

Abstract
Multiple physiologic estrogens (estradiol, estriol, and estrone), as well as xenoestrogenic compounds (including alkylphenols and bisphenol A), can act via nongenomic signaling initiated by liganding of the plasma membrane estrogen receptor-α (mERα). We examined heterotrimeric G protein involvement leading to extracellular-regulated kinase (ERK) activation in GH3/B6/F10 rat anterior pituitary tumor cells that express abundant mERα, and smaller amounts of mERβ and GPR30. A combination of microarrays, immunoblots, and quantitative immunoassays demonstrated the expression of members of all α, β, and γ G protein classes in these cells. Use of selective inhibitors showed that the Gαi subtype was the primary initiator of downstream ERK signaling. Using antibodies against the GTP-bound form of Gα protein subtypes i and s, we showed that xenoestrogens (bisphenol A, nonylphenol) activated Gαi at 15-30 sec; all alkylphenols examined subsequently suppressed activation by 5 min. GTP-activation of Gαi for all estrogens was enhanced by irreversible cumulative binding to GTPγS. In contrast, Gαs was neither activated nor deactivated by these treatments with estrogens. ERα and Gαi co-localized outside nuclei and could be immuno-captured together. Interactions of ERα with Gαi and caveolin I were demonstrated by epitope proximity ligation assays. An ERα/β antagonist (ICI182780) and a selective disruptor of caveolar structures (nystatin) blocked estrogen-induced ERK activation.
Conclusions
Xenoestrogens, like physiologic estrogens, can evoke downstream kinase signaling involving selective interactions of ERα with Gαi and caveolin I, but with some different characteristics, which could explain their disruptive actions.
Keywords: membrane estrogen receptor, GPR30, nongenomic, GH3 cells, bisphenol A, alkylphenols
Introduction
The actions of the physiologic estrogen estradiol (E2) in high pM to nM concentration ranges are most often associated with reproductive function, and with the development of cancers in reproductive tissues. Other endogenous estrogenic compounds can be more prevalent during other life phases where they can have significant effects on tissue development, function, and disease states. Estrone (E1) is a significant estrogenic hormone contributor in both reproductive (~0.5-1 nM) and postmenopausal (150-200 pM) women and in men; estriol (E3) levels are much higher in pregnant women (~10-100 nM) than in nonpregnant women (<7 nM) [1], and changes in free E3 levels in pregnancy have been associated with complications of eclampsia [2] and the incidence of Down’s syndrome in offspring [3]. All three of these physiologic estrogens are also produced by aromatases in a number of nonreproductive tissues where their effects may extend beyond reproductive functions [4,5]. Therefore, loss or enhancement of these physiological estrogenic effects due to interference by xenoestrogenic compounds could affect human and animal health in a large number of tissues and life stages. Nongenomic signaling actions of E1, E2, and E3 at physiologic concentrations have been demonstrated [6-8], and we have shown that xenoestrogens can interfere with their activity via this mode of action [9,10].
Alkylphenol (AP) xenoestrogens are structurally related variants (in aliphatic side-chain lengths) and are commercially useful in a variety of processes requiring surfactants. The related bisphenol A (BPA) has a substituted phenol group instead of a long side chain, and is a widely used and environmentally distributed plastics monomer [11]. We have previously shown that these compounds also signal via nongenomic estrogen receptor pathways, but with distinct timing and nonmonotonic dose-response characteristics compared to that observed for physiological estrogens [12-14], suggesting a mechanism for the deleterious effects of xenoestrogens.
Rapid nongenomic actions can be triggered by estrogens and other steroids to generate a variety of second messengers and diverse pathway activities in many cell types [15]. One of the signaling paradigms known to be utilized by estrogens are those involving G proteins, as demonstrated in vascular tissues [16-18] and neurons [19]. The particular Gα protein involved in most of those responses is of the “i” subclass. Membrane versions of ERs (mERs) are located in caveolar membrane specializations in some tissues [20-22], making them available to partner with many other signaling proteins including G proteins involved in propagating nongenomic signals, leading to further downstream activation of kinases and phosphatases, including the mitogen-activated protein kinases (MAPKs) [23,24].
Traditionally, receptors that associate with heterotrimeric G proteins are seven-transmembrane receptors, such as the G protein-coupled receptor 30 (GPR30 or GPER) which binds estrogens and mineralocorticoids [25]. In oocytes and in ovarian and breast cancer cells GPR30 has been shown to activate Gαs or Gβγ [26,27]. However, the classical “nuclear” ERα, has also been shown to interact with Gαi [16-19] in endothelial and brain tissues. In one case ERβ was specifically studied for this role, but was not found to be involved [18]. In our GH3/B6/F10 rat pituitary cells nongenomic estrogenic signaling operates via a membrane version of estrogen receptor-α (mERα) [28,29], with some mostly inhibitory participation by the other membrane ERs (mERβ and GPR30) shown by use of selective ligands in selective (nM) concentration ranges [10].
The functional endpoints for nongenomic signaling are varied, dependent upon cell type, and often involve rapidly generated second messengers (reviewed in [30]). All three mER types (mERα, mERβ, GPR30) are known to have some nuclear effects downstream of their initial perimembrane signaling activations (examples in [31-33]). Nongenomic and genomic actions of estrogens are thus a continuum beginning at the plasma membrane and sometimes culminating in nuclear actions when sustained signaling warrants a commitment of the cell to new synthesis of macromolecules [34], though some nongenomic actions such as peptide secretion can terminate in actions devoid of nuclear participation [35]. We selected the GH3/B6/F10 rat pituitary tumor cell line for study based on its robust expression of mERα and previous demonstrations of a variety of rapid signaling responses to estrogens that culminate in functional changes (reviewed in [24]) such as prolactin release, cell proliferation, apoptosis, and changes in cell shape and attachment to substrate. We have previously shown that all MAPKs can be rapidly phospho-activated by a variety of estrogens in these cells and that resulting functional responses can be blocked when extracellular-regulated kinases (ERKs) are inhibited ([9,24], and references therein). Changes in the activation of MAPKs are generally associated with functions such as changes in cell number and the mechanisms leading to that outcome (cell division, cell differentiation, cellular apoptosis) (reviewed in [36,37]). Though prolactin release in these cells is initiated by Ca++ signaling [14], other aspects of secretion (eg. vesicle docking and refilling) probably involve other signaling cascades including those of kinases [38].
Our current studies examine if the actions of these three physiologic estrogens (E1, E2, E3), and the APs/BPA operate proximally via mERα and G protein-coupled signaling mechanisms. We relate downstream activation of one class of MAPKs, the ERKs, to interactions with a specific G protein subclass – αi.
Experimental
Reagents and cell culture
We purchased phenol red-free Dulbecco modified Eagle medium (DMEM, high glucose) from Mediatech (Herndon, VA); horse serum from Gibco BRL (Grand Island, NY); defined supplemented calf sera and fetal bovine sera from Hyclone (Logan, UT); penicillin-streptomycin and trypsin EDTA from Mediatech (Manassas, VA); charcoal and Triton X-100 from Sigma (St. Louis, MO). NF023 and NF449 inhibitors were purchased from CalBiochem (San Diego, CA). All other materials were purchased from Fisher Scientific (Pittsburgh, PA) or Sigma-Aldrich (St. Louis, MO).
GH3/B6/F10 cells were routinely cultured in phenol red-free DMEM containing 12.5% horse serum, 2.5% defined-supplemented calf serum, and 1.5% fetal bovine serum with penicillin-streptomycin (50 U/ml). Cells were used between passages 13 and 20 to stably maintain the robust mERα expression levels [35,39] needed for our assessment of these nongenomic responses. Because serum levels of steroids can mask the responses we monitor, we removed small hydrophobic molecules, including steroids, from serum by stripping 4 times with dextran-coated charcoal; cells were grown in welled plates pre-coated with poly-D-lysine in these media for 48 hrs before treatments. For some treatments (BPA) we used multiple concentrations to avoid discrepancies that exist in the literature about activating vs. inhibiting effects of xenoestrogens (for example [40,41]) due to complex nonmonotonic concentration-responses that we have seen previously.
Antibodies (Abs) and Ab assay reagents
Abs to GTP-Gαi and GTP-Gas were from NewEast Biosciences (Malvern, MA); Abs to ERα (MC-20) and caveolin-1 (N-20) were from Santa Cruz (Santa Cruz, CA); ERα C542 Ab was from Assay Designs, Enzo Life Sciences International (Plymouth Meeting, PA). Our other G protein Abs were from Santa Cruz or CalBiochem: Gαs (k20); Gαq (E-17); Gα/1213 (A-20); several anti-Gαi subtypes [3 (C-10), o/t/z (D-15)]; Gβ3 (C-16). Vectastain kits with biotin-conjugated secondary Abs and ABC-AP color development reagents were from Vector Laboratories (Burlingame, CA); fish gelatin from Bio-Rad (Hercules, CA); FITC and Cy3 conjugated secondary Abs from Jackson ImmunoResearch Laboratories (West Grove, PA); Duolink reagents were from Olink Bioscience (Uppsala, Sweden).
pERK plate immunoassay
GH3/B6/F10 cells were treated with nM concentrations of all estrogens (plus 10fM BPA) for 5 min, emphasizing this earlier time point as a known nongenomic response [12,42]. Briefly, 10,000 cells were plated in poly-D-lysine-coated wells of 96-well plates, deprived of serum steroids, and then treated with physiologic estrogens (E1,E2 or E3), xenoestrogens (APs or BPA), 12-O-tetradecanoylphorbol 13-acetate (TPA, 20nM) as a positive control, or ethanol vehicle as a negative control. The cells were then fixed with 2% paraformaldehyde (PFA)/0.2% picric acid at 4°C for 48hr, permeabilized with 0.1% Triton X-100 for 1hr at RT, blocked with 0.2% fish gelatin, and exposed to Ab for phospho-ERKs 1 and 2 overnight at 4°C. Biotin-conjugated secondary Ab was then applied, followed by washing, development with Vectastain kit avidin-conjugated alkaline phosphatase, 0.1% Triton X-100 washes, and the addition of alkaline phosphatase substrate paranitrophenol phosphate (pNpp). The yellow product pNp was monitored at A405 nm in a model 1420 Wallac microplate reader (Victor, Perkin Elmer, Waltham, MA). The plates were then washed, dried, and stained with crystal violet solution as previously described [39,43] to estimate cell numbers for normalization. For inhibitor studies, selective G protein inhibitors used were: pertussis toxin (a Gαi + Gαo + Gαz inhibitor, 100ng/ml or 9.5 nM), NF449 (Gαs inhibitor, 50 M), and NF023 (Gαi/o inhibitor, 50 M). The ICI182780 selective inhibitor for ERsα/β was used at 1nM, and the caveolae-disrupting chemical nystatin [44] at 50μg/ml. Replicates of 7-8 wells were performed for pERK assays in each of 2-3 separate experiments.
Gαi and Gαs activation assays
The activation of Gαi and Gαs was determined by Ab recognition of the levels of these proteins that were GTP-bound in a quantitative plate immunoassay, adapted from our previously developed protocols [39]. This assay was optimized for cell permeabilization conditions, saturating amounts of Ab producing a signal above that of using secondary Ab alone, and for production of a differential signal for total vs. activated Gαi, according to NewEast Biosciences company recommendations. Cells were plated at 10,000-13,000 cells/well in 96-well plates, and serum steroid-deprived (were incubated in DMEM without serum) for at least 2 hrs before treatment. Cells were then treated with physiologic estrogens or xenoestrogens for 15 sec to 8 min. The cells were then processed as described above for the pERK plate assays, except that cells were fixed with 4% PFA for 10 min, and 100μM GTPγS was additionally added, when required, with the permeabilization solution. The fixed cells were incubated with the primary Abs (anti-GTP-Gαi or anti-GTP-Gαs) overnight at 4°C. Replicates of 7-8 wells were performed for GTP-G proteins over 2-3 separate experiments. When we originally developed these types of plate immunoassays for quantitating various receptor and activated signaling proteins, we used nonspecific IgGs instead of specific primary Abs as negative controls, to determine the specificity of this technique for identifying the protein of interest [39], and thus have not repeated those studies again here. We have now utilized this assay format to quantify a variety of proteins in GH3/B6/F10 cell membranes or cell interiors including: ERs and β, GPR30, clathrin, three activated MAPK subtypes, and the activated transcription factors Elk-1 and ATF-2 [32,39,42].
Co-immunocapture (Co-IC) and immunoblots
For the results shown in Fig. 4, after serum deprivation, cells were washed in fresh DMEM/1% stripped serum and treated with either 10nM E2 or vehicle control (EtOH) for 10 min. To prepare whole cell lysates cells were washed in ice cold PBS and lysed by scraping from plates into 1ml lysis buffer (0.25M sucrose, 20mM Tris, 1mM EDTA, 1mM PMSF, 1mM DTT, protease inhibitors, pH 7.8) followed by freezing and thawing three times. Lysates were microfuged at 10,000g for 10 min, the pellet discarded, and the supernatant saved on ice for immediate analysis.
Fig. 4. ERα and Gαi proteins interact in whole-cell lysates.
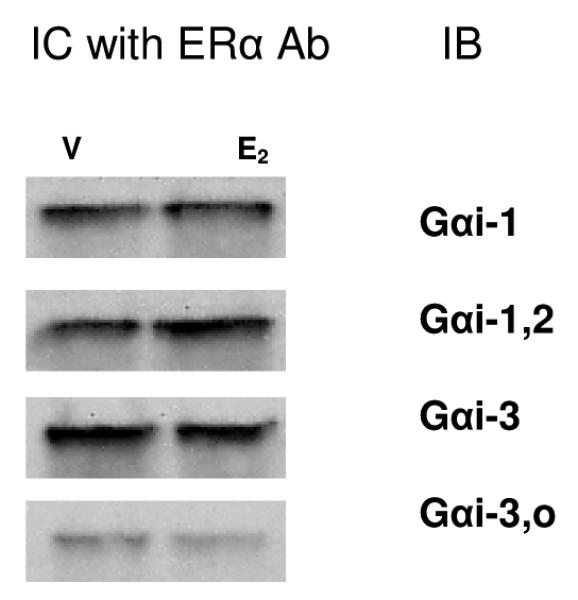
Lysates from cells treated with either vehicle (V) or 10nM E2 (E2) for 10 min were bound to specific Ab to ERα (MC-20 recognizing the C-terminus) and captured on Protein G agarose beads. Immuno-captured (IC) proteins were subsequently eluted, electrophoresed, blotted, and probed with four different subtype-selective Gαi Abs (IB), as indicated. All captured G proteins were ~48 MW, and no changes were noted due to E2 treatment. A representative immunoblot from 3 experiments is shown.
Protein-G agarose beads were conjugated to ERα Ab by combining 30 ls of a 50% bead slurry and 1 g of the ERα Ab (MC-20) used for capture. The suspension was tumbled for at least 4 hrs at 4°C, then microfuged for 2 sec to retrieve the beads, which were then washed 3 times in lysis buffer. Then equal amounts of lysate protein [assessed by Pierce Bicinchoninic Acid (BCA) Protein Assays, Rockford, IL] were added to the Ab-conjugated protein-G beads and the mixture was tumbled overnight at 4°C. The suspension was microfuged for 5 sec, washed 3 times with cold lysis buffer, and proteins released by adding SDS-PAGE loading buffer (40 ls) and boiling for 10 min. Proteins were then electrophoresed and immunoblotted with different selective Gαi protein Abs (using a kit from CalBiochem) by standard procedures. As E2 treatment did not appear to change the levels by visual inspection, the bands were not evaluated by densitometry. Cell compartment-specific antigen preparations (isolating cytosolic and membrane fractions) along with additional Abs recognizing ERα, and more sensitive detection schemes (Odyssey fluorescent Ab system; Li-Cor Biosciences, Lincoln NE) were tried, but we were unable to consistently detect these protein interactions under these more dissociating conditions. Nonspecific IgGs were included as a negative control in some experiments, instead of the primary Ab of interest, and no proteins in the size range of interest for G proteins or ERs were observed (data not shown).
Co-localization by immunofluorescence
GH3/B6/F10 cells were cultured on cover slips, washed twice with PBS, and then fixed with 4% PFA for 20 min. The cells were then blocked with Duolink II blocking buffer for 30 min at 37°C followed by incubating overnight at 4°C with primary Ab (MC-20) for the protein expected to be exposed outside of the plasma membrane of the cell (ERα). Then the cells were refixed with 2% PFA for 5 min, permeabilized with 0.1% Triton X-100 for 10 min, and then incubated with primary Ab for the second protein (Gαi; C-10 Ab) for 2 hrs. Cells were then incubated with secondary Abs for each primary Ab conjugated to either FITC or Cy3 (Jackson ImmunoResearch Laboratories, West Grove, PA) for 2 hrs at 37°C, followed by washing and mounting in 4μl of Duolink II Mounting Medium with DAPI. The slides were kept at −20°C before being viewed with an Olympus BX51 fluorescence microscope with an Olympus DP71 camera (at 10x and 60x magnifications for Fig. 5).
Fig. 5. ERα and Gαi proteins partially co-localize at non-nuclear sites.

The subcellular location of ERα was assessed with Ab MC-20 followed by Cy3-tagged secondary Ab (red staining) in unpermeabilized cells. After permeabilization with 0.1% Triton X-100 for 10 min the subcellular location of Gαi was probed with Ab C-10 followed by an FITC-tagged secondary Ab (green staining). The top row shows a multi-cell field at 10X magnification and the bottom row shows a single cell at 60X magnification. The merged images display colocalization of ERα and G i as a yellow signal, and nuclei (blue) DAPI staining for comparison. Representative images from 2 experiments are shown.
Co-localization by epitope Proximity Ligation Assay (PLA)
We used a relatively new technique to determine the distance between two epitopes of interest in our studies. The revised PLA protocol [45] determines the nearness of partnered protein epitopes within 35nm [46]. Basically the technique detects nearby epitopes tagged with primary Abs of choice made in two different species, recognized in turn by two different anti-species Abs having attached oligonucleotides. When the two epitopes are sufficiently close ( 35 nm) the attached oligonucleotides can hybridize, producing a template for a rolling circle DNA amplification, which can then be probed efficiently with fluorescent oligonucleotide probes that will appear as discreet dots by fluorescence imaging. To identify a single protein, or to use as a control for the analysis of epitope nearness. epitopes representing two parts of the same protein are chosen. In our assays GH3/B6/F10 cells were cultured on cover slips overnight and washed twice with PBS before fixation with 4% PFA for 20 min, which does not permeabilize cells [47]. The cells were then blocked with Duolink blocking buffer for 30 min at 37°C, followed by incubation with primary Ab (i.e. in unpermeabilized cells for proteins such as ERα expected to be on the outside of the plasma membrane) overnight at 4°C and washed. Then the cells were re-fixed with 2% PFA for 5 min and permeabilized with 0.1% Triton X-100 for 10 min. Subsequent incubation with primary Ab for proteins inside the plasma membrane (Gαi, caveolin-1) was for 2 hrs at RT, followed by washing. Appropriate anti-species secondary Abs to which oligonucleotides had been conjugated (anti-rabbit PLA probe PLUS and anti-mouse PLA probe MINUS) were then incubated with the preparation for 2 hrs at 37°C, followed by treatment with Duolink ligation-ligase solution for 30 min at 37°C. Finally, cells were incubated with the Duolink amplification-polymerase solution for 100 min at 37°C and then labeling oligonucleotides, followed by washing and mounting on slides with 4μl of Duolink 2 Mounting Medium containing DAPI fluorescent dye for staining nuclei. The slides were kept at −20°C before being viewed with a Zeiss 510 Meta confocal microscope (63× magnification with an additional 2.4 × zoom magnification).
Statistics
Data from plate assays for pERK and GTP-charged G proteins were analyzed by one-way analysis of variance (ANOVA) followed by multiple comparisons vs. control group (Holm-Sidak method) using Sigma Stat v.3 (Systat Software, Inc.) Significance was accepted at p<0.05.
Results
All G protein classes are present in GH3/B6/F10 cells
First we asked which G protein subtypes are present in our subcloned (F10) cell line, as a first step in determining which and how many interactions with ERα might be possible. At the RNA level, microarray analysis demonstrated that members of all Gα protein families plus several β and subtype RNAs were consistently present (found in repeat analyses). We then confirmed the presence of all major subtypes by either immunoblots, plate immunoassays, or both, using commercially available Abs. Table 1 (supplemental materials) summarizes these findings. Some subclass-specific Abs can recognize more than one of the class members (eg. Gαi-3,o recognizes both i subtypes 3 and o). Correctly sized G proteins were observed in all immunoblots. Multiple members of most classes were detected.
G protein role in signal generation by physiologic and nonphysiologic estrogens
We then determined whether the major subclasses of the G proteins present were involved in the signaling pathways leading to ERK activation (measured by quantitative 96-well plate immunoassay, Fig. 1). For this we used several G protein class-selective inhibitors. These data were all consistent with the involvement of the Gαi subclass, as responses for all estrogens that activated ERK at this concentration were inhibited by both PTX and NF023 (panels A and B respectively). Because the responses to all physiologic and nonphysiologic estrogens were similarly affected by these inhibitors, it suggests that Gαi is involved in all of their actions. As the G s inhibitor NF449 was inconsistently effective (panel C, 4/8 estrogens), this isoform does not appear to be as clearly involved in mediating ERK activation via ERs by all of these estrogens. Because there are many different signaling streams that culminate in ERK activation, it is possible that some estrogens could engage Gαs or other G proteins and then secondarily activate ERK pathways by 5 min, and this could be yet another distinction between the actions of different estrogens.
Fig. 1. G protein involvement in signaling leading to ERK activation triggered by three physiologic estrogens and alkylphenols.

Cells were incubated with 1nM concentrations of estrogens except where an additional 10fM concentration is noted for BPA. Two concentrations of BPA were tested due to its consistent highly nonmonotonic concentration-response curve. ERK activation was measured using the pERK quantitative plate immunoassay. A. Cells were pretreated with 100ng/ml pertussis toxin (PT) overnight and then stimulated with estradiol (E2), estrone (E1), estriol (E3), bisphenol A (BPA), ethylphenol (EP), propylphenol (PP), octylphenol (OP), nonylphenol (NP), or vehicle (V) for 5 min. Tetradecanoylphorbol acetate (TPA, or phorbol 12-myristate 13-acetate) was used as a positive control for post-receptor activation of pathways leading to ERK phosphorylation. B. Cells were pretreated with 50μM of the Gαi-selective inhibitor NF023 for 30 min, and then stimulated by all estrogens named above for 5 min. C. As in B except cells were pretreated with 50μM of the Gαs-selective inhibitor NF449. *=statistical significance (P<0.05) for estrogen treatments alone vs. vehicle control (100%); #=statistical significance (P<0.05) for inhibitor pretreatment before estrogen treatment vs. no inhibitor treatment. These results are mean ± SEM values of 24 samples assessed over 3 separate experiments.
For further studies on Gα proteins subcategories “i” and “s” we employed a relatively recently developed experimental tool -- Abs that recognize G proteins that are in an active state due to the binding of GTP [48]. Abs for both unmodified and GTP-bound Gαi recognized the expected 48MW protein in immunoblots of cell lysates from cells treated with either vehicle or 1nM E2 for 5 min (data not shown). We then adapted our quantitative plate immunoassay for use with these Abs recognizing activated Gαi and Gαs proteins. We demonstrated that Gαi could be activated at 15-30 sec (Fig. 2) by low concentrations of estrogens (BPA, nonylphenol, and E2 as a trend but not significantly). However, after 5 min (300 sec) of treatment (Figs. 2 and 3A) this GTP-activation of Gαi was significantly suppressed, even below vehicle-treated levels, for those samples treated with AP estrogens only. Because GTP-binding can apparently fluctuate very rapidly, and may differ for different estrogens, it could therefore be difficult to demonstrate the time course for each estrogen without many more experiments, or techniques more amenable to elucidating such very rapid responses. Therefore, we decided to add GTPγS to these samples, to potentially trap the GTP-bound form by irreversible cumulative binding to this modified GTP (Fig. 3A & B). As all values for GTP-Gαi were higher after adding GTPγS, it suggests that the Ab to activated Gαi recognizes both GTP-bound and GTPγS-bound G proteins. While the reductions due to treatment with estrogenic APs were eliminated by this GTPγS treatment, Gαi 5 min activation levels were enhanced for treatment with the physiologic estrogens (E1, E2, E3) and BPA, presumably by trapping of the activated forms so they could not be rapidly down-regulated (obscuring the response) after early activation. However, Gαs (Fig. 3B) was neither activated nor deactivated by any estrogens tested, and trapping of activated forms with GTP-γS did not increase any of these responses.
Fig. 2. Time-dependent activation of Gαi by a physiologic estrogen and two xenoestrogens.

Cells were treated with 1nM estradiol (E2), nonylphenol (NP), and bisphenol A (BPA). GTP-bound Gαi was measured with an Ab specific for the complex at various times shown in sec. n=24 samples (over 3 experiments) for each condition; *= statistical significance (P<0.05) for each estrogen/xenoestrogen treatment vs. the vehicle control (shown at time 0).
Fig. 3. Estrogens cause Gαi deactivation at 5 min and GTPγS binding shows cumulative activations; there are no such effects on Gαs.

Cells were treated with vehicle (V), estradiol (E2), estrone (E1), estriol (E3), bisphenol A (BPA), ethylphenol (EP), propylphenol (PP), octylphenol (OP) and nonylphenol (NP) for 5 min, all estrogens at 1nM, except that BPA was additionally tested at 10 fM. GTP-bound Gαi (A) and Gαs (B) were measured by recognition with an Ab selective for these GTP-bound forms in the absence (white bar) or presence (dark bar) of 100μM GTPγS. Total Gαi and Gαs were also measured, but did not differ under any conditions of estrogen treatments (data not shown). For statistical annotations: For A *= statistical significance (P<0.05) from paired V control for the no GTPγS group. #= GTPγS-treated group statistical significance (P<0.05) from paired estrogen-treated levels; n=48 without, and n=32 with GTPγS treatment over 3 experiments. For B n=32 without and n=16 with GTPγS treatment over 2 experiments.
Complex formation between ERα, Gαi, and caveolin-1 in pituitary cells
We tested whether G i proteins from GH3/B6/F10 cells could be co-immuno-captured with ERα (Fig. 4). Ab-bound ERα and its companion proteins were pulled out of cell lysates on Protein G beads and we detected the presence of Gαi in the eluted, electrophoresed and immunoblotted proteins. E2 treatment for 10 min did not influence the association of ERα with Gαi. We also isolated subcompartment-resident ERα (cytosol or membrane fractions) to try to determine if a compartmentalized subset of ERs associate with G proteins (data not shown). While these results supported the presence of these proteins in our cells (see supplementary material Table I), we could not consistently demonstrate co-capture. Therefore, we conclude that the association between ERα and G proteins is fragile, and that further manipulations that are required to isolate subcellular fractions carried out in dissociating conditions (centrifugations, washes, etc.) can easily destroy this association.
We examined our cells with imaging techniques that might demonstrate these protein associations in situ, where they would be less susceptible to disruption by dissociating experimental conditions. ERα and Gαi proteins visualized by Ab binding partially co-localized at non-nuclear sites (Fig. 5). Abs to ERα were applied before permeabilization (Ab MC-20 and Cy3-tagged secondary Ab, red staining) to favor the labeling of the membrane (and not the more plentiful nuclear) version of this receptor. Because these images are produced by conventional fluorescence micrography, we cannot determine whether the signals are on or underneath the cell membrane. Membrane and cytoplasmic staining over the nuclear region of these cells is usually sparse as the large nuclei of these cells protrude and thin the cytoplasm in that area, and place the membrane in a different focal plane [49]. After permeabilization of the cells, Gαi was stained with Ab C-10 and an FITC-tagged secondary Ab (green staining). These antigens gave a largely similar staining pattern. The merged image shows that their locations can overlap (yellow) in the non-nuclear compartment (nuclei stained blue with DAPI), but with the more abundant Gαi protein being the predominant unpaired signal, as expected.
Finally, we examined an alternative method for visualizing protein partners in combination with confocal imaging -- Duolink epitope PLAs (Fig. 6). We examined ERα using Ab C542 (applied before cell permeabilization) interacting with Gαi (using Ab C-10) or caveolin-1 (using Ab N-20), both latter Abs applied after cell permeabilization. Signals with this technique are visualized by Ab proximity-based ligation. Ab-attached oligonucleotides hybridize wherever the Abs are near enough to each other to allow this. The hybridized oligos are then amplified by rolling circle DNA synthesis. Finally, the amplified DNA is probed with complementary Cy3-labeled oligos. We display individual mid-cell optical sections (left panels), and 3D reconstructions combining all optical slices for each sample (right panels). For orientation to the subcellular compartments, the nuclei were stained by DAPI (blue). The differences between positive and negative controls are remarkably clear. In addition, the positive control of ERα alone is an interesting visualization of the total population of mERα and its position on the cell, regardless of whether it is partnered with other proteins. The signals indicating ERα partnering with Gαi and caveolin-1 are both nonnuclear (C & D, respectively), and mostly coincident with the cell periphery. These cells usually have a small and irregular cytoplasmic rim around a large nucleus, as we have observed previously with other types of staining and 3D confocal reconstruction of images [47,49]. Inspection of the rotated orthogonal view of these images showed that signal dots that appear to be over nuclei in the 3D panels are really adjacent (and not inside) nuclei when viewed from the side (not shown). Note that the unpaired ERα signals in A are more plentiful and have a smaller diameter compared to the partnered proteins in C and D.
Fig. 6. ERα, and Gαi are in the same complex at or near the plasma membrane.

Duolink epitope proximity assays for two different epitopes amplify a signal (red; cy3) due to the adjacency (<35nm) of epitopes. Each sample was also counterstained for nuclei (blue; DAPI). For each treatment, a representative image from 2 experiments is shown in the left panels as a single confocal slice through the middle of several cells. The representations in the right panels are 3D reconstructions of confocal slices into a composite image. The white bar in the lower left image measures 10 μM and the images were obtained using a 63X objective with a 2X optical zoom. A. Positive control: Two different ERα epitopes were assayed including one at the amino terminus (Ab ERα 21-32) and one at the carboxy terminus (Ab C542). B. Negative control: No primary Ab was included and there was no signal. C. Before permeabilizing the cells, the ERα Ab (C452) and a secondary Ab-linked Duolink probe was applied; after permeabilizing the cells the Gαi Ab (C-10) and its secondary Ab-linked Duolink probe was applied. The signal generated demonstrates close proximity of these two proteins in a non-nuclear pattern. D. ERα Ab (C542) and its probe were applied pre-permeabilization, and caveolin-1 Ab (N-20) and its probe were applied after cell permeabilization, again showing an interaction.
Caveolae and ERs are both involved in xenoestrogen and physiologic estrogen signaling endpoints (ERK)
Finally, we addressed the significance of ERs in caveolar locations, by perturbing various factors with selective inhibitors and monitoring ERK activation (Fig. 7). Interrupting caveolar structure with nystatin [50] destroyed the ability of all competent estrogens, both physiologic and nonphysiologic, to mediate ERK activation. Likewise, inhibiting the actions of ERα/β with the selective (at this concentration) inhibitor ICI182780 also destroyed the ability of most of these estrogens to elicit an ERK response. Where significant responses were not elicited by OP and NP, those concentrations have been previously shown to not cause ERK activation, due to the nonmonotonic nature of the concentration dependence of these signaling responses [14]. In the case of E1 eliciting a response that the ICI compound was unable to inhibit, it is possible that there is involvement of GPR30 estrogen receptors, though we have seen the GPR30 selective ligand G1 enhance ERK responses only at very low (10−13 to 10−11M) concentrations while inhibiting responses at nM or higher concentrations [10]. Though the ICI compound has also been shown by others to elicit actions via GPR30 [51,52], this was only at μM concentrations, well above the 10nM concentration used here.
Fig. 7. Intact caveolae and ERs are involved in the ERK responses.

Cells were pretreated (30 min) with 50μg/ml nystatin (A) which disrupts caveolar structure or (B) the selective ERα/β antagonist at this concentration, ICI 182780 (ICI, 10nM). Then cells were treated for 5 min with various estrogens before assessing ERK activation via the plate immunoassay. Physiologic estrogens at 1nM used to evoke an ERK response were estradiol (E2), estrone (E1), estriol (E3). Xenoestrogens were bisphenol A (BPA), ethylphenol (EP), propylphenol (PP), octylphenol (OP), nonylphenol (NP). Tetradecanoylphorbol acetate (TPA) was used as a post-receptor positive control for ERK activation. Vehicle (V) treatment was the negative control. n=16 samples for each condition done in 2 separate experiments; *= statistical significance (P<0.05) for ERK activators vs. vehicle control; #= statistical significance (P<0.05) for inhibition of the stimulated level by inhibitors.
Discussion
Both physiological estrogens and xenoestrogens cause nongenomic signaling leading to functional responses (reviewed in [24]), and we have again demonstrated that ability in these studies. However, while several downstream signaling components have been clearly linked to the actions of xenoestrogens, the question remained as to the proteins involved in organizing these actions at a site more proximal to membrane receptor stimulation. Here we have demonstrated the involvement of specific G proteins in the initiation of signaling by a variety of physiological and environmental estrogens. Our evidence for the interaction of ERα with both G i and caveolin proteins by multiple approaches further supports the idea that these proximal protein partners participate in the actions of both estrogens and xenoestrogens initiated at the membrane.
While G proteins of every major class are expressed in many cell types (including in our pituitary tumor cell line used in these studies), it appears that the Gαi subclass is so far, the one most often linked with the actions of mERα [16-18] with some exceptions (Gαq; [18]). Our data agree with these ERβGαi signaling partnerships demonstrated in other cell types. Even though Gαs does not appear to be consistently involved in downstream ERK signaling in the present studies, we have preliminary evidence (using inhibitors) that both Gαi and Gαs are involved in Ca++ signaling evoked by E2 in this cell type (Bulayeva & Watson, unpublished observation). Therefore, it may be that different Gα proteins can partner with ERs selectively to mediate different downstream effects, and these other associations and activations may be on different time scales and affected differently by distinct estrogens.
We also examined the nature of the physical association of ERα with Gαi proteins, as well as the influence of different estrogens on this G protein’s activation. The 15-30 sec GTP-activation of Gαi was subsequently (at 5 min) suppressed by some estrogens (the APs), even below vehicle-treated levels. This could indicate a compensatory response more robust for xenoestrogens than for physiologic estrogens. Because GTPγS increased measurable levels of GTP-activated protein in all samples, this Ab directed at “loaded” Gαi apparently recognizes GTPγS-bound Gαi as an activated form. Because activation of Gαi was not statistically significant in our time course beginning at 15 sec by E2, actions by this physiological estrogen could be more rapid, and only observable when all activated G proteins were trapped at 5 min with irreversible and thus cumulative binding of GTPγS to Gαi. This trapping of the active form essentially erased the compensatory down-regulation of the GTP-bound form seen with the AP treatments. Because G protein activation appears to be a very dynamic response over such a short time frame, it is difficult to determine optimal activation and deactivation profiles for each estrogen separately given the expense of these reagents. However, it is interesting to see that the APs appear to regulate GTP activation/deactivation cycles differently (slower/more robustly) than the physiologic estrogens and BPA. We recently saw a similar separation between these two subsets of estrogens and their ability to temporally shift ERK response phases [10].
Co-immuno-capture experiments were done under a variety of conditions, some providing evidence that ERα and Gαi can partner in these cells. However, we were only reliably successful when using whole cell lysates, perhaps because such weak protein-protein interactions are vulnerable to the extra experimental manipulations required when preparing membrane and cytosolic fractions. However, the co-localization immunocytochemistry and the PLAs were done in fixed cells that did not undergo the same disruptions, which perhaps allowed these weaker protein complexes to survive [53]. Our experiments do not distinguish between direct vs. indirect protein interactions, as both immunocapture and a distance of <35 nM in PLAs could allow for the participation of other proteins as “adaptors” for formation of this complex. It is interesting that the PLA signals were qualitatively different when detecting ERα alone (a large number of dispersed small signals), vs. ERα partnered with other proteins (fewer, larger signals). Partnered signals should be a subset of the total number and thus be fewer. Because receptors and other signaling proteins are known to cluster in caveolae, it is possible that we observed pools of these proteins (and thus larger diameter signals) [54,55]. The clustering of mERα signals is an immuno-staining pattern that we have also previously seen after treatment of cells with estrogens [56].
Conclusions
We showed that xenoestrogens of the AP class are potent activators of nongenomic signaling via G proteins. Because such responses are normally mediated by a spectrum of physiologic estrogens (E1, E2, and E3) each having different life stage-specific significance, the actions of these xenoestrogens could disrupt many different normal estrogenic functions including fertility/cycling, pregnancy, post-menopausal signaling, and non-reproductive tissue signaling (e.g. bone, cardiovascular, and behavioral functions). Actions on these cells representing the anterior pituitary suggest that xenoestrogen disruptions could affect a wide variety of endocrine axis tissues that are regulated from the pituitary. Altogether, our results contribute new strategies and evidence for both physiologic and nonphysiologic estrogens acting via mERα to selectively interact with and activate Gαi. This Gαi response precedes and is necessary for the downstream activation of ERKs in these cells. Though nonphysiologic xenoestrogens also cause these responses, they cause them with distinct differences in timing and magnitude. Therefore, xenoestrogens are potent nongenomic signal activators, but they induce defective (altered) estrogenic responses.
Supplementary Material
Gαi initiates ERK activation by physiologic and xenoestrogens in pituitary cells.
Estrogens activate Gαi (cause binding of GTP) by 15 seconds.
Xenoestrogen-induced compensatory deactivation of Gαi is blocked by GTPγS.
ERα physically partners with Gαi and caveolin I.
ERs and caveoli are necessary for ERK signaling via estrogens.
Acknowledgements
The authors would like to thank Alicia Goldberg for excellent editing of our manuscript. These studies were supported by funding from NIHR01 ES015292.
List of abbreviations
- AP
alkylphenol
- Ab
antibody
- BPA
bisphenol A
- GPR30 or GPER
G protein-coupled estrogen receptor
- E1
estrone
- E2
estradiol
- E3
estriol
- ERK
extracellular-regulated kinase
- mERα
membrane estrogen receptor-α
- mERβ
(membrane estrogen receptor-β
- MAPK
mitogen-activated protein kinase
- PFA
paraformaldehyde
- PLA
proximity ligation assay
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Greenspan FS, Gardner DG. Basic and Clinical Endocrinology. 7th edn Lange Medical Books, McGraw Hill; New York: 2004. [Google Scholar]
- 2.Shenhav S, Gemer O, Volodarsky M, Zohav E, Segal S. Midtrimester triple test levels in women with severe preeclampsia and HELLP syndrome. Acta Obstet Gynecol Scand. 2003;82:912–915. doi: 10.1034/j.1600-0412.2003.00250.x. [DOI] [PubMed] [Google Scholar]
- 3.Chard T, Macintosh MC. Screening for Down’s syndrome. J Perinat Med. 1995;23:421–436. doi: 10.1515/jpme.1995.23.6.421. [DOI] [PubMed] [Google Scholar]
- 4.Meinhardt U, Mullis PE. The essential role of the aromatase/p450arom. Semin Reprod Med. 2002;20:277–284. doi: 10.1055/s-2002-35374. [DOI] [PubMed] [Google Scholar]
- 5.Jansson L, Holmdahl R. Enhancement of collagen-induced arthritis in female mice by estrogen receptor blockage. Arthritis Rheum. 2001;44:2168–2175. doi: 10.1002/1529-0131(200109)44:9<2168::aid-art370>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 6.Morley P, Whitfield JF, Vanderhyden BC, Tsang BK, Schwartz J. A new, nongenomic estrogen action: The rapid release of intracellular calcium. Endocr. 1992;131:1305–1312. doi: 10.1210/endo.131.3.1505465. [DOI] [PubMed] [Google Scholar]
- 7.Selles J, Polini N, Alvarez C, Massheimer V. Novel action of estrone on vascular tissue: regulation of NOS and COX activity. Steroids. 2005;70:251–256. doi: 10.1016/j.steroids.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 8.Watson CS, Jeng YJ, Kochukov MY. Nongenomic actions of estradiol compared with estrone and estriol in pituitary tumor cell signaling and proliferation. FASEB J. 2008;22:3328–3336. doi: 10.1096/fj.08-107672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeng YJ, Kochukov M, Watson CS. Combinations of physiologic estrogens with xenoestrogens alter calcium and kinase responses, prolactin release, and membrane estrogen receptor trafficking in rat pituitary cells. Environ Health. 2010;9:61. doi: 10.1186/1476-069X-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeng YJ, Watson CS. Combinations of physiologic estrogens with xenoestrogens alter ERK phosphorylation profiles in rat pituitary cells. Environ Health Perspect. 2011;119:104–112. doi: 10.1289/ehp.1002512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lakind JS, Naiman DQ. Bisphenol A (BPA) daily intakes in the United States: estimates from the 2003-2004 NHANES urinary BPA data. J Expo Sci Environ Epidemiol. 2008;18:608–615. doi: 10.1038/jes.2008.20. [DOI] [PubMed] [Google Scholar]
- 12.Bulayeva NN, Watson CS. Xenoestrogen-induced ERK-1 and ERK-2 activation via multiple membrane-initiated signaling pathways. Environ Health Perspect. 2004;112:1481–1487. doi: 10.1289/ehp.7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wozniak AL, Bulayeva NN, Watson CS. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-alpha-mediated Ca2+ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Environ Health Perspect. 2005;113:431–439. doi: 10.1289/ehp.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kochukov MY, Jeng Y-J, Watson CS. Alkylphenol xenoestrogens with varying carbon chain lengths differentially and potently activate signaling and functional responses in GH3/B6/F10 somatomammotropes. Env Health Perspect. 2009;117:723–730. doi: 10.1289/ehp.0800182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watson CS, Gametchu B. Proteins of multiple classes participate in nongenomic steroid actions. Exp Biol Med. 2003;228:1272–1281. doi: 10.1177/153537020322801106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wyckoff MH, Chambliss KL, Mineo C, Yuhanna IS, Mendelsohn ME, Mumby SM, et al. Plasma membrane estrogen receptors are coupled to endothelial nitric-oxide synthase through Galpha(i) J Biol Chem. 2001;276:27071–27076. doi: 10.1074/jbc.M100312200. [DOI] [PubMed] [Google Scholar]
- 17.Kumar P, Wu Q, Chambliss KL, Yuhanna IS, Mumby SM, Mineo C, et al. Direct Interactions with G alpha i and G betagamma mediate nongenomic signaling by estrogen receptor alpha. Mol Endocrinol. 2007;21:1370–1380. doi: 10.1210/me.2006-0360. [DOI] [PubMed] [Google Scholar]
- 18.Lin AH, Leung GP, Leung SW, Vanhoutte PM, Man RY. Genistein enhances relaxation of the spontaneously hypertensive rat aorta by transactivation of epidermal growth factor receptor following binding to membrane estrogen receptors-alpha and activation of a G protein-coupled, endothelial nitric oxide synthase-dependent pathway. Pharmacol Res. 2011;63:181–189. doi: 10.1016/j.phrs.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Navarro CE, Saeed SA, Murdock C, Martinez-Fuentes AJ, Arora KK, Krsmanovic LZ, et al. Regulation of cyclic adenosine 3′,5′-monophosphate signaling and pulsatile neurosecretion by Gi-coupled plasma membrane estrogen receptors in immortalized gonadotrophin-releasing hormone neurons. Mol Endocrinol. 2003;17:1792–1804. [PubMed] [Google Scholar]
- 20.Razandi M, Oh P, Pedram A, Schnitzer J, Levin ER. ERs associate with and regulate the production of caveolin: Implications for signaling and cellular actions. Mol Endocrinol. 2002;16:100–115. doi: 10.1210/mend.16.1.0757. [DOI] [PubMed] [Google Scholar]
- 21.Zivadinovic D, Watson CS. Membrane estrogen receptor-alpha levels predict estrogen-induced ERK1/2 activation in MCF-7 cells. Breast Cancer Res. 2005;7:R130–R144. doi: 10.1186/bcr959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chambliss KL, Yuhanna IS, Mineo C, Liu P, German Z, Sherman TS, et al. Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ Res. 2000;87:E44–E52. doi: 10.1161/01.res.87.11.e44. [DOI] [PubMed] [Google Scholar]
- 23.Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008;22:954–965. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 24.Watson CS, Jeng YJ, Kochukov MY. Nongenomic signaling pathways of estrogen toxicity. Toxicol Sci. 2010;115:1–11. doi: 10.1093/toxsci/kfp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feldman RD, Gros R. Unraveling the mechanisms underlying the rapid vascular effects of steroids: sorting out the receptors and the pathways. Br J Pharmacol. 2011;163:1163–1169. doi: 10.1111/j.1476-5381.2011.01366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocr. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 27.Peyton C, Thomas P. Involvement of Epidermal Growth Factor Receptor Signaling in Estrogen Inhibition of Oocyte Maturation Mediated Through the G Protein-Coupled Estrogen Receptor (Gper) in Zebrafish (Danio rerio) Biol Reprod. 2011 doi: 10.1095/biolreprod.110.088765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watson CS, Zivadinovic D, Bulayeva N, Hawkins B, Campbell CH, Gametchu B. Edited by Watson CS. Kluwer Academic Publishers; Boston: 2003. A membrane form of estrogen receptor-α mediates estrogenic, nongenomic effects. In The Identitities of Membrane Steroid Receptors; pp. 11–19. [Google Scholar]
- 29.Watson CS, Bulayeva NN, Wozniak AL, Finnerty CC. Signaling from the membrane via membrane estrogen receptor-alpha: estrogens, xenoestrogens, and phytoestrogens. Steroids. 2005;70:364–371. doi: 10.1016/j.steroids.2005.03.002. PMCID:15862819. [DOI] [PubMed] [Google Scholar]
- 30.Watson CS, Gametchu B. Membrane-initiated steroid actions and the proteins that mediate them. Proc Soc Exp Biol Med. 1999;220:9–19. doi: 10.1046/j.1525-1373.1999.d01-2.x. [DOI] [PubMed] [Google Scholar]
- 31.Madeo A, Maggiolini M. Nuclear alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer Res. 2010;70:6036–6046. doi: 10.1158/0008-5472.CAN-10-0408. [DOI] [PubMed] [Google Scholar]
- 32.Jeng YJ, Watson CS. Proliferative and anti-proliferative effects of dietary levels of phytoestrogens in rat pituitary GH3/B6/F10 cells - the involvement of rapidly activated kinases and caspases. BMC Cancer. 2009;9:334. doi: 10.1186/1471-2407-9-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bjornstrom L, Sjoberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. doi: 10.1210/me.2004-0486. [DOI] [PubMed] [Google Scholar]
- 34.Pietras RJ, Szego CM. Cell membrane estrogen receptors resurface. Nature Medicine. 1999;5:1330. doi: 10.1038/70877. [DOI] [PubMed] [Google Scholar]
- 35.Pappas TC, Gametchu B, Yannariello-Brown J, Collins TJ, Watson CS. Membrane estrogen receptors in GH3/B6 cells are associated with rapid estrogen-induced release of prolactin. Endocrine. 1994;2:813–822. [Google Scholar]
- 36.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 37.Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 38.Bulayeva NN, Wozniak A, Lash LL, Watson CS. Mechanisms of membrane estrogen receptor-{alpha}-mediated rapid stimulation of Ca2+ levels and prolactin release in a pituitary cell line. Am J Physiol Endocrinol Metab. 2005;288:E388–E397. doi: 10.1152/ajpendo.00349.2004. [DOI] [PubMed] [Google Scholar]
- 39.Campbell CH, Watson CS. A comparison of membrane vs. intracellular estrogen receptor-alpha in GH(3)/B6 pituitary tumor cells using a quantitative plate immunoassay. Steroids. 2001;66:727–736. doi: 10.1016/s0039-128x(01)00106-4. [DOI] [PubMed] [Google Scholar]
- 40.Adlercreutz H. Phytoestrogens: epidemiology and a possible role in cancer protection. Environ Health Perspect. 1995;103(Suppl 7):103–112. doi: 10.1289/ehp.95103s7103. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang YC, Nair MG, Nitiss JL. Metabolites of daidzein and genistein and their biological activities. Journal of Natural Products. 1995;58:1901–1905. doi: 10.1021/np50126a016. [DOI] [PubMed] [Google Scholar]
- 42.Bulayeva NN, Gametchu B, Watson CS. Quantitative measurement of estrogen-induced ERK 1 and 2 activation via multiple membrane-initiated signaling pathways. Steroids. 2004;69:181–192. doi: 10.1016/j.steroids.2003.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gillies RJ, Didier N, Denton M. Determination of cell number in monolayer cultures. Anal Biochem. 1986;159:109–113. doi: 10.1016/0003-2697(86)90314-3. [DOI] [PubMed] [Google Scholar]
- 44.Anderson HA, Chen Y, Norkin LC. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol Biol Cell. 1996;7:1825–1834. doi: 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pisconti A, Cornelison DD, Olguin HC, Antwine TL, Olwin BB. Syndecan-3 and Notch cooperate in regulating adult myogenesis. J Cell Biol. 2010;190:427–441. doi: 10.1083/jcb.201003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gullberg M, Andersson A-C. Visualization and quantification of protein-protein interactions in cells and tissues. Nature Methods. 2010;6:641–647. [Google Scholar]
- 47.Pappas TC, Gametchu B, Watson CS. Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J. 1995;9:404–410. doi: 10.1096/fasebj.9.5.7896011. [DOI] [PubMed] [Google Scholar]
- 48.Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, et al. Beta-arrestin-but not G protein-mediated signaling by the “decoy” receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107:628–632. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watson CS, Pappas TC, Gametchu B. The other estrogen receptor in the plasma membrane: implications for the actions of environmental estrogens. Environ Health Perspect. 1995;103(Suppl 7):41–50. doi: 10.1289/ehp.95103s741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ushio-Fukai M, Hilenski L, Santanam N, Becker PL, Ma Y, Griendling KK, et al. Cholesterol depletion inhibits epidermal growth factor receptor transactivation by angiotensin II in vascular smooth muscle cells: role of cholesterol-rich microdomains and focal adhesions in angiotensin II signaling. J Biol Chem. 2001;276:48269–48275. doi: 10.1074/jbc.M105901200. [DOI] [PubMed] [Google Scholar]
- 51.Filardo EJ, Quinn JA, Bland KI, Frackelton AR. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14:1649–1660. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 52.Meyer MR, Baretella O, Prossnitz ER, Barton M. Dilation of epicardial coronary arteries by the G protein-coupled estrogen receptor agonists G-1 and ICI 182,780. Pharmacology. 2010;86:58–64. doi: 10.1159/000315497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leuchowius KJ, Weibrecht I, Soderberg O. In situ proximity ligation assay for microscopy and flow cytometry. Curr Protoc Cytom. 2011 doi: 10.1002/0471142956.cy0936s56. Chapter 9: Unit9. [DOI] [PubMed] [Google Scholar]
- 54.Marin R, Ramirez C, Morales A, Gonzalez M, Alonso R, Diaz M. Modulation of Abeta-induced neurotoxicity by estrogen receptor alpha and other associated proteins in lipid rafts. Steroids. 2008;73:992–996. doi: 10.1016/j.steroids.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 55.Chambliss KL, Shaul PW. Rapid activation of endothelial NO synthase by estrogen: evidence for a steroid receptor fast-action complex (SRFC) in caveolae. Steroids. 2002;67:413–419. doi: 10.1016/s0039-128x(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 56.Campbell CH, Bulayeva N, Brown DB, Gametchu B, Watson CS. Regulation of the membrane estrogen receptor-alpha: role of cell density, serum, cell passage number, and estradiol. FASEB J. 2002;16:1917–1927. doi: 10.1096/fj.02-0182com. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.