disease is due to mutations in Cx32. More than 300 mutations have now been identified in patients, with clinical phenotype characterized by an accruing weakness in the peripheral parts of the lower extremities with progression to the more proximal muscles. Because a single Schwann cell forms the entire part of a functional unit in peripheral myelin, the so called internode, there is obviously no need for intercellular coupling. The idea arose and was later confirmed that gap junctions in peripheral myelin consist mainly of reflexive junctions. Such reflexive junctions are common, and were mentioned above in the case of astrocytes. Since reflexive junctions may serve as short-cuts to allow fast diffusion of ions or metabolites between extremely tortuous cytoplasmic compartments of the same cell, in the Schwann cell they may provide radial conduits at sites of the myelin sheath where cytoplasmic inclusions are localized such as the Schmidt-Lantermann incisures and the paranodal processes. The radial orientation allows fast diffusion of nutrients from the outer circumference of the myelin sheath, where the soma of the Schwann cell is localized to its central adaxonal cytoplasmic collar. Loss of function mutations of the Cx32 gene could thus result in malnutrition of the myelin sheath in particular of its cytoplasmic portions. Because single axons emanating from motoneurons of the lumbar spinal cord span a distance more than a meter from their origin in humans, it is no surprise that deficits in nutritive supply and clearance of metabolic waste of the myelin sheath leads to initial degeneration in the most distal muscles.
In central myelin, the oligodendrocytes are arranged a little differently. Here, more than one cell contributes to the formation of a single internode and in addition to that, a single oligodendrocyte contributes to the formation of multiple internodes. Nevertheless, the central myelin sheath also harbors a number of gap junctions, especially in its outer circumference, where the connexins forming gap junctions are Cx32, Cx29 and Cx47 (Nagy et al. 2004; Theis et al. 2005). Besides interconnecting oligodendrocytes, gap junctions are also formed between oligodendrocytes and astrocytes. The use of transgenic mice carrying reporter genes and deletions of different connexin genes provided evidence that most of the junctions consist of either of the oligodendrocytic/astrocytic (O/A) combination Cx47 and Cx43, or the O/O homotypic Cx32/Cx32 configuration. Likewise O/A junctions of Cx32 and Cx30 also exist. The extensive heterotypic coupling of both classes of glial cells has led to the concept of a panglial syncytium, indicating that astrocytes and oligodendrocytes form a functional network in terms of connectivity.
Mutations in the oligodendrocytic Cx47 may cause a devastating leukodystrophy called Pelizaeus-Merzbacher-like disease or a milder spastic paraplegia (Scherer and Wrabetz 2008). It also is the cause of lymphedema, in which lymph flow is disrupted. In addition, CNS demyelination may be caused by defects in genes expressing astrocytic gap junction proteins, which are essential for O/A coupling. In mice, mutants carrying a deletion of both the oligodendrocytic genes encoding for Cx32 and Cx47 severe deficits in myelination occur in central myelin resulting in thin or absent myelin sheaths and extensive vacuolation, signs of degeneration.
Astroglial regulation of the neurovascular unit
Astrocytes, endothelial cells, neurons and pericytes comprise the neurovascular unit, in which glia play major regulatory roles [for recent review, see (Dermietzel and Spray 2012)]. One such role is induction of proteins involved in polarized distribution of functional components of the blood-brain barrier, a notable example being aquaporin 4 expression in glial endfeet enwrapping neural vessels (shown schematically in Fig. 5). Glial induction of both adhesive and tight junction protein expression in endothelial cells creates a permeation boundary that is a target for both delivery of therapeutic pharmacological agents and for breach by infectious organisms. A second function of glial cells in neurovascular coupling is the control of local blood flow, thereby matching tissue oxygenation to demand. Here, astrocytic release of vasoactive factors mediates the additional delivery of metabolic molecules from the glial endfeet throughout the panglial syncytium, as illustrated by arrows showing flow routes in Fig. 5.
Astroglial regulation of synaptic plasticity
There is a growing body of evidence demonstrating a role for astrocytes in regulating long-term synaptic plasticity at central synapses. Such a glial contribution to changes in synaptic strength involves the release of transmitters, including glutamate, ATP and D-serine, from astrocytes. These substances have been reported to act pre- and/or postsynaptically at nearby synapses affecting transmission in a transient or long-lasting manner [e.g. (Panatier et al. 2006; Serrano et al. 2006; Jourdain et al. 2007)]. These astrocytic regulations can be classified in two groups according to the type of synaptic plasticity concerned: i) those that are well established and have been extensively studied or ii) those that were not described before the contribution of astrocytes was investigated.
The most common forms of synaptic plasticity found in the brain depend on the activation of NMDA receptors (NMDARs). This includes NMDAR-mediated long term potentiation (LTP) and long term depression (LTD) which cause an insertion or a removal of AMPA receptors from the postsynaptic membrane, respectively (Malenka and Bear 2004). NMDARs are peculiar glutamate receptors since they require the binding of an agonist, glutamate, and a co-agonist that was first identified as glycine (Johnson and Ascher 1987). In the last decade, however, several studies have challenged this idea by showing that another amino acid, D-serine, was indeed serving as an NMDAR endogenous co-agonist (Mothet et al. 2000) and that it was released from astrocytes (Yang et al. 2003; Mothet et al. 2005) and neurons (Kartvelishvily et al. 2006; Rosenberg et al. 2010). Recordings obtained from cultures (Yang et al. 2003; Mothet et al. 2005) and from acute slices (Panatier et al. 2006; Henneberger et al. 2010; Fossat et al. 2011) have demonstrated that astrocytes were important for supporting NMDAR activity at glutamatergic synapses through the supply of D-serine, thereby enabling NMDAR-mediated synaptic plasticity (Fig. 6). In the hypothalamus, the glial withdrawal that occurs in the supraoptic nucleus during lactation (Theodosis et al. 2008) causes an impairment of NMDAR activity that is completely accounted for by decreased D-serine levels within the cleft (Panatier et al. 2006). As a consequence the activity-dependence of NMDAR-mediated synaptic plasticity is dramatically altered since less NMDARs are available for activation. In the CA1 region of the hippocampus, where NMDAR-dependent plasticity has been the subject of numerous studies, a similar phenomenon prevails (Henneberger et al. 2010). Impairing intracellular Ca2+ in an individual astrocyte leads to a reduced supply of D-serine, thereby inhibiting NMDAR activity and LTP induction at synapses located within the glial anatomical territory. These findings indicate that astrocytes are key determinant to NMDAR-mediated plasticity and that each astrocyte controls NMDAR activity at independent sets of synapses.
Figure 6.
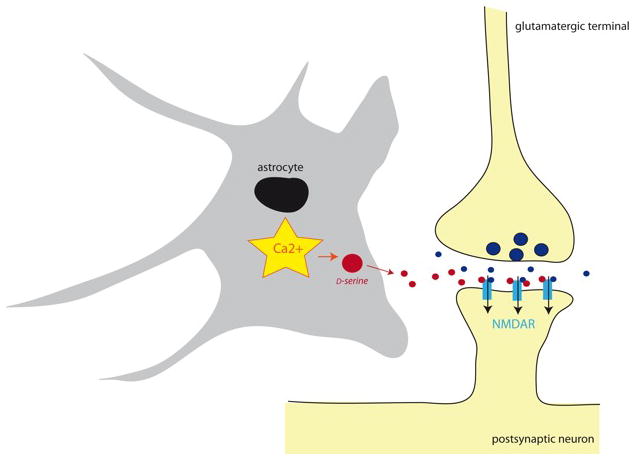
Increase in intracellular Ca2+ levels triggers exocytotic release of D-serine from astrocytes. As D-serine (red) reaches the synaptic cleft, it binds to post-synaptic NMDA receptors (NMDARs), which also binds glutamate (blue) released from the pre-synaptic terminal. This co-incidental NMDAR detection favors NMDAR activation and induction of the most common forms of synaptic plasticity. Astrocytic nucleus is drawn as a black oval.
Another well-described short-term synaptic plasticity is the heterosynaptic depression of glutamatergic inputs occurring in the CA1 region in response to tetanic stimulation (Manzoni et al. 1994). This form of plasticity was later shown to involve the activation of interneurons and the consecutive stimulation of neighboring astrocytes through GABA-B receptors (Pascual et al. 2005; Serrano et al. 2006). This protocol of stimulation activates astrocytes, leading to the release of ATP with its metabolite adenosine, converted extracellularly, and then acting presynaptically on A1 receptors to inhibit excitatory transmission. The overall effect is an increased contrast when comparing synaptic strength at the glutamatergic inputs that are directly tetanized and potentiated with those neighboring excitatory terminals that are not tetanized, but that endure the adenosine-mediated heterosynaptic depression (Serrano et al. 2006).
Astrocytes have been also involved in the regulation of other forms of synaptic plasticity in the CNS that were not described beforehand in the literature. In the hypothalamus, for example, ATP released from astrocytes promotes the insertion of AMPA receptors (Gordon et al. 2005). This plasticity differs from that depending on NMDARs since it is not input specific, causing a general synaptic scaling, a phenomenon known as homeostatic plasticity. Such non-Hebbian plasticity also occurs at other central synapses in response to glial tumor necrosis factor-α (Stellwagen and Malenka 2006). This phenomenon appears to be important not only for increasing synaptic strength but also for preserving it (Beattie et al. 2002).
In the hippocampus, a form of LTP, different from NMDA receptor-dependent LTP, that involves glia has been described when postsynaptic depolarization was combined with astrocytic stimulation through Ca2+ uncaging (Perea and Araque 2007). This protocol causes a long lasting enhancement of minimal glutamatergic synaptic responses evoked in CA1 neurons. It is mediated by astrocyte-derived glutamate that acts on presynaptic metabotropic GluRs (mGluRs). Interestingly, mGluR activation by glial glutamate can be triggered by endocannabinoids originating from neighboring neurons and acting on CB1 receptors located on astrocytes (Navarrete and Araque 2010). This, however, only generates a short-lasting enhancement of glutamatergic transmission. In the dentate gyrus, glutamate released from astrocytes activates presynaptic NMDA receptors, thereby transiently facilitating excitatory transmission onto granule cells (Jourdain et al. 2007).
These few chosen examples illustrate the major problem that the field of neuron-glia interaction is facing regarding the astrocytic regulation of synaptic plasticity. Not only phenomena that have been widely investigated in many laboratories appear to depend on astrocytes, but also new types of short- and long-term plasticity that were never reported before seem to depend on glia. While the relevance of all these processes for network activity and cerebral communication remains to be unraveled, they all point to a pivotal role played by astrocytes in the regulation of synaptic strength.
Astroglial role in control of adenosine tone: Consequences on synaptic plasticity and sleep homeostat
Santiago Ramón y Cajal had numerous great insights into brain function. Another has recently been realized. In 1895 he proposed that astrocytes control sleep and waking states. He specifically proposed that astrocytic processes are electrical insulators that, when extended between neurons, act as circuit breakers to facilitate sleep but, when retracted, allow neuronal circuits to communicate, facilitating wakefulness. Now, following work of the past five years we know that his intuition was generally correct because we have been able to demonstrate that astrocytes regulate the extracellular accumulation of adenosine a factor that is known to control sleep homeostasis.
It has been known for some time that adenosine levels rise during wakefulness, that infusion of adenosine can promote sleep and that adenosine receptor antagonists promote wakefulness (Basheer et al., 2004). However, the source of adenosine was unclear. A study designed to address a different question fortuitously led to the discovery that astrocytes control adenosine, and as a consequence sleep (Halassa et al., 2009).
One of the difficulties with identifying the roles of astrocytes in the regulation of synaptic transmission, circuits and behavior is that pharmacological studies do not allow a discrimination of glial versus neuronal mechanisms of actions. The explosion of mouse molecular genetics is beginning to give new insights into the role of astrocytes in brain and behavior. In 2005 a landmark study demonstrated that astrocytes regulate adenosine (Pascual et al., 2005). In this study conditional, astrocyte specific molecular genetics was used to inhibit the release of bioactive compounds from astrocytes. In this study the cytosolic SNARE domain of synaptobrevin 2 was expressed in adult astrocytes. By expressing this truncated protein, which acted as dominant negative SNARE (dnSNARE), the formation of the core SNARE complex that is required for exocytosis was prevented. In a control experiment the investigators asked whether basal hippocampal synaptic transmission was modulated through glial expression of dnSNARE. They were surprised to find that the magnitude of CA3-CA1 synaptic transmission was augmented, and that the mechanism was mediated by the removal of a tonic presynaptic inhibition that is normally mediated by basal extracellular adenosine acting on presynaptic A1 receptors. The extracellular adenosine was generated by the action of ecto-nucleotidases on extracellular ATP released from astrocytes in the SNARE-dependent manner.
The consequences of this adenosine-mediated astrocyte-dependent synaptic modulation are multi-fold. Foremost, the ability to induce synaptic plasticity, in the form of LTP following theta-burst stimuli, is reduced. For many years, the mechanism of this glial modulation of synaptic plasticity was unknown. However, recently it has been demonstrated that the tonic activation of postsynaptic purinergic A1 receptors lead to a src family kinase dependent tyrosine phosphorylation of NR2A and NR2B subunits of the NMDA receptor (Deng et al., 2011). Phosphorylation of these subunits leads to a reduced rate of their endocytosis. In the astrocytic dnSNARE mice, reduced phosphorylation led to a reduction in surface expression of NMDA receptor subunits and consequently to reduced synaptic NMDA receptor currents. Since NMDA receptors are critical for the induction of LTP it is not surprising then that the magnitude of hippocampal LTP is reduced in dnSNARE mice.
In addition to effects on synaptic plasticity, it has been discovered that the astrocytic source of adenosine is essential for the process of sleep homeostasis and for responses to sleep deprivation (Halassa et al., 2009). Sleep can be considered to be controlled by at least two processes: i) the circadian oscillator that sets the timing of sleep and wakefulness, and ii) the sleep homeostat that integrates the amount of wakefulness and promotes the drive to sleep. Astrocytic adenosine is critical for the control of sleep homeostasis. When an animal sleeps the power of slow wave activity (SWA) during non rapid eye movement (NREM) sleep is proportional to sleep drive. Thus, when one is sleep deprived, the power of the SWA is increased in proportion to the sleep debt that was incurred. When electroencephalography and electromyography recordings were performed from dnSNARE mice, it was found that the power of SWA during NREM sleep was significantly attenuated at the onset of the sleep period. Additionally, on a subsequent day, sleep deprivation caused an attenuated increase in the power of SWA in astrocytic dnSNARE mice. Another consequence of sleep deprivation is that there is compensatory increase in sleep time. Surprisingly, astrocytic dnSNARE mice did not exhibit altered sleep times following sleep deprivation. These effects of astrocytic dnSNARE expression were phenocopied in wild type mice by intracerebroventricular administration of an A1 receptor antagonist indicating that these glia, as suggested by Cajal, exert powerful control over sleep.
There are many questions left unanswered and studies that are still to be performed. It is essential that the mechanism of adenosine accumulation be understood. Our current conjecture is that astrocytes release ATP through a dnSNARE sensitive exocytotic mechanism and that the released ATP is hydrolyzed to adenosine in the extracellular space. Direct measurements of ATP and adenosine are now needed in these glial specific dnSNARE mice. What regulates the daily rise and fall of adenosine in response to wakefulness? Is there a wakefulness dependent modulation of the release or uptake of adenosine? Does the glial modulation of sleep homeostasis contribute to disorders of brain function? So many brain disorders exhibit sleep co-morbidities. Depressed patients either sleep little or sleep too much. Bipolar patients exhibit a limited need to sleep during mania. Do inactivated glia contribute to these psychiatric conditions? Recovering alcoholics exhibit highly fragmented sleep and this sleep fragmentation is the most reliable predictor of relapse. Additionally, sleep deprivation increases the probability of breakthrough seizures in epileptics. While we do not propose for one moment that glia are the cause of all of these disorders, it will be fascinating to discern whether one can develop new therapeutics for these disorders based on emerging knowledge of glial-based targets.
Astroglia in the neuropathology of Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disorder, which is clinically characterized by progressive memory loss and dysfunction of higher cognitive domains. Pathologically, the aggregation and deposition of β-amlyoid (Aβ) peptides and the formation of neurofibrillary tangles represent the classical hallmarks of the disease. Considerable evidence now suggests that Aβ deposition in the brain precedes neurofibrillary tangle formation, neuronal cell death and subsequent deterioration of brain function (Jack et al. 2010). The familiar type of AD is caused by mutations in genes involved in processing of the amyloid precursor protein (APP), with an increased Aβ production or change in the ratio of Aβ peptides to favor the overall Aβ1–42 content. In contrast, the sporadic form of AD seems not to be caused by overproduction of Aβ peptides, but rather occurs in response to dysfunctional degradation and clearance (Mawuenyega et al. 2010). In both cases however, deposition of Aβ and clinical appearance of first cognitive dysfunction may lie decades apart.
Only recently brain inflammation has been identified as third important component of the disease pathogenesis (Wyss-Coray 2006; Heneka and O’Banion 2007). Increased cerebral levels of A β peptides and their subsequent deposition in so called A β plaques lead to the activation of the surrounding microglia and astrocytes (Fig. 7), which can even be detected in AD patients (Cagnin et al. 2001) using positron emission tomography ligands targeting the peripheral benzodiazepine receptor located on activated microglial cells. Upon activation, both, microglial cells and astrocytes release a variety of pro- and anti-inflammatory mediators thereby establishing a chronic parenchymal inflammation in the brain. While inflammatory signals have been originally designed for host defense again invading microorganisms and are, after successful execution of this task, usually terminated, the chronic accumulation of Aβ during the course of AD represents an ongoing and sustained stimulus for inflammatory activation. Of note, Aβ induced activation of microglia and astrocytes may well be mediated by the very same receptor signaling cascade as the activation by bacterial or viral cell wall components, namely through the toll receptor family members. Importantly, inflammation will not occur as reaction to Aβ deposition only, but also establishes feedback mechanisms that further increase A β generation and at the same time compromise A β clearance mechanisms. Thus it has been shown that proinflammatory cytokines increase levels of APP and also increase the transcription of β-secretase-1, which is the rate limiting enzyme in amyloidogenic A β production (Sastre et al. 2003; Sastre et al. 2006). Furthermore, phagocytosis and degradation of A β is decreased once cells have been challenged with inflammatory cytokines derived from astrocytes and microglia.
Figure 7.

Astroglia assembling around a plaque in an amyloid precursor protein/presenilin 1 transgenic mouse. Astrocytes (red) are labeled using indirect immunochemistry and antibody against glial fibrillary acidic protein, while the core and periphery of the β-amlyoid (Aβ) plaque are stained using methoxy-XO4 [binding Aβ fibrils; (Klunk et al. 2002)] and monoclonal antibody IC16 [binding Aβ dimer, with the epitope at residues 2–8 of Aβ; (Muller-Schiffmann et al. 2011)], respectively. Courtesy of Drs. Markus Kummer and Michael Heneka.
The presence of glia-derived, inflammatory molecules in the brain has both, functional as well as structural consequences. Thus, cytokines themselves can suppress LTP, a critical factor for hippocampal memory formation and consolidation (Tancredi et al. 1990; Tancredi et al. 1992; Murray and Lynch 1998). Likewise, chronic inflammatory stimulation of astrocytes reduces the glial capacity to generate and release neurotrophic mediators such as glial cell-derived neurotrophic factor for support of bystander neurons (Nagatsu and Sawada 2005). Both actions may well be involved in early cognitive dysfunction. At later stages, inflammation may become directly damaging to the brain and glial cytokines and chemokines may lead to the destruction of axons, dendrites and synapses.
Astrocytes are in the center of this chronic inflammatory scene as they react to and also release cytokines, thereby fueling the above described vicious cycles between neurodegenerative and neuroinflammatory processes. Even more directly and with special relevance to the sporadic form of AD, astrocytes are involved into the degradation and clearance of Aβ from the brain (Koistinaho et al. 2004; Pihlaja et al. 2011). These astroglial involvements may occur at various levels and involve direct and indirect actions. Thus, it has been shown that astrocytes may be directly involved in the uptake and degradation of Aβ (Koistinaho et al. 2004). Indirectly, but equally important, astrocytes may function as key regulators of microglial phagocytosis through the generation and release of apolipoprotein E (ApoE) and the modulation of its lipidation status by ATP-binding cassette transporter subfamily A member 1 (ABCA1). Both, ApoE and ABCA1 are expressed by astrocytes in response to liver X receptor (LXR) activation. In vitro, microglial phagocytosis of aggregated Aβ is more effective in the presence of supernatants derived from astrocytes (Terwel et al. 2011) stimulated with LXR activators. Interestingly, this effect depends on the presence of ApoE and it was absent when supernatants from ApoE knock-out astrocytes were applied. Of the two known LXR subtypes, LXR-α seems to be critical for the observed regulation of astrocyte-guided microglial Aβ uptake.
As astrocytes themselves may undergo functional and structural changes in response to chronic neuroinflammation and degeneration, as recently suggested by Olabarria et al. (Olabarria et al. 2010), and in particular show substantial atrophy, one may speculate that the above described beneficial functions of glia are consecutively compromised.
Astrocytes, however, do not only act as a source of cytokines and are involved in clearance mechanisms, they are also the major site of expression of the inducible nitric oxide synthase (iNOS, also referred to as NOS2), followed by neurons and microglial cells, in human AD (Heneka et al. 2001) and related mouse models (Heneka et al. 2005). Immunostimulated and sustained release of nitric oxide (NO) has miscellaneous consequences for the nearby environment. Once released, NO can interact with signaling cascades and regulate gene transcription, impair mitochondrial respiration or directly induce cell death by apoptosis or necrosis. One important downstream product of iNOS activation is peroxynitrite which is generated through the reaction of NO with oxygen. Peroxynitrite then can exert similar functions as described for NO and additionally lead to posttranslational modifications of tyrosine and cysteine residues in proteins, nitration and nitrosylation, respectively. There is ample evidence from post-mortem tissue and CSF analysis that suggests that NO-dependent peroxynitrite formation and subsequent nitration takes place during AD. Only recently Aβ itself has been identified as a prominent target of nitration in AD. Aβ, which contains a single tyrosine at position 10 is being nitrated by iNOS derived NO and peroxynitrite (Kummer et al. 2011). Importantly, this phenomenon can be observed in human AD brain samples as well as in the APP/presenilin 1(PS1) transgenic mouse model. The nitration of the tyrosine at position 10 however is not only a new pathological finding, but it itself changes the biophysical behavior of the Aβ peptide, potentiating the self-aggregation and deposition of the peptide. Nitrated Aβ is being found in the core, of each plaque and seems to represent the seed for plaque deposition. This assumption has been further supported by experiments, where nitrated and non-nitrated Aβ was used to seed subsequent plaque formation in APP/PS1 transgenic mice at an age, well before the usual occurrence of Aβ plaques. Nitrated, but not non-nitrated Aβ was effective in seeding Aβ plaques. Importantly, the exchange of the tyrosine 10 for an alanine, completely prevented the occurrence of Aβ plaques.
Together, astrocytes are intimately and at multiple levels involved in the pathogenesis of AD. They may be driving neurodegeneration by the release of various mediators, but also try to resolve pathological stimuli, such as Aβ; the latter is accomplished by modifying the uptake and clearance of a stimulus by microglia. Further studies are needed to investigate at which stage of the disease and through which pathways, the modulation of astrocytes may be therapeutically used.
Glial neoplasm: Impact of endothelial and microglial cells on invasive glioma growth
Primary brain tumors are intracranial solid neoplasms that grow and originate within the brain or central spinal canal. Many different types of primary brain tumors exist and each gets its name from the cell type from which they originate. Gliomas derive from glial cells or their precursors [reviewed in (Siebzehnrubl et al. 2011)], and comprise the large majority of malignant brain tumors, with the most common type being classified as astrocytoma grade IV, and traditionally referred to as a glioblastoma multiforme (GBM). In spite of therapy, GBM remains among the deadliest tumors. The main obstacle to successful GBM eradication is its reappearance within a few centimeters from the surgery lesion or the formation of satellite (secondary) tumors in distant parts of the brain, both indicating an infiltrative nature of the tumor spreading along myelinated nerve fibers, subependymal layers and vasculature (Zagzag et al. 2008). Signals generated at the GBM interface with the brain surroundings in this invasion process are not well understood. However, recent evidence implicates that microglial and endothelial cells contribution to invasive migration of GBMs as discussed below (Fig. 8).
Figure 8.

Intricate interactions of glioblastoma multiforme (GBM) tumor edge with the brain microenvironment. Cells within the core GBM mass are heterogeneous with ~30% microglia residing within. Tumor cells secrete various motility factors that maintain the process of invasion and express receptors for factors produced by normal brain cells to smooth the progress of tumor infiltration, e.g., bradykinin concentration gradient formed predominantly from endothelial cells attracts tumor cells and increases their motility directing them towards blood vessels. Glial cells in surrounding also respond to bradykinin, i.e., astrocytes by releasing a number of signaling molecules, and increasing matrix metalloproteinase (MMP)-9 expression and cell migration, while microglia by increasing mobility. Microglia further facilitates glioma invasion by releasing membrane type 1 MMP/MMP-14 essential for activation of an inactive MMP-2 pro-form secreted by glioma cells. Migration of glioma cells away from the core GBM mass may lead to the formation of the satellite tumors. Drawing is not to scale.
Tumor cells produce various autocrine motility factors to help maintain the process of invasion (Lyons et al. 2007). Similarly, they express receptors for an array of paracrine factors secreted by normal cells in the brain, such as growth factors, neurotrophins, chemokines, cytokines, and kinins, (Hoelzinger et al. 2007). Some of these receptors in glioma cells can be up-regulated due to hypoxic conditions within the tumor (Zagzag et al. 2008). Similarly, hypoxia in the brain surrounding tumor causes various changes in otherwise normal cells. One such change is an increase in bradykinin (BK) production predominantly by endothelial cells. Increased expression of bradykinin receptor 2 (B2R), one of two classes of receptors for bradykinin and dominantly expressed by gliomas (Montana and Sontheimer 2011) has been previously correlated with higher tumor grade (Raidoo et al. 1999). Recently, Montana and Sontheimer (Montana and Sontheimer 2011) investigated the role of B2R activation on glioma cell migration and invasion. Data from a series of in vitro experiments suggested significant increase in velocity, distance and directionality of GBM cell motility tracks with, and towards, the higher BK concentrations. The most intriguing finding, however, was generated using acute brain slices, where the presence of BK gradient greatly enhanced GBM penetration into the tissue, and significantly increased the number of glioma cells attached to blood vessels. All effects were attenuated in the presence of B2R antagonists or in B2R knock-down GBM cells, further suggesting an important role for B2R in BK mediated attraction of glioma cells to blood vessels (Montana and Sontheimer 2011). Owing to the GBM overexpression of B2R when compared to normal brain cellular milieu, and recent availability of the clinically approved B2R blocker icatibant (Cicardi et al. 2010), it is tempting to speculate that this novel B2R-mediated GBM-blood vessel signaling pathway may represent a point for adjuvant therapy intervention.
Indeed, normal glial cells can respond to bradykinin adding further complexity to interactions of normal and tumor cells at a tumor advancing edge. After BK stimulation, both astrocytes and microglia can increase mobility (Ifuku et al. 2007; Hsieh et al. 2008). Additionally, BK-evoked astrocytes can release gliotransmitters using a Ca2+-dependnet exocytosis (e.g. glutamate, D-serine, ATP) (Parpura et al. 1994; Verderio and Matteoli 2001; Montana et al. 2004; Montana et al. 2006; Martineau et al. 2008) and their secretion of matrix metalloproteinase (MMP) is increased as well (Hsieh et al. 2008). MMPs are essential for GBM invasion process as they degrade the extracellular matrix (ECM). Besides the membrane type 1 MMP (MT1-MMP), gliomas also secrete gelatinases MMP-2 and MMP-9 (Stylli et al. 2008). Expression and enzymatic activity of MMPs correlate with invasive behavior and/or malignancy of the tumor. It has been observed beforehand that microglia, attracted by cytokines released from glioma, comprise substantial portion of tumor. Although microglial cells appeared activated based on their morphological characteristics, they failed in providing their immune functions (Graeber et al. 2002). To a surprise, microglia supported glioma growth and spread, as evidenced in elegant experiments by Markovic et al (Markovic et al. 2005). They used cultured mouse brain slices with and without microglia, seeded with glioma cells to invade slices. In slices lacking microglia, glioma cells invaded significantly less in comparison to control slices with microglia present, suggesting that microglia enhanced glioma invasion (Markovic et al. 2005). The mechanism underlying this surprising finding includes glioma-microglia signaling (Markovic et al. 2009). Glioma cells released soluble factors which bound to toll-like receptors on microglia, and through activation of the p38 MAPK pathway, up-regulated microglial expression and activity of MT1-MMP/MMP14. They further showed that microglial MT1- MMP was essential for conversion of an inactive pro-MMP-2 secreted by glioma cells, to its active form. Consequently, degradation of ECM and alteration in local architecture allows glioma cells to move further from the tumor mass and facilitate the pathway of invasion. This finding was corroborated in vivo using a mouse with the deletion of the toll-like receptor adapter protein MyD88 in which glioma expansion was attenuated; similar in vivo result was obtained with microglial depletion in wild-type mouse (Markovic et al. 2009). Clinically approved antibiotic minocycline blocked glioma-mediated increase in MT1-MMP expression and activity in microglia and led to reduction of glioma growth in an experimental mouse model (Markovic et al. 2011), indicating a possibility for a new therapeutic intervention in treatment of gliomas.
Clearly, interactions between glioma cells and their environment are numerous and very complex (Fig. 8). Detached glioma cells are exposed to and respond to a wide range of signaling molecules. The emerging evidence that microglial and endothelial cells support invasive glioma growth draws attention to new potential therapeutic targets. In order to improve upon current therapies and increase patient survival, perhaps we should start considering how to complement existing treatments of gliomas with novel therapeutics that could affect bidirectional interactions between glioma and normal brain cells.
Concluding remarks
In this review we summarized up to date findings describing (astro)glial contributions to physiology and pathophysiology in the brain. Astrocytes play a positive role in neurogenic niche, showing some characteristics similar to those of stem cells. Their intimate structural-functional relationship with neurons in adult brain is dynamic, especially at the tripartite synapse. It is at these sites that astrocytes can metabolically support neurons and detect synaptic activity. Synaptically or otherwise evoked, as well as spontaneous, changes in astrocytic intracellular Na+ and Ca2+ dynamics can lead to an increased lactate production and release of gliotransmitters via Ca2+-dependent exocytosis, respectively. Subsequently, astrocytes can modulate neuronal activity, by modulating synaptic transmission and plasticity, and by affecting sleep behavior. Besides these physiological tasks, astrocytes can play role in various pathological conditions in the brain. We have discussed AD, as an example of a disease in which astrocytes can contribute to etiology. Glial derived tumors bidirectionally signal with normal cells (astrocytes, microglia and endothelial cells) within the brain; using trickery, the GBM employs normal brain cells to expand its own entity. Whilst this review sheds new light on the brain (dys)function it also presents many unknowns. Consequently, it is clear that much more work still needs to be done in order to solve the puzzle of how the brain operates in health and disease.
Acknowledgments
Discussion about this review between the authors took place at the International Society for Neurochemistry Satellite Meeting “Glia in (patho)physiology”, held in August, 2011 in Ljubljana, Slovenia. This work is supported by: National Science Foundation (CBET 0943343) to VP and IKERBASQUE, The Basque Science Foundation to AV and VP; NINDS (NS037585) and NIDA (DA025967) to PGH; the Deutsche Forschungsgemeinschaft (PA615/2-1) to TP; the Deutsche Forschungsgemeinschaft (GRK 1097) and EduGlia ITN EU grant to AR, AV, MPy, RZ and SO; by the Swedish Medical Research Council (project 11548 to MPy and 20116 to MPa), AFA (MPy and MPa), ALF Göteborg (11392 to MPy and 11267 to MPa) the Free Mason Foundation (MPy), and NanoNet COST (MPy, AR and RZ); P3 310, J3 4051, J3 3632 and J3 4146 grants from the Slovenian Research Agency (ARRS) to RZ; the Alzheimer’s Research Trust (UK) Programme Grant (ART/PG2004A/1), the Grant Agency of the Czech Republic (GACR 305/08/1381 and GACR 305/08/1384).
Abbreviations
- Aβ
β-amlyoid
- ABCA1
ATP-binding cassette transporter sub-family A member 1
- AD
Alzheimer’s disease
- AMPA
α-amino-3-hydroxy-5-methyl-isoxazole propionate
- ApoE
apolipoprotein E
- APP
amyloid precursor protein
- ATP
adenosine 5′-triphosphate
- BK
bradykinin
- B2R
BK receptor 2
- [Ca2+]i
cytosolic Ca2+ levels
- CSF
cerebrospinal fluid
- CNS
central nervous system
- Cx
connexin
- dnSNARE
dominant negative SNARE
- ECM
extracellular matrix
- ECS
extracellular space
- ER
endoplasmic reticulum
- GABA
γ-amino butyric acid
- GBM
glioblastoma multiforme
- GFAP
glial fibrillary acidic protein
- GluR
glutamate receptors
- GSC
glial synaptic current
- [Na+]i
cytosolic Na+ concentration
- NO
nitric oxide
- iNOS
inducible nitric oxide synthase
- InsP3
inositol 1,4,5-trisphosphate
- LTD
long-term depression
- LTP
long-term potentiation
- LXR
liver X receptor
- mGluRs
metabotropic glutamate receptors
- MMP
matrix metalloproteinase
- MT1-MMP
membrane type 1 MMP
- NCX
Na+/Ca2+ exchanger
- NMDA
N-methyl-D-aspartate
- NMDAR
NMDA receptor
- NREM
non rapid eye movement
- O/A
oligodendrocytic/astrocytic
- PAP
peripheral astrocyte process
- PS1
presenilin 1
- SNARE
soluble N-ethyl maleimide-sensitive fusion protein attachment protein receptor
- SNAP-23
synaptosome-associated protein of 23 kDa
- SOCE
store-operated Ca2+ entry
- SWA
slow wave activity
- TRPC
canonical transient receptor potential
- VGCCs
voltage-gated Ca2+ channels
- V-ATPase
vacuolar type of proton ATPase
- VGLUT
vesicular glutamate transporter
- VNUT
vesicular nucleotide transporter
References
- Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science. 2010;327:1250–1254. doi: 10.1126/science.1184821. [DOI] [PubMed] [Google Scholar]
- Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, Casper KB, Fiacco TA, McCarthy KD. What is the role of astrocyte calcium in neurophysiology? Neuron. 2008;59:932–946. doi: 10.1016/j.neuron.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriezen WL. The neuroglia elements of the brain. Brit Med J. 1893;2:227–230. doi: 10.1136/bmj.2.1700.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anlauf E, Derouiche A. Astrocytic exocytosis vesicles and glutamate: a high-resolution immunofluorescence study. Glia. 2005;49:96–106. doi: 10.1002/glia.20094. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci. 1998;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Bak LK, Schousboe A, Sonnewald U, Waagepetersen HS. Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J Cereb Blood Flow Metab. 2006;26:1285–1297. doi: 10.1038/sj.jcbfm.9600281. [DOI] [PubMed] [Google Scholar]
- Bal-Price A, Moneer Z, Brown GC. Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia. 2002;40:312–323. doi: 10.1002/glia.10124. [DOI] [PubMed] [Google Scholar]
- Barkho BZ, Song H, Aimone JB, Smrt RD, Kuwabara T, Nakashima K, Gage FH, Zhao X. Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cells Dev. 2006;15:407–421. doi: 10.1089/scd.2006.15.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Beck A, Nieden RZ, Schneider HP, Deitmer JW. Calcium release from intracellular stores in rodent astrocytes and neurons in situ. Cell Calcium. 2004;35:47–58. doi: 10.1016/s0143-4160(03)00171-4. [DOI] [PubMed] [Google Scholar]
- Bennay M, Langer J, Meier SD, Kafitz KW, Rose CR. Sodium signals in cerebellar Purkinje neurons and Bergmann glial cells evoked by glutamatergic synaptic transmission. Glia. 2008;56:1138–1149. doi: 10.1002/glia.20685. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhauser C, Pilati E, Volterra A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci. 2004;7:613–620. doi: 10.1038/nn1246. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Bouzier-Sore AK, Voisin P, Canioni P, Magistretti PJ, Pellerin L. Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J Cereb Blood Flow Metab. 2003;23:1298–1306. doi: 10.1097/01.WCB.0000091761.61714.25. [DOI] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. Two forms of single-vesicle astrocyte exocytosis imaged with total internal reflection fluorescence microscopy. Proc Natl Acad Sci U S A. 2007;104:4212–4217. doi: 10.1073/pnas.0607625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Sickmann HM, Fosgerau K, Lund TM, Schousboe A, Waagepetersen HS, Ransom BR. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J Neurosci Res. 2005;79:74–80. doi: 10.1002/jnr.20335. [DOI] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP, Mori T, Gotz M. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008;105:3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Chao TI, Rickmann M, Wolff JR. The synapse-astrocyte boundary: anatomical basis for an integrative role of glia in synaptic transmission. In: Volterra A, Magistretti P, Haydon PG, editors. Tripartite synapses: Synaptic transmission with glia. Oxford University Press; Oxford-New York: 2002. pp. 3–23. [Google Scholar]
- Cho KS, Yang L, Lu B, Feng Ma H, Huang X, Pekny M, Chen DF. Reestablishing the regenerative potential of central nervous system axons in postnatal mice. J Cell Sci. 2005;118:863–872. doi: 10.1242/jcs.01658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres BA, Prince DA. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A. 2010;107:7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicardi M, Banerji A, Bracho F, Malbran A, Rosenkranz B, Riedl M, Bork K, Lumry W, Aberer W, Bier H, Bas M, Greve J, Hoffmann TK, Farkas H, Reshef A, Ritchie B, Yang W, Grabbe J, Kivity S, Kreuz W, Levy RJ, Luger T, Obtulowicz K, Schmid-Grendelmeier P, Bull C, Sitkauskiene B, Smith WB, Toubi E, Werner S, Anne S, Bjorkander J, Bouillet L, Cillari E, Hurewitz D, Jacobson KW, Katelaris CH, Maurer M, Merk H, Bernstein JA, Feighery C, Floccard B, Gleich G, Hebert J, Kaatz M, Keith P, Kirkpatrick CH, Langton D, Martin L, Pichler C, Resnick D, Wombolt D, Fernandez Romero DS, Zanichelli A, Arcoleo F, Knolle J, Kravec I, Dong L, Zimmermann J, Rosen K, Fan WT. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. 2010;363:532–541. doi: 10.1056/NEJMoa0906393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colucci-Guyon E, Portier MM, Dunia I, Paulin D, Pournin S, Babinet C. Mice lacking vimentin develop and reproduce without an obvious phenotype. Cell. 1994;79:679–694. doi: 10.1016/0092-8674(94)90553-3. [DOI] [PubMed] [Google Scholar]
- Condorelli DF, Conti F, Gallo V, Kirchhoff F, Seifert G, Steinhauser C, Verkhratsky A, Yuan X. Expression and functional analysis of glutamate receptors in glial cells. Adv Exp Med Biol. 1999;468:49–67. doi: 10.1007/978-1-4615-4685-6_5. [DOI] [PubMed] [Google Scholar]
- Conti F, Minelli A, Brecha NC. Cellular localization and laminar distribution of AMPA glutamate receptor subunits mRNAs and proteins in the rat cerebral cortex. J Comp Neurol. 1994;350:241–259. doi: 10.1002/cne.903500208. [DOI] [PubMed] [Google Scholar]
- Conti F, DeBiasi S, Minelli A, Melone M. Expression of NR1 and NR2A/B subunits of the NMDA receptor in cortical astrocytes. Glia. 1996;17:254–258. doi: 10.1002/(SICI)1098-1136(199607)17:3<254::AID-GLIA7>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Dai H, Shin OH, Machius M, Tomchick DR, Sudhof TC, Rizo J. Structural basis for the evolutionary inactivation of Ca2+ binding to synaptotagmin 4. Nat Struct Mol Biol. 2004;11:844–849. doi: 10.1038/nsmb817. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Progr Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Deitmer JW, Rose CR. Ion changes and signalling in perisynaptic glia. Brain Res Rev. 2010;63:113–129. doi: 10.1016/j.brainresrev.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Spray DC. Blood-brain barrier and the neural vascular unit. In: Scemes E, Spray DC, editors. Astrocytes: Wiring the brain. CRC Press; Boca Raton, FL: 2012. pp. 179–203. [Google Scholar]
- Dienel GA, Ball KK, Cruz NF. A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: implications for glycogen turnover. J Neurochem. 2007;102:466–478. doi: 10.1111/j.1471-4159.2007.04595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson C, Sahlgren C, Berthold CH, Stakeberg J, Celis JE, Betsholtz C, Eriksson JE, Pekny M. Intermediate filament protein partnership in astrocytes. J Biol Chem. 1999;274:23996–24006. doi: 10.1074/jbc.274.34.23996. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J, McCarthy KD. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatorysynaptic activity. Neuron. 2007;54:611–626. doi: 10.1016/j.neuron.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci. 2006;7:423–436. doi: 10.1038/nrn1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossat P, Turpin FR, Sacchi S, Dulong J, Shi T, Rivet JM, Sweedler JV, Pollegioni L, Millan MJ, Oliet SH, Mothet JP. Glial D-Serine Gates NMDA Receptors at Excitatory Synapses in Prefrontal Cortex. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr130. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH. The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci U S A. 2002;99:14488–14493. doi: 10.1073/pnas.222546799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadea A, Lopez-Colome AM. Glial transporters for glutamate, glycine and GABA I. Glutamate transporters. J Neurosci Res. 2001;63:453–460. doi: 10.1002/jnr.1039. [DOI] [PubMed] [Google Scholar]
- Gallo V, Ghiani CA. Glutamate receptors in glia: new cells, new inputs and new functions. Trends Pharmacol Sci. 2000;21:252–258. doi: 10.1016/s0165-6147(00)01494-2. [DOI] [PubMed] [Google Scholar]
- Gandhi GK, Cruz NF, Ball KK, Dienel GA. Astrocytes are poised for lactate trafficking and release from activated brain and for supply of glucose to neurons. J Neurochem. 2009;111:522–536. doi: 10.1111/j.1471-4159.2009.06333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs ME, Anderson DG, Hertz L. Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia. 2006;54:214–222. doi: 10.1002/glia.20377. [DOI] [PubMed] [Google Scholar]
- Goldman WF, Yarowsky PJ, Juhaszova M, Krueger BK, Blaustein MP. Sodium/calcium exchange in rat cortical astrocytes. J Neurosci. 1994;14:5834–5843. doi: 10.1523/JNEUROSCI.14-10-05834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golgi C. Suella struttura della sostanza grigia del cervello (comunicazione preventiva) Gazzetta Medica Italiana, Lombardia. 1873;33:244–246. [Google Scholar]
- Golgi C. Opera Omnia. Hoepli; Milano: 1903. [Google Scholar]
- Golovina VA. Visualization of localized store-operated calcium entry in mouse astrocytes. Close proximity to the endoplasmic reticulum. J Physiol. 2005;564:737–749. doi: 10.1113/jphysiol.2005.085035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GR, Baimoukhametova DV, Hewitt SA, Rajapaksha WR, Fisher TE, Bains JS. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat Neurosci. 2005;8:1078–1086. doi: 10.1038/nn1498. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia. 2002;40:252–259. doi: 10.1002/glia.10147. [DOI] [PubMed] [Google Scholar]
- Griffin LD, Gelb BD, Adams V, McCabe ER. Developmental expression of hexokinase 1 in the rat. Biochim Biophys Acta. 1992;1129:309–317. doi: 10.1016/0167-4781(92)90508-w. [DOI] [PubMed] [Google Scholar]
- Grosche J, Matyash V, Moller T, Verkhratsky A, Reichenbach A, Kettenmann H. Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci. 1999;2:139–143. doi: 10.1038/5692. [DOI] [PubMed] [Google Scholar]
- Habbas S, Ango F, Daniel H, Galante M. Purinergic signaling in the cerebellum: Bergmann glial cells express functional ionotropic P2X7 receptors. Glia. 2011;59:1800–1812. doi: 10.1002/glia.21224. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. Tripartite synapses: roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology. 2009;57:343–346. doi: 10.1016/j.neuropharm.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton GI. Glial-neuronal interactions in the mammalian brain. Adv Physiol Educ. 2002;26:225–237. doi: 10.1152/advan.00038.2002. [DOI] [PubMed] [Google Scholar]
- Heja L, Barabas P, Nyitrai G, Kekesi KA, Lasztoczi B, Toke O, Tarkanyi G, Madsen K, Schousboe A, Dobolyi A, Palkovits M, Kardos J. Glutamate uptake triggers transporter-mediated GABA release from astrocytes. PLoS One. 2009;4:e7153. doi: 10.1371/journal.pone.0007153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer’s disease. J Neuroimmunol. 2007;184:69–91. doi: 10.1016/j.jneuroim.2006.11.017. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Wiesinger H, Dumitrescu-Ozimek L, Riederer P, Feinstein DL, Klockgether T. Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:906–916. doi: 10.1093/jnen/60.9.906. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Dewachter I, Walter J, Klockgether T, Van Leuven F. Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation. 2005;2:22. doi: 10.1186/1742-2094-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of D-serine from astrocytes. Nature. 2010;463:232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L, Zielke HR. Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends Neurosci. 2004;27:735–743. doi: 10.1016/j.tins.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Hertz L, Peng L, Dienel GA. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab. 2007;27:219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- Hertz L, Dringen R, Schousboe A, Robinson SR. Astrocytes: glutamate producers for neurons. J Neurosci Res. 1999;57:417–428. [PubMed] [Google Scholar]
- Hirrlinger J, Hulsmann S, Kirchhoff F. Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur J Neurosci. 2004;20:2235–2239. doi: 10.1111/j.1460-9568.2004.03689.x. [DOI] [PubMed] [Google Scholar]
- Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99:1583–1593. doi: 10.1093/jnci/djm187. [DOI] [PubMed] [Google Scholar]
- Hsieh HL, Wu CY, Yang CM. Bradykinin induces matrix metalloproteinase-9 expression and cell migration through a PKC-delta-dependent ERK/Elk-1 pathway in astrocytes. Glia. 2008;56:619–632. doi: 10.1002/glia.20637. [DOI] [PubMed] [Google Scholar]
- Hua X, Malarkey EB, Sunjara V, Rosenwald SE, Li WH, Parpura V. Ca2+-dependent glutamate release involves two classes of endoplasmic reticulum Ca2+ stores in astrocytes. J Neurosci Res. 2004;76:86–97. doi: 10.1002/jnr.20061. [DOI] [PubMed] [Google Scholar]
- Ifuku M, Farber K, Okuno Y, Yamakawa Y, Miyamoto T, Nolte C, Merrino VF, Kita S, Iwamoto T, Komuro I, Wang B, Cheung G, Ishikawa E, Ooboshi H, Bader M, Wada K, Kettenmann H, Noda M. Bradykinin-induced microglial migration mediated by B1-bradykinin receptors depends on Ca2+ influx via reverse-mode activity of the Na+/Ca2+ exchanger. J Neurosci. 2007;27:13065–13073. doi: 10.1523/JNEUROSCI.3467-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti B, Parpura V, Haydon PG. Imaging extracellular waves of glutamate during calcium signaling in cultured astrocytes. J Neurosci. 2000;20:1800–1808. doi: 10.1523/JNEUROSCI.20-05-01800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci. 2007;10:331–339. doi: 10.1038/nn1849. [DOI] [PubMed] [Google Scholar]
- Kartvelishvily E, Shleper M, Balan L, Dumin E, Wolosker H. Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. J Biol Chem. 2006;281:14151–14162. doi: 10.1074/jbc.M512927200. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK. The problem of astrocyte identity. Neurochem Int. 2004;45:191–202. doi: 10.1016/j.neuint.2003.08.015. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Pang S, Treble DH. Excitatory amino acid-stimulated uptake of 22Na+ in primary astrocyte cultures. J Neurosci. 1989;9:1141–1149. doi: 10.1523/JNEUROSCI.09-04-01141.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinouchi R, Takeda M, Yang L, Wilhelmsson U, Lundkvist A, Pekny M, Chen DF. Robust neural integration from retinal transplants in mice deficient in GFAP and vimentin. Nat Neurosci. 2003;6:863–868. doi: 10.1038/nn1088. [DOI] [PubMed] [Google Scholar]
- Kirchhoff F, Mulhardt C, Pastor A, Becker CM, Kettenmann H. Expression of glycine receptor subunits in glial cells of the rat spinal cord. J Neurochem. 1996;66:1383–1390. doi: 10.1046/j.1471-4159.1996.66041383.x. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Kettenmann H, Verkhratsky A. Na+/Ca2+ exchanger modulates kainate-triggered Ca2+ signaling in Bergmann glial cells in situ. FASEB J. 1997;11:566–572. doi: 10.1096/fasebj.11.7.9212080. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Kettenmann H, Verkhratsky A. Membrane currents and cytoplasmic sodium transients generated by glutamate transport in Bergmann glial cells. Pflugers Arch. 2007;454:245–252. doi: 10.1007/s00424-007-0207-5. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Tuschick S, Verkhratsky A, Kettenmann H. Calcium signalling in mouse Bergmann glial cells mediated by α1-adrenoreceptors and H1 histamine receptors. Eur J Neurosci. 1996a;8:1198–1208. doi: 10.1111/j.1460-9568.1996.tb01288.x. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Moller T, Voitenko N, Kettenmann H, Verkhratsky A. ATP-induced cytoplasmic calcium mobilization in Bergmann glial cells. J Neurosci. 1995;15:7861–7871. doi: 10.1523/JNEUROSCI.15-12-07861.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk S, Kirchhoff F, Matyash V, Kettenmann H, Verkhratsky A. Glutamate-triggered calcium signalling in mouse Bergmann glial cells in situ: role of inositol-1,4,5-trisphosphate-mediated intracellular calcium release. Neuroscience. 1999;92:1051–1059. doi: 10.1016/s0306-4522(99)00067-6. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Matiash V, Kulik A, Voitenko N, Kostyuk P, Verkhratsky A. Activation of P2-purino-, α1-adreno and H1-histamine receptors triggers cytoplasmic calcium signalling in cerebellar Purkinje neurons. Neuroscience. 1996b;73:643–647. doi: 10.1016/0306-4522(96)00205-9. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med. 2004;10:719–726. doi: 10.1038/nm1058. [DOI] [PubMed] [Google Scholar]
- Kölliker A. Handbuch der Gewebelehre des Menschen. Wilhelm Engelmann; Leipzig: 1889. [Google Scholar]
- Kreft M, Stenovec M, Rupnik M, Grilc S, Krzan M, Potokar M, Pangrsic T, Haydon PG, Zorec R. Properties of Ca(2+)-dependent exocytosis in cultured astrocytes. Glia. 2004;46:437–445. doi: 10.1002/glia.20018. [DOI] [PubMed] [Google Scholar]
- Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, Konig S, Roeber S, Jessen F, Klockgether T, Korte M, Heneka MT. Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron. 2011;71:833–844. doi: 10.1016/j.neuron.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Parpura V, Verkhratsky A. Ionotropic receptors in neuronal-astroglial signalling: what is the role of “excitable” molecules in non-excitable cells. Biochim Biophys Acta. 2011a;1813:992–1002. doi: 10.1016/j.bbamcr.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Kirchhoff F, North RA, Verkhratsky A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci. 2006;26:2673–2683. doi: 10.1523/JNEUROSCI.4689-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, Palygin O, North RA, Verkhratsky A, Pankratov Y. Age-dependent remodelling of ionotropic signalling in cortical astroglia. Aging Cell. 2011b;10:392–402. doi: 10.1111/j.1474-9726.2011.00682.x. [DOI] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Wichert SP, Rossner MJ, North RA, Kirchhoff F, Verkhratsky A. P2X1 and P2X5 subunits form the functional P2X receptor in mouse cortical astrocytes. J Neurosci. 2008;28:5473–5480. doi: 10.1523/JNEUROSCI.1149-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer J, Rose CR. Synaptically induced sodium signals in hippocampal astrocytes in situ. J Physiol. 2009;587:5859–5877. doi: 10.1113/jphysiol.2009.182279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer J, Stephan J, Theis M, Rose CR. Gap junctions mediate intercellular spread of sodium between hippocampal astrocytes in situ. Glia. 2011;60:239–252. doi: 10.1002/glia.21259. [DOI] [PubMed] [Google Scholar]
- Laywell ED, Rakic P, Kukekov VG, Holland EC, Steindler DA. Identification of a multipotent astrocytic stem cell in the immature and adult mouse brain. Proc Natl Acad Sci U S A. 2000;97:13883–13888. doi: 10.1073/pnas.250471697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhossek Mv. Zur Kenntnis der Neuroglia des menschlichen Ruckenmarkes. Verh Anat Ges. 1891;5:193–221. [Google Scholar]
- Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28:1722–1732. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Liebner S, Czupalla CJ, Wolburg H. Current concepts of blood-brain barrier development. Int J Dev Biol. 2011;55:467–476. doi: 10.1387/ijdb.103224sl. [DOI] [PubMed] [Google Scholar]
- Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, Goldman SA, Nedergaard M. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci. 2007;27:12255–12266. doi: 10.1523/JNEUROSCI.3404-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch RM, Fogarty KE, Fay FS. Modulation of hexokinase association with mitochondria analyzed with quantitative three-dimensional confocal microscopy. J Cell Biol. 1991;112:385–395. doi: 10.1083/jcb.112.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67:9463–9471. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006;209:2304–2311. doi: 10.1242/jeb.02208. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ. Role of glutamate in neuron-glia metabolic coupling. Am J Clin Nutr. 2009 doi: 10.3945/ajcn.2009.27462CC. [DOI] [PubMed] [Google Scholar]
- Malarkey EB, Parpura V. Temporal characteristics of vesicular fusion in astrocytes: examination of synaptobrevin 2-laden vesicles at single vesicle resolution. J Physiol. 2011;589:4271–4300. doi: 10.1113/jphysiol.2011.210435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkey EB, Ni Y, Parpura V. Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia. 2008;56:821–835. doi: 10.1002/glia.20656. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Manzoni OJ, Manabe T, Nicoll RA. Release of adenosine by activation of NMDA receptors in the hippocampus. Science. 1994;265:2098–2101. doi: 10.1126/science.7916485. [DOI] [PubMed] [Google Scholar]
- Marchaland J, Cali C, Voglmaier SM, Li H, Regazzi R, Edwards RH, Bezzi P. Fast subplasma membrane Ca2+ transients control exo-endocytosis of synaptic-like microvesicles in astrocytes. J Neurosci. 2008;28:9122–9132. doi: 10.1523/JNEUROSCI.0040-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic DS, Glass R, Synowitz M, Rooijen N, Kettenmann H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J Neuropathol Exp Neurol. 2005;64:754–762. doi: 10.1097/01.jnen.0000178445.33972.a9. [DOI] [PubMed] [Google Scholar]
- Markovic DS, Vinnakota K, van Rooijen N, Kiwit J, Synowitz M, Glass R, Kettenmann H. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain Behav Immun. 2011;25:624–628. doi: 10.1016/j.bbi.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, Sliwa M, Lehmann S, Kalin R, van Rooijen N, Holmbeck K, Heppner FL, Kiwit J, Matyash V, Lehnardt S, Kaminska B, Glass R, Kettenmann H. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A. 2009;106:12530–12535. doi: 10.1073/pnas.0804273106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martineau M, Galli T, Baux G, Mothet JP. Confocal imaging and tracking of the exocytotic routes for D-serine-mediated gliotransmission. Glia. 2008;56:1271–1284. doi: 10.1002/glia.20696. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Takuma K, Nishiguchi E, Hashimoto H, Azuma J, Baba A. Involvement of Na+-Ca2+ exchanger in reperfusion-induced delayed cell death of cultured rat astrocytes. Eur J Neurosci. 1996;8:951–958. doi: 10.1111/j.1460-9568.1996.tb01582.x. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna MC, Dienel GA, Sonnewald U, Waagepetersen HS, Schousboe A. Energy metabolism in the brain. In: Siegel GJ, Albers RW, Brady ST, Price DL, editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 8. Elsevier-Academic Press; London: 2012. pp. 223–258. [Google Scholar]
- Menet V, Prieto M, Privat A, Gimenez y Ribotta M. Axonal plasticity and functional recovery after spinal cord injury in mice deficient in both glial fibrillary acidic protein and vimentin genes. Proc Natl Acad Sci U S A. 2003;100:8999–9004. doi: 10.1073/pnas.1533187100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minelli A, Castaldo P, Gobbi P, Salucci S, Magi S, Amoroso S. Cellular and subcellular localization of Na+-Ca2+ exchanger protein isoforms, NCX1, NCX2, and NCX3 in cerebral cortex and hippocampus of adult rat. Cell Calcium. 2007;41:221–234. doi: 10.1016/j.ceca.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Miyazaki I, Asanuma M, Diaz-Corrales FJ, Miyoshi K, Ogawa N. Direct evidence for expression of dopamine receptors in astrocytes from basal ganglia. Brain Res. 2004;1029:120–123. doi: 10.1016/j.brainres.2004.09.014. [DOI] [PubMed] [Google Scholar]
- Montana V, Sontheimer H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J Neurosci. 2011;31:4858–4867. doi: 10.1523/JNEUROSCI.3825-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana V, Ni Y, Sunjara V, Hua X, Parpura V. Vesicular glutamate transporter-dependent glutamate release from astrocytes. J Neurosci. 2004;24:2633–2642. doi: 10.1523/JNEUROSCI.3770-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana V, Malarkey EB, Verderio C, Matteoli M, Parpura V. Vesicular transmitter release from astrocytes. Glia. 2006;54:700–715. doi: 10.1002/glia.20367. [DOI] [PubMed] [Google Scholar]
- Mothet JP, Pollegioni L, Ouanounou G, Martineau M, Fossier P, Baux G. Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc Natl Acad Sci U S A. 2005;102:5606–5611. doi: 10.1073/pnas.0408483102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Parent AT, Wolosker H, Brady RO, Jr, Linden DJ, Ferris CD, Rogawski MA, Snyder SH. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Schiffmann A, Andreyeva A, Horn AH, Gottmann K, Korth C, Sticht H. Molecular Engineering of a Secreted, Highly Homogeneous, and Neurotoxic Aβ Dimer. ACS Chem Neurosci. 2011;2:242–248. doi: 10.1021/cn200011h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CA, Lynch MA. Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci. 1998;18:2974–2981. doi: 10.1523/JNEUROSCI.18-08-02974.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsu T, Sawada M. Inflammatory process in Parkinson’s disease: role for cytokines. Curr Pharm Des. 2005;11:999–1016. doi: 10.2174/1381612053381620. [DOI] [PubMed] [Google Scholar]
- Nagler K, Mauch DH, Pfrieger FW. Glia-derived signals induce synapse formation in neurones of the rat central nervous system. J Physiol. 2001;533:665–679. doi: 10.1111/j.1469-7793.2001.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Dudek FE, Rash JE. Update on connexins and gap junctions in neurons and glia in the mammalian nervous system. Brain Res Brain Res Rev. 2004;47:191–215. doi: 10.1016/j.brainresrev.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Navarrete M, Araque A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron. 2010;68:113–126. doi: 10.1016/j.neuron.2010.08.043. [DOI] [PubMed] [Google Scholar]
- Newman EA. New roles for astrocytes: regulation of synaptic transmission. Trends Neurosci. 2003;26:536–542. doi: 10.1016/S0166-2236(03)00237-6. [DOI] [PubMed] [Google Scholar]
- Ni Y, Parpura V. Dual regulation of Ca2+-dependent glutamate release from astrocytes: vesicular glutamate transporters and cytosolic glutamate levels. Glia. 2009;57:1296–1305. doi: 10.1002/glia.20849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y, Malarkey EB, Parpura V. Vesicular release of glutamate mediates bidirectional signaling between astrocytes and neurons. J Neurochem. 2007;103:1273–1284. doi: 10.1111/j.1471-4159.2007.04864.x. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Wang X, Goldman S, Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006;29:547–553. doi: 10.1016/j.tins.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia. 2010;58:831–838. doi: 10.1002/glia.20967. [DOI] [PubMed] [Google Scholar]
- Oliveira JF, Riedel T, Leichsenring A, Heine C, Franke H, Krugel U, Norenberg W, Illes P. Rodent cortical astroglia express in situ functional P2X7 receptors sensing pathologically high ATP concentrations. Cereb Cortex. 2011;21:806–820. doi: 10.1093/cercor/bhq154. [DOI] [PubMed] [Google Scholar]
- Paco S, Margeli MA, Olkkonen VM, Imai A, Blasi J, Fischer-Colbrie R, Aguado F. Regulation of exocytotic protein expression and Ca2+-dependent peptide secretion in astrocytes. J Neurochem. 2009;110:143–156. doi: 10.1111/j.1471-4159.2009.06116.x. [DOI] [PubMed] [Google Scholar]
- Paluzzi S, Alloisio S, Zappettini S, Milanese M, Raiteri L, Nobile M, Bonanno G. Adult astroglia is competent for Na+/Ca2+ exchanger-operated exocytotic glutamate release triggered by mild depolarization. J Neurochem. 2007;103:1196–1207. doi: 10.1111/j.1471-4159.2007.04826.x. [DOI] [PubMed] [Google Scholar]
- Palygin O, Lalo U, Pankratov Y. Distinct pharmacological and functional properties of NMDA receptors in mouse cortical astrocytes. Br J Pharmacol. 2011;163:1755–1766. doi: 10.1111/j.1476-5381.2011.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palygin O, Lalo U, Verkhratsky A, Pankratov Y. Ionotropic NMDA and P2X1/5 receptors mediate synaptically induced Ca2+ signalling in cortical astrocytes. Cell Calcium. 2010;48:225–231. doi: 10.1016/j.ceca.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Pangrsic T, Potokar M, Stenovec M, Kreft M, Fabbretti E, Nistri A, Pryazhnikov E, Khiroug L, Giniatullin R, Zorec R. Exocytotic release of ATP from cultured astrocytes. J Biol Chem. 2007;282:28749–28758. doi: 10.1074/jbc.M700290200. [DOI] [PubMed] [Google Scholar]
- Parpura V, Zorec R. Gliotransmission: Exocytotic release from astrocytes. Brain Res Rev. 2010;63:83–92. doi: 10.1016/j.brainresrev.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Baker BJ, Jeras M, Zorec R. Regulated exocytosis in astrocytic signal integration. Neurochem Int. 2010;57:451–459. doi: 10.1016/j.neuint.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M. Astrocyte intermediate filaments in CNS pathologies and regeneration. J Pathol. 2004;204:428–437. doi: 10.1002/path.1645. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Pekny M, Wilhelmsson U. GFAP and astrocyte intermediate filaments. In: Lajtha A, editor. Handbook of neurochemistry and molecular neurobiology: neuroactive proteins and peptides. Springer; Berlin: 2006. pp. 289–314. [Google Scholar]
- Pekny M, Lane EB. Intermediate filaments and stress. Exp Cell Res. 2007;313:2244–2254. doi: 10.1016/j.yexcr.2007.04.023. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M, Wilhelmsson U, Chen DF. Astroglial cells sitting at the controls? Trends Neurosci. 2004;27:243–244. doi: 10.1016/j.tins.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Pekny M, Leveen P, Pekna M, Eliasson C, Berthold CH, Westermark B, Betsholtz C. Mice lacking glial fibrillary acidic protein display astrocytes devoid of intermediate filaments but develop and reproduce normally. Embo J. 1995;14:1590–1598. doi: 10.1002/j.1460-2075.1995.tb07147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekny M, Johansson CB, Eliasson C, Stakeberg J, Wallen A, Perlmann T, Lendahl U, Betsholtz C, Berthold CH, Frisen J. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol. 1999;145:503–514. doi: 10.1083/jcb.145.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Astrocytes potentiate transmitter release at single hippocampal synapses. Science. 2007;317:1083–1086. doi: 10.1126/science.1144640. [DOI] [PubMed] [Google Scholar]
- Pihlaja R, Koistinaho J, Kauppinen R, Sandholm J, Tanila H, Koistinaho M. Multiple cellular and molecular mechanisms are involved in human Abeta clearance by transplanted adult astrocytes. Glia. 2011;59:1643–1657. doi: 10.1002/glia.21212. [DOI] [PubMed] [Google Scholar]
- Prebil M, Vardjan N, Jensen J, Zorec R, Kreft M. Dynamic monitoring of cytosolic glucose in single astrocytes. Glia. 2011;59:903–913. doi: 10.1002/glia.21161. [DOI] [PubMed] [Google Scholar]
- Raidoo DM, Sawant S, Mahabeer R, Bhoola KD. Kinin receptors are expressed in human astrocytic tumour cells. Immunopharmacology. 1999;43:255–263. doi: 10.1016/s0162-3109(99)00097-1. [DOI] [PubMed] [Google Scholar]
- Ramón y Cajal S. Histologie du système nerveux de l’homme et des vertébrés; Maloine, Paris. 1911. [Google Scholar]
- Ramón y Cajal S. Sobre un nuevo proceder de impregnacion de la neuroglia y sus resultados en los centros nerviosos del hombre y animales. Trab Lab Invest Biol Univ Madrid. 1913;11:219–237. [Google Scholar]
- Ransom B, Behar T, Nedergaard M. New roles for astrocytes (stars at last) Trends Neurosci. 2003;26:520–522. doi: 10.1016/j.tins.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Fern R. Does astrocytic glycogen benefit axon function and survival in CNS white matter during glucose deprivation? Glia. 1997;21:134–141. [PubMed] [Google Scholar]
- Reichenbach A. Attempt to classify glial cells by means of their process specialization using the rabbit retinal Muller cell as an example of cytotopographic specialization of glial cells. Glia. 1989;2:250–259. doi: 10.1002/glia.440020406. [DOI] [PubMed] [Google Scholar]
- Reyes RC, Parpura V. Mitochondria modulate Ca2+-dependent glutamate release from rat cortical astrocytes. J Neurosci. 2008;28:9682–9691. doi: 10.1523/JNEUROSCI.3484-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes RC, Parpura V. Modulation of Ca2+-Dependent Glutamate Release in Rat Cortical Astrocytes by the Na+/Ca2+ Exchanger. J Neurochem. 2009;108:116–117. [Google Scholar]
- Reyes RC, Perry G, Lesort M, Parpura V. Immunophilin deficiency augments Ca2+-dependent glutamate release from mouse cortical astrocytes. Cell Calcium. 2011;49:23–34. doi: 10.1016/j.ceca.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas H, Ramos M, Benaim G, Caputo C, DiPolo R. The activity of the Na+/Ca2+ exchanger largely modulates the Ca2+i signal induced by hypo-osmotic stress in rat cerebellar astrocytes. The effect of osmolarity on exchange activity. J Physiol Sci. 2008;58:277–279. doi: 10.2170/physiolsci.RP009208. [DOI] [PubMed] [Google Scholar]
- Rojas H, Colina C, Ramos M, Benaim G, Jaffe EH, Caputo C, DiPolo R. Na+ entry via glutamate transporter activates the reverse Na+/Ca2+ exchange and triggers Cai2+-induced Ca2+ release in rat cerebellar Type-1 astrocytes. J Neurochem. 2007;100:1188–1202. doi: 10.1111/j.1471-4159.2006.04303.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg D, Kartvelishvily E, Shleper M, Klinker CM, Bowser MT, Wolosker H. Neuronal release of D-serine: a physiological pathway controlling extracellular D-serine concentration. Faseb J. 2010;24:2951–2961. doi: 10.1096/fj.09-147967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–1555. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23:9796–9804. doi: 10.1523/JNEUROSCI.23-30-09796.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A. 2006;103:443–448. doi: 10.1073/pnas.0503839103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, Omote H, Yamamoto A, Moriyama Y. Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci U S A. 2008;105:5683–5686. doi: 10.1073/pnas.0800141105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scemes E, Suadicani SO, Dahl G, Spray DC. Connexin and pannexin mediated cell-cell communication. Neuron Glia Biol. 2007;3:199–208. doi: 10.1017/S1740925X08000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia. 2008;56:1578–1589. doi: 10.1002/glia.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipke CG, Kettenmann H. Astrocyte responses to neuronal activity. Glia. 2004;47:226–232. doi: 10.1002/glia.20029. [DOI] [PubMed] [Google Scholar]
- Schipke CG, Haas B, Kettenmann H. Astrocytes discriminate and selectively respond to the activity of a subpopulation of neurons within the barrel cortex. Cereb Cortex. 2008;18:2450–2459. doi: 10.1093/cercor/bhn009. [DOI] [PubMed] [Google Scholar]
- Schousboe A, Sickmann HM, Walls AB, Bak LK, Waagepetersen HS. Functional importance of the astrocytic glycogen-shunt and glycolysis for maintenance of an intact intra/extracellular glutamate gradient. Neurotox Res. 2010;18:94–99. doi: 10.1007/s12640-010-9171-5. [DOI] [PubMed] [Google Scholar]
- Schousboe A, Sickmann HM, Bak LK, Schousboe I, Jajo FS, Faek SA, Waagepetersen HS. Neuron-glia interactions in glutamatergic neurotransmission: roles of oxidative and glycolytic adenosine triphosphate as energy source. J Neurosci Res. 2011;89:1926–1934. doi: 10.1002/jnr.22746. [DOI] [PubMed] [Google Scholar]
- Seifert G, Steinhauser C. Ionotropic glutamate receptors in astrocytes. Prog Brain Res. 2001;132:287–299. doi: 10.1016/S0079-6123(01)32083-6. [DOI] [PubMed] [Google Scholar]
- Seifert G, Steinhauser C. Neuron-astrocyte signaling and epilepsy. Exp Neurol. 2011 doi: 10.1016/j.expneurol.2011.08.024. [DOI] [PubMed] [Google Scholar]
- Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A. Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci. 2001;21:7153–7160. doi: 10.1523/JNEUROSCI.21-18-07153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26:5370–5382. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman RG, Hyder F, Rothman DL. Cerebral energetics and the glycogen shunt: neurochemical basis of functional imaging. Proc Natl Acad Sci U S A. 2001;98:6417–6422. doi: 10.1073/pnas.101129298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann HM, Walls AB, Schousboe A, Bouman SD, Waagepetersen HS. Functional significance of brain glycogen in sustaining glutamatergic neurotransmission. J Neurochem. 2009;109(Suppl 1):80–86. doi: 10.1111/j.1471-4159.2009.05915.x. [DOI] [PubMed] [Google Scholar]
- Siebzehnrubl FA, Reynolds BA, Vescovi A, Steindler DA, Deleyrolle LP. The origins of glioma: E Pluribus Unum? Glia. 2011;59:1135–1147. doi: 10.1002/glia.21143. [DOI] [PubMed] [Google Scholar]
- Song H, Stevens C, Gage FH. Astroglia induce neurogenesis from adult neural stem cells. Nature. 2002;417:39–44. doi: 10.1038/417039a. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Stylli SS, Kaye AH, Lock P. Invadopodia: at the cutting edge of tumour invasion. J Clin Neurosci. 2008;15:725–737. doi: 10.1016/j.jocn.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Suh SW, Bergher JP, Anderson CM, Treadway JL, Fosgerau K, Swanson RA. Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP-316,819 ([R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmet hyl)propyl]-1H-indole-2-carboxamide) J Pharmacol Exp Ther. 2007;321:45–50. doi: 10.1124/jpet.106.115550. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–823. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA. Physiologic coupling of glial glycogen metabolism to neuronal activity in brain. Can J Physiol Pharmacol. 1992;70(Suppl):S138–144. doi: 10.1139/y92-255. [DOI] [PubMed] [Google Scholar]
- Swanson RA, Choi DW. Glial glycogen stores affect neuronal survival during glucose deprivation in vitro. J Cereb Blood Flow Metab. 1993;13:162–169. doi: 10.1038/jcbfm.1993.19. [DOI] [PubMed] [Google Scholar]
- Takamori S. VGLUTs: ‘exciting’ times for glutamatergic research? Neurosci Res. 2006;55:343–351. doi: 10.1016/j.neures.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Takemura H, Putney JW., Jr Capacitative calcium entry in parotid acinar cells. The Biochemical journal. 1989;258:409–412. doi: 10.1042/bj2580409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tancredi V, Zona C, Velotti F, Eusebi F, Santoni A. Interleukin-2 suppresses established long-term potentiation and inhibits its induction in the rat hippocampus. Brain Res. 1990;525:149–151. doi: 10.1016/0006-8993(90)91331-a. [DOI] [PubMed] [Google Scholar]
- Tancredi V, D’Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett. 1992;146:176–178. doi: 10.1016/0304-3940(92)90071-e. [DOI] [PubMed] [Google Scholar]
- Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson JA, Holtzman DM, Heneka MT. Critical role of astroglial apolipoprotein E and liver X receptor-alpha expression for microglial Abeta phagocytosis. J Neurosci. 2011;31:7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Sohl G, Eiberger J, Willecke K. Emerging complexities in identity and function of glial connexins. Trends Neurosci. 2005;28:188–195. doi: 10.1016/j.tins.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Theodosis DT, Poulain DA, Oliet SH. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol Rev. 2008;88:983–1008. doi: 10.1152/physrev.00036.2007. [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–6906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Matteoli M. ATP mediates calcium signaling between astrocytes and microglial cells: modulation by IFN-gamma. J Immunol. 2001;166:6383–6391. doi: 10.4049/jimmunol.166.10.6383. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. Physiology of neuronal-glial networking. Neurochem Int. 2011;57:332–343. doi: 10.1016/j.neuint.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Kirchhoff F. NMDA Receptors in Glia. Neuroscientist. 2007;13:28–37. doi: 10.1177/1073858406294270. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Orkand RK, Kettenmann H. Glial calcium: homeostasis and signaling function. Physiol Rev. 1998;78:99–141. doi: 10.1152/physrev.1998.78.1.99. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Parpura V, Rodriguez JJ. Where the thoughts dwell: the physiology of neuronal-glial “diffuse neural net”. Brain Res Rev. 2011;66:133–151. doi: 10.1016/j.brainresrev.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Virchow R. Die Cellularpathologie in ihrer Begründung auf physiologische and pathologische Gewebelehre. Zwanzig Vorlesungen gehalten während der Monate Februar, März und April 1858 im pathologischen Institut zu Berlin. 1. Hirschwald; Berlin: 1858. Aug, p. 440. [Google Scholar]
- Vogt C, Vogt O. Sitz und Wesen der Krankheiten im Lichte der topistischen Hirnforschung und des Variierens der Tiere. Erster Teil: Befunde der topistischen Hirnforschung als Beitrag zur Lehre vom Krankheitssitz. Journal für Psychologie und Neurologie. 1937;47:238–457. [Google Scholar]
- Waagepetersen HS, Westergaard N, Schousboe A. The effects of isofagomine, a potent glycogen phosphorylase inhibitor, on glycogen metabolism in cultured mouse cortical astrocytes. Neurochem Int. 2000;36:435–440. doi: 10.1016/s0197-0186(99)00146-1. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Sonnewald U, Schousboe A. Energy and amino acid neurotransmitter metabolism in astrocytes. In: Parpura V, PHH, editors. Astrocytes in (Patho)physiology of the Nervous System. Springer; Boston: 2009. pp. 177–199. [Google Scholar]
- Walls AB, Heimburger CM, Bouman SD, Schousboe A, Waagepetersen HS. Robust glycogen shunt activity in astrocytes: Effects of glutamatergic and adrenergic agents. Neuroscience. 2009;158:284–292. doi: 10.1016/j.neuroscience.2008.09.058. [DOI] [PubMed] [Google Scholar]
- Walls AB, Sickmann HM, Brown A, Bouman SD, Ransom B, Schousboe A, Waagepetersen HS. Characterization of 1,4-dideoxy-1,4-imino-d-arabinitol (DAB) as an inhibitor of brain glycogen shunt activity. J Neurochem. 2008;105:1462–1470. doi: 10.1111/j.1471-4159.2008.05250.x. [DOI] [PubMed] [Google Scholar]
- Wender R, Brown AM, Fern R, Swanson RA, Farrell K, Ransom BR. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J Neurosci. 2000;20:6804–6810. doi: 10.1523/JNEUROSCI.20-18-06804.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergaard N, Drejer J, Schousboe A, Sonnewald U. Evaluation of the importance of transamination versus deamination in astrocytic metabolism of [U-13C]glutamate. Glia. 1996;17:160–168. doi: 10.1002/(SICI)1098-1136(199606)17:2<160::AID-GLIA7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Widestrand A, Faijerson J, Wilhelmsson U, Smith PL, Li L, Sihlbom C, Eriksson PS, Pekny M. Increased neurogenesis and astrogenesis from neural progenitor cells grafted in the hippocampus of GFAP−/− Vim−/− mice. Stem Cells. 2007;25:2619–2627. doi: 10.1634/stemcells.2007-0122. [DOI] [PubMed] [Google Scholar]
- Wilhelm A, Volknandt W, Langer D, Nolte C, Kettenmann H, Zimmermann H. Localization of SNARE proteins and secretory organelle proteins in astrocytes in vitro and in situ. Neurosci Res. 2004;48:249–257. doi: 10.1016/j.neures.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE, Pekny M. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci. 2004;24:5016–5021. doi: 10.1523/JNEUROSCI.0820-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff JR, Stuke K, Missler M, Tytko H, Schwarz P, Rohlmann A, Chao TI. Autocellular coupling by gap junctions in cultured astrocytes: a new view on cellular autoregulation during process formation. Glia. 1998;24:121–140. doi: 10.1002/(sici)1098-1136(199809)24:1<121::aid-glia12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Wolosker H, Sheth KN, Takahashi M, Mothet JP, Brady RO, Jr, Ferris CD, Snyder SH. Purification of serine racemase: biosynthesis of the neuromodulator D-serine. Proc Natl Acad Sci U S A. 1999;96:721–725. doi: 10.1073/pnas.96.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, Poo M, Duan S. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc Natl Acad Sci U S A. 2003;100:15194–15199. doi: 10.1073/pnas.2431073100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagzag D, Esencay M, Mendez O, Yee H, Smirnova I, Huang Y, Chiriboga L, Lukyanov E, Liu M, Newcomb EW. Hypoxia- and vascular endothelial growth factor-induced stromal cell-derived factor-1alpha/CXCR4 expression in glioblastomas: one plausible explanation of Scherer’s structures. Am J Pathol. 2008;173:545–560. doi: 10.2353/ajpath.2008.071197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Fukuda M, Van Bockstaele E, Pascual O, Haydon PG. Synaptotagmin IV regulates glial glutamate release. Proc Natl Acad Sci U S A. 2004a;101:9441–9446. doi: 10.1073/pnas.0401960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Pangrsic T, Kreft M, Krzan M, Li N, Sul JY, Halassa M, Van Bockstaele E, Zorec R, Haydon PG. Fusion-related release of glutamate from astrocytes. J Biol Chem. 2004b;279:12724–12733. doi: 10.1074/jbc.M312845200. [DOI] [PubMed] [Google Scholar]