

Abstract
Self-assembly of proteins on surfaces is utilized in many fields to integrate intricate biological structures and diverse functions with engineered materials. Controlling proteins at bio-solid interfaces relies on establishing key correlations between their primary sequences and resulting spatial organizations on substrates. Protein self-assembly, however, remains an engineering challenge. As a novel approach, we demonstrate here that short dodecapeptides selected by phage display are capable of self-assembly on graphite and form long-range ordered biomolecular nanostructures. Using atomic force microscopy and contact angle studies, we identify three amino-acid domains along the primary sequence that steer peptide ordering and lead to nanostructures with uniformly displayed residues. The peptides are further engineered via simple mutations to control fundamental interfacial processes, including initial binding, surface aggregation and growth kinetics, and intermolecular interactions. Tailoring short peptides via their primary sequence offers versatile control over molecular self-assembly, resulting in well-defined surface properties essential in building engineered, chemically rich, bio-solid interfaces.
Keywords: molecular self-assembly, inorganic binding peptides, nanotechnology, sequence mutation, atomic force microscopy, molecular recognition, liquid-solid interface
In organisms, interfaces in hard tissues are controlled by proteins via molecular recognition of specific mineral faces in, e.g., bones, spicules, shells, and teeth,1–3 where they initiate nucleation and regulate growth of specific phases to form intricate solid architectures.4–6 Inspired by biology, self-assembly of proteins onto solid surfaces is an enabling methodology for developing molecular surface coatings for a wide range of biological applications, e.g., biocompatible implants,7 controlled biofilms,8 and, more recently, the development of molecular biosensors.9 In these applications, protein functions on solids, such as molecular recognition10 and self-assembly,11 derives from inherently rich chemistry and complex molecular conformations coded by their amino acid sequences. Understanding the relationship between protein sequences and surface functions during self-assembly would establish them as highly programmable molecular constructs to tailor structure and chemistry of bio-solid interfaces. To this end, unique 2D organizations of proteins have been exploited on solid surfaces using, e.g., bacterial surface-layer proteins,12 linear amyloid structures13–15 and ordered films of de novo designed peptides.16, 17 However, the correlation between amino acid sequences and detailed 2D molecular ordering remains largely unknown, due to complex biomolecule-solid and intermolecular interactions that accompany surface growth processes.
Recently developed solid-binding peptides (7–30 amino acids),18–22 due to their short length and ease of chemical synthesis, offer a more comprehensive interrogation and, hence, control over their interactions with solids in contrast to larger protein systems (100 amino acids or more). These peptides are screened to preferentially bind to inorganic compounds from combinatorial phage libraries, generated by randomized oligonucleotides inserted in genes for the M13 phage coat protein19, 23. Biocombinatorially selected solid-binding peptides are shown to be selective, with high affinity, to a variety of solids such as metals, oxides, semiconductors as well as minerals,18, 19, 24 analogous in function to natural mineral-binding proteins. For example, a 42-amino acid gold binding peptide forms supramolecular nanostructures aligned to Au(111) via charged amino acids.21, 25 Surface functions of peptides have also been shown to be dependent on their molecular conformation.26 Previously, carbon-nanotube-binding peptides have been identified by biocombinatorial selection methods.22 Peptide self-assembly on graphite, however, remains largely unexplored although its atomically flat surface could be ideal for direct molecular investigations. Here, we utilize a graphite-binding dodecapeptide (GrBP) which forms long range ordered structures on graphite (0001). By simple substitution of amino acids in the dodecapeptide, we show that not only solid binding but, more importantly, key molecular interactions and surface phenomena can be probed, leading to control over the spatial organization and surface chemistry of self assembled peptide nanostructures on graphite.
RESULTS
In this molecular self-assembly study, we utilize GrBP5 (IMVTESSDYSSY, Fig. 1a), the strongest solid binding member of the biocombinatorially selected sixty different sequences, designated as the wild-type (WT) peptide. A combinatorial library of ~109 random 12-mer peptides fused to the minor coat protein (pIII) of M13 phage was used to select sequences with affinity to graphite flakes. Four selection rounds were carried out in the panning experiment, where each round consisted of (i) Panning the phage library against graphite powder, (ii) Rinsing unbound phage, (iii) Elution of specifically bound phages and (iv) Amplification of the enriched selection library. Affinity of the final selected clones were then quantified by spectrophotometric absorbance of depleted phage solutions after long exposure to graphite (see SI, Supplementary Methods S1-S3). We first characterize assembly of WT GrBP5 on highly ordered pyrolytic graphite (HOPG), incubated with 2 μM peptide solution in distilled water for three hours and scanned with an atomic force microscope (AFM) in air. The images reveal the unique capability of GrBP5 to form uniformly ordered molecular structures over several micrometers in dimension on the HOPG surface (Fig. 1b). The peptide film displays highly ordered nanostructures that display six-fold symmetry; seen as discrete maxima in the fast Fourier transform (FFT) power spectra taken by using the AFM image (Fig 1c). The symmetry in the peptide film is likely to be guided by the molecular recognition of the underlying (0001) hexagonal graphite lattice, leading to preferred growth along specific crystallographic directions during the assembly process. The measured height of ~1.4 nm implies that the film is monomolecular thick, well below the longest dimension of a stretched single peptide (with a stretched end-to-end distance of ~4.2 nm). The considerably short height implies that peptides conform into a more compact, folded, structure during molecular ordering on graphite.
Figure 1.
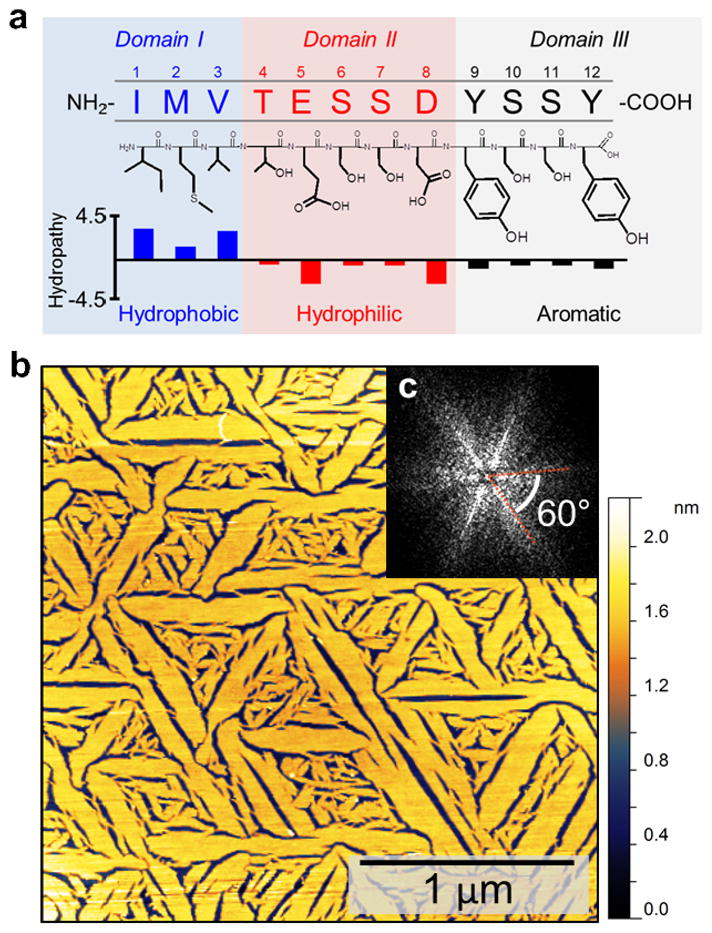
Chemical properties of GrBP5 sequence and its self-assembled, ordered, nanostructure on graphite (0001) lattice. (a) The three chemically distinct domains of GrBP5. The mean hydropathy (defined by Kyte and Doolittle)42 value of Domain-I is 3.53 (on an increasing scale from -4.5 to 4.5) and -1.86 for Domain-II, making the latter considerably more hydrophilic. (b) AFM image of GrBP5 on graphite showing ordering of the WT peptide over several micrometers displaying six-fold symmetrical self-assembled nanostructures, as observed in (c) the FFT of the AFM image.
To understand the formation of peptide nanostructures, graphite surfaces were exposed to GrBP5 in a time-lapsed series of experiments (Fig. 2a and b). Initially, upon 10 minutes of exposure to HOPG, peptides form discrete clusters ~10–50 nm in diameter with an average height of ~1.2 nm. At 60 minutes, two distinct phases are present, revealing that surface bound peptides undergo a dynamic morphological transformation from an amorphous (disordered) to an ordered state. The thicker, amorphous phase (AP) is ~1.8 nm in height, and the thinner, flat, ordered phase (OP) is ~1.3 nm in height. Phase-lag imaging by AFM signify a large difference between AP and OP structures (See SI, Supplementary Fig. 1). In the AP, surface-bound peptides likely crowd together randomly to form a topologically rough, disordered and porous structure (Fig. 2a, 60 min). By 180 minutes, the disordered phase has fully transformed into a flat ~1.3-nm thick OP monolayer with ordered morphology covering the HOPG. Pseudo three-dimensional renderings of the AFM images (Fig. 2b) better highlight the higher topography of disordered molecules among the ordered regions, showing a distinctive color for the OP (red strips) from the higher AP regions (yellow porous film). As seen in Fig. 2d, growing OP edges are dotted with peaks (in yellow, separated by ~30-nm average distance), suggesting that loosely ordered peptides are captured and incorporated into growing OP structures (also confirmed by in situ AFM experiments, See SI, Supplementary Fig. 2).
Figure 2.

Time-lapsed AFM of GrBP5 assembly. (a) Height contrast AFM images, for 10, 60 and 180 minutes, display structural evolution beginning with (left) discrete peptide clusters; (middle) growth of both amorphous (AP), and ordered (OP) phases as respectively labeled; and (right) complete OP monolayer. (b) Psuedo-3D representations of boxed regions showing height contrast among the phases formed: discrete (red), higher AP (yellow) and flat OP (orange), which are labeled below (c) on cross- sections of height taken across *---* in (a). Inset (d) shows lateral growth of OP including a cross sectional height taken between two peaks of AP peptides on either side. (e) Plot of percent total disordered/ordered peptide vs total coverage showing ordering transition and (f) Schematic of peptide self-assembly process highlighting surface phenomena: (i) Aggregation involving binding, diffusion, and clustering processes and (ii) Ordering involving self-assembly.
In contrast to the previous observations of peptide assembly on other atomically flat solid surfaces in the literature,21, 25, 27 we discovered in this work that the two phases formed by the peptides coexist until an eventual full ordering of a GrBP5 monolayer. This unique disordered-to-ordered peptide phase transformation takes place at about 60% total surface coverage as shown in Fig. 2e, where percent total coverage of disordered and ordered peptides are plotted against total surface coverage (See SI, Supplementary Methods S4). Depicted in Fig. 2f, the assembly process of GrBP5 can be divided into two broad regimes: (i) Surface aggregation, comprised of binding to and clustering of peptides on graphite, and (ii) A gradual densification and ordering process, accompanied by a phase transformation that occurs at ~60% total peptide coverage.
To demonstrate that the surface processes of GrBP5 self-assembly can be interrogated through rational mutations of the peptide, we first classify the sequence into three chemically distinct domains, as depicted in Fig. 1a: (I) hydrophobic (IMV), (II) hydrophilic (TESSD) and (III) aromatic (YSSY). Aromatic residues such as tyrosine (Y) are known to strongly interact with graphitic surfaces through a coupling of π-electrons.28, 29 Two of the four residues at the C-terminus of GrBP5 are aromatic-containing tyrosines (Y), defining YSSY as Domain-III. Located at the C-terminus of the peptide, this aromatic domain may function as an anchor for initial binding and possible diffusion during the aggregation regime of GrBP5 on graphite. On the other hand, intermolecular interactions necessary for long-range order may be driven by the amphiphilic tail comprised of hydrophobic Domain-I and hydrophilic Domain-II. Domain-I contains three purely aliphatic residues isoleucine (I), methionine (M) and valine (V), located at the N-terminus, while the Domain-II spans residues 4–8: threonine (T), glutamic acid (E), two serines (S), and aspartic acid (D) in the center of the peptide. The prevalence of amphiphilic motifs in many self-assembling molecular systems13, 14, 30 leads us to assume that the tail component of the peptide may play a key role in the AP to OP ordering transformation. Overall, therefore, we hypothesize that each domain can be correlated to the observed framework of the three regimes in the interfacial processes, i.e., binding, diffusion and self-assembly (Fig. 1f) which were then individually addressed using five mutants (labeled M1 through M5) generated specifically for this purpose.
To test the function of the presumed binding domain, first the aromatic content of Domain-III was knocked out by replacing both of the tyrosine residues at the C-terminus with alanine, A. This mutant peptide, named M1 (IMVTESSDASSA), is expected to maintain minimal interactions with graphite by displaying only methyl groups. Not unexpectedly, AFM analysis demonstrates that the HOPG surfaces remain bare even after exposure to 1 μM of M1 for up to 3 hours (Fig. 3a). From this simple mutation, the binding capability of GrBP5 to graphite is largely eliminated, indicating that the roles of hydrophobic residues in Domain-I and hydrophilic residues in Domain-II are not sufficient to promote an interaction with graphite. More importantly, these results show that Domain-III (YSSY), provides anchoring of the peptide to the surface, and can be addressed independently of the remaining sequence to alter peptide binding and, possibly, surface diffusion leading to aggregation.
Figure 3.

Time-lapse behavior of the peptides with Domain-III mutations. (a) In Mutant 1 the aromatic residues, Tyrosine (Y), of GrBP5 are eliminated and replaced by Alanine (A) resulting in no bound molecules on surface. (b) Tryptophan and (c) Phenylalanine replace WT-Tyrosine in Mutants 2 (M2) and 3 (M3), respectively. The resultant peptides, respectively, are either strongly bound to the surface forming percolated, but finely porous, film (M3) or weakly bound peptides forming isolated islands, each over the course of 3 hours. (d) Fractional coverage trends from time-lapse AFM of WT, M1, M2, and M3; (e) Particle count of each of the peptides; and (f) Average particle size over time; Error bars represent standard deviation of 3 different images from the sample surface, totaling an area of 16 μm2.
To examine the binding characteristics of the peptide further, the tyrosine residues in Domain-III were replaced with either tryptophan (W) or phenylalanine (F), two other natural amino acids containing aromatic moieties. Previous studies suggest that, W, containing an extra indole-ring, provides a more conformal and stable π-interaction with graphite surfaces, giving it a higher affinity over Y. On the other hand, F, lacking an OH- group, was found to maintain the weakest affinity towards graphite.28, 29, 31 Thus, the designed two mutant sequences, M2 (IMVTESSDWSSW) and M3 (IMVTESSDFSSF) may have different binding and aggregation from those of GrBP5. Systematic time-lapsed AFM experiments show that the type of aromatic residues in the anchoring domain significantly influences the formation of peptide nanostructures on graphite (Fig. 3b and c). M2 and M3 display either a highly porous disordered film or only fine peptide clusters, respectively. The effect of aromatic residues on binding, as well as unbinding, is evident when the initial deposition rates of peptides, D, are quantified and compared among samples prepared at the earliest exposure times: 5 seconds for WT, 10 min for M2 and M3 (See SI, Supplementary Methods S7). Such an analysis reveals that WT peptides arrive at the surface with an estimated D of ~4235±43 s−1, while M2 arrives at ~73±9 s−1 and M3 at 22±8 s−1 over a 1 μm2 area (Fig. 3d). Thus, there is a ~60x discrepancy in initial binding between WT and M2, while M3 remains the slowest.
In addition to binding, the anchoring domain may also have a fundamental role in cluster formation during the initial aggregation stages of interfacial processes on solid surfaces. Measuring the number and size of peptide cluster features on graphite over time provides means to quantitatively track peptide surface kinetics in response to Domain-III mutations. In this scheme, surface aggregation is manifested as a decrease in the number density of clusters while the cluster size increases on the average. AFM analyses of early adsorption by WT and M2 reveal that, as total area coverage increases over time, the number of WT clusters decreases at a rate of ~1 s−1 compared to a rate of ~1.3×10−2 s−1 for M2 over an area of 1 μm2; a 75-fold difference (determined from the initial slope from Fig. 3e and See SI, Supplementary Fig. 3). While the number of clusters decreases, as expected, the average cluster size increases over time for both WT and M2 peptides (Fig. 3f); clusters of WT grow 24x faster than those of M2. These observations signify that WT has a higher aggregation rate than M2. In contrast, the number of clusters in M3 increases over time while the size remains the same, implying limited diffusion and, hence, lack of aggregation. The ~58x discrepancy in deposition rate between WT and M2 might explain the higher aggregation rate of WT over M2. The relative binding and aggregation rates of mutant peptides can, therefore, be ranked in decreasing order as: WT, highest binding and aggregation; M2, low binding and low aggregation; M3, low binding and no aggregation; and M1, no binding.
The aggregation behavior of peptides is likely to be the key in determining their final, ordered nanostructures on graphite. This phenomenon was further studied by exposing graphite samples to three concentrations of peptide solutions for 3 hours each, as shown in Figure 4. Upon this incubation period, WT formed long-range ordered self-assembled peptide nanostructures in all three conditions, i.e., 0.1, 1.0 and 5.0 μM. The mutant M2, with the second highest rate of aggregation, remains disordered at 0.1 μM and 1.0 μM concentrations; interestingly, however, at 5.0 μM it forms very finely ordered structures after 3 hours of incubation (See Fig. 4 and SI, Supplementary Fig. 5). Since the density of peptide clusters increases monotonically with total surface coverage, a critical density of clusters is likely required for ordering. This is apparent for the WT peptide, where the film undergoes transformation from AP to OP at ~60% total surface coverage (Fig. 2e). At 60%, half of the adsorbed WT peptide exists in the ordered phase, indicating threshold coverage for transformation. M2, on the other hand, remains entirely disordered at the same total surface coverage. Since M2 eventually orders at near 100% coverage, it likely undergoes transformation at a higher total coverage than WT. We speculate that the slower aggregation kinetics of M2 impedes peptide clusters from crowding on the surface and results in finer AP features, defining the small dimension of the ordered features observed at 5 μM. On the other hand, M3 remains too sparsely clustered at all three concentrations used here, never reaching a coverage threshold and remaining discretely bound even after 3 hours and at the highest concentrations. The trends in peptide aggregation rates were also verified by quantifying differences in values of peptide affinity constants, K, which are quantitatively estimated using Langmuir-like treatments (Fig. 4, and Supplementary Methods S9). Here, the Y-containing WT displayed the highest K of 3.78 μM−1, while the WSSW mutant had a K of 1.17 μM−1; an affinity to graphite of about one third of the WT value. Lastly, the mutant M3 shows a significant loss of affinity with a low K of ~ 0.1 μM−1. The high K of WT peptide may be an indication that the highly ordered structures are the most stable on graphite surfaces due to favorable intermolecular interactions as well as their strong surface binding.
Figure 4.

Quantification of graphite affinity for aromatic mutants. (a) AFM images of HOPG exposed to 0.5 and 5.0 μM of WT, M1, M2, and M3 peptides for 3 hours. (b) Graph of surface coverage for each peptide plotted against concentration, and fitted using a Langmuir adsorption model for affinity constant, K. Error bars represent standard deviation of 3 different images from the sample surface, totaling an area of 16 μm2.
To probe the domain that directs the ordering seen in the WT GrBP5 on graphite, Domain-I at the N-terminus was mutated by modifying its hydrophobic nature. For this purpose, both negative and positive sequence knock-outs were prepared. In the design of a negative knock-out sequence, we replaced IMV with three similarly sized hydrophilic amino acids: threonine (T), glutamine (Q) and serine (S). The resulting sequence of mutant M4, therefore, is entirely hydrophilic. In contrast to the highly ordered structures of WT peptide on the surface (Fig. 5a), the M4 peptide forms highly porous and disordered structures (Fig. 5b). Next, the positive knock-out mutant M5 was designed to restore the hydrophobic characteristics of Domain-I and, therefore, the amphiphilic nature of the overall tail, presuming this mutant might allow the formation of ordered structures on graphite. Here IMV was replaced with three other aliphatic amino acids; leucine (L), isoleucine (I) and alanine (A) which results in slightly higher hydropathy than that of WT. As expected, the mutant M5 is also found to bind strongly to graphite (See SI, Supplementary Fig. 4), and, upon assembly, maintains ordering similar to that by WT (Fig. 5a and c). This result, therefore, proves that the hydrophobic nature of Domain-I, and the amphiphilic tail it forms with Domain-II, are essential for long-range order of the dodecapeptide on graphite.
Figure 5.

Chemical properties of Domain-I mutants and their assembly behavior. (Left column) Domain-I mutant sequences and hydropathy values; (Right column) AFM images of hydropathic mutants on HOPG, and (insets) contact angle measurements of imaged surfaces. (a) WT GrBP5 forms long-range ordered nanometer-scale structure and a high contact angle, θCA, of 65.3°. (b) Hydrophilic mutant M4 does not form an observable long range order and displays a low contact angle of 34.7°; while (c) Hydrophobic mutant M5 forms an ordered peptide film, similar to that of WT, with a much greater θCA of 88.9°.
These hydropathic mutations can also be utilized to control the chemical characteristics of the graphite surface, such as wettability. For this, contact angle (θCA) of aqueous droplets were measured on HOPG coated with confluent peptide films, each at a comparable surface coverage (Fig. 5 insets, see Methods and Materials). The ordered WT and M5 films display an average contact angle of 65.3±0.8° and 88.9±0.7°, respectively, indicating that peptides with hydrophobic Domain-I exposed display surfaces with low wettability. In contrast, graphite covered by disordered peptides, e.g., the case of M4, exhibit a significant drop in θCA with a contact angle of 34.7±1.2°, forming a wetting surface. A simple knock out of hydrophobic Domain-I in M4, therefore, considerably lowers θCA as compared to those in WT and M5. These results demonstrate that the chemical properties of graphite surface are tunable by rational amino acid mutations, governed by specific peptide conformations brought about by the self-assembly processes.
DISCUSSION
The correlation between sequence and self-assembly, as established, provides key insight into the transformation from disordered to ordered nanostructures of the peptide, GrBP5, on graphite. An analysis of the amphiphilic behavior of our peptides, manifested by a change in concurrent wetting angle and film height over time, permits us to propose a unique transition in the molecular conformation during assembly on the solid surface. Contact angle measurements of samples scanned by AFM containing AP peptide reveal that the θCA values for WT, M4 and M5 decrease linearly with surface coverage, becoming hydrophilic. The linear trend of cos(θCA) shown in Fig. 6a (blue dotted line) for all three peptides implies that the AP displays chemistries, and conformations, are similar in both discrete and confluent states.32 As seen in Fig. 5, ordered films present a shift to greater hydrophobicity. In the low coverage regime, the terminal hydrophobic amino acid domain of the WT peptide is likely first buried towards graphite while hydrophilic residues are exposed to water, as depicted in Fig. 6b. When coverage reaches a certain threshold, however, a drastic shift is observed from hydrophilic towards more hydrophobic values (black arrow in Fig. 6a). This phenomenon suggests that the hydrophobic domain emerges from its buried state and is, then, exposed to water, effectively switching the amphiphile’s conformation. The plausible changes in the molecular structure of peptides between the disordered and ordered states are concurrent with the height changes of the peptides, as determined by AFM (colored as “pink” in Fig 5a). Here, the height of WT and M5 peptide films exhibit a drastic contraction by 33% and 50%, respectively. The range of height changes observed for the OP is only ~0.1 nm, while it is ~1.0 nm for the AP. Thus, the switch in surface wetting properties, accompanied by a physical molecular contraction, leads us to believe that amphiphilic peptides undergo folding to reach their energetically favored, and uniform, final state in the ordered phase on the surface. The purely hydrophilic M4 peptide, by contrast, exhibits no folding, or ordering, that can be measured by AFM or contact angle, as plotted in Fig. 6a and schematically illustrated in Fig. 6c.
Figure 6.
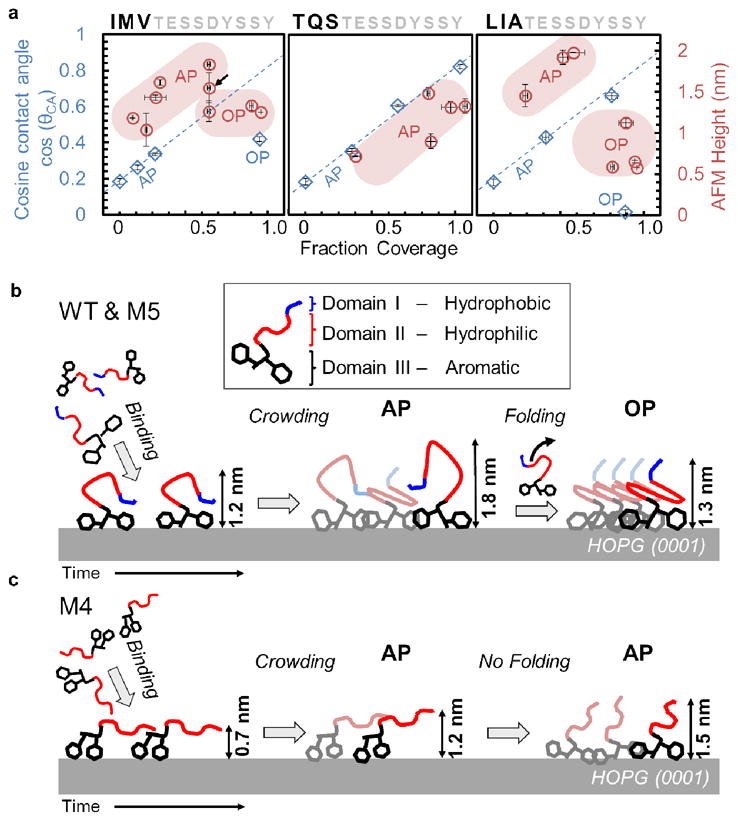
Behavior of peptides during molecular self-assembly. (a) AFM height (red) and cosine of contact angle, cos(θCA), (blue) both plotted against surface coverage, respectively. Data from AP and OP images labeled accordingly. Blue dotted lines represent guides for the AP of peptides to demonstrate their shared linear behavior, i.e., chemistry, as defined by Cassie’s Law.43 Black arrow in WT indicates heights averaged from an image containing both AP and OP. Horizontal error bars represents standard deviation from 3 different analyzed images on the sample surface, totaling an area of 16 μm2. Vertical error bars are the standard deviation from two droplets on duplicate samples (b) A schematic of WT and M5 self-assembly mechanism where peptides undergo binding and diffusion via Domain-III, first aggregating randomly to form rough AP and, finally, rearranging Domains-I and -II while folding into OP. (c) Mechanism of M4 in the absence of hydrophobic Domain-I; here there is neither retraction in height nor change in surface chemistry.
CONCLUSION
Detailed investigation of molecular self-assembly by a graphite-binding peptide, GrBP5, on graphite studied by AFM imaging revealed a strong correlation between the amino acid composition and sequence to the resulting self-assembled nanostructures. Formation of peptide nanostructures on graphite involves first the formation of a disordered film that eventually transforms into an ordered structure. This is accompanied by height changes in the film as well as various wetting characteristics, as determined by contact angle measurements. Analysis of the sequence reveals three distinct chemical domains which play a role in the binding, diffusion, and assembled organization of the peptide on graphite: aromatic, hydrophilic, and hydrophobic. Mutations of the aromatic domain at the C-terminus significantly modifies the binding characteristics of GrBP5. Tyrosine (Y) residues replaced by alanine (A) largely eliminates the ability for the peptide to bind to graphite, while replacing tyrosine with tryptophan (W) or phenylalanine (F) tunes the peptide’s affinity to graphite from strong to weak or moderate binding, respectively. Mutation of the tail at the N-terminus from hydrophobic to hydrophilic eliminates the amphiphilic character of the peptide, disrupting intermolecular interactions on the surface, and prevents bound peptides from forming long range ordered nanostructures. Disordered and ordered nanostructures of peptide mutants on graphite display either hydrophilic or hydrophobic domains, respectively, effectively tuning the contact angle and, thus, giving the film wetting or non-wetting characteristics.
A wide-range of surface phenomena exhibited by peptides during assembly, e.g., binding, clustering, and ordering, share large similarities with well-established epitaxial growth processes of atomic systems on surfaces, e.g., in molecular beam epitaxy (MBE),33, 34 the foundation for modern semiconducting devices. As demonstrated (Figs. 3 and 4), the growth behavior of peptides can be controlled by varying the peptide concentration and incubation time, which effectively changes their rate of arrival to the surface and growth on graphite. Unique to peptides, however, a simple sequence of amino acids, as found here, can be further engineered to contain programmable segments for independently controlling multiple surface and intermolecular interactions. The ability to address peptide domains provides an opportunity for the predictable control over biomolecular self-assembly in the formation of complex, novel nanoarchitectures i.e., nanoislands (M2), nanowires (WT), and amorphous and ordered confluent films (M5 and WT, respectively). Self-assembled peptides (SAPs), with the capability to form ordered nanostructures and controlled surface chemistries, therefore, have the potential to be the foundation of future peptide-based hybrid molecular technologies such as protein chips,35, 36 peptide-molecular circuits,37 and designer multifunctional proteins38 and enzymes,39 that can be genetically engineered to perform diverse, addressable functions.
METHODS
Peptide synthesis
Peptides were prepared on an automated solid-phase peptide synthesizer (CS336X, CSBio Inc., Menlo Park, CA) employing standard batch wise Fmoc chemistry procedures as reported previously.21 Peptides were verified by MALDI-TOF mass spectrometry. The monomeric state of peptides in solution was also verified via size-exclusion chromatography (See SI, Supplementary Methods S12).
Sample Preparation for Microscopy
For ex situ imaging, it was essential to prevent the reorganization of peptide structures during the drying process. We found that removing incubation solutions by applying a flash freeze and freeze-dry technique, common in biological electron microscopy specimen preparation,40, 41 preserved surfaces adequately. Freshly cleaved HOPG surfaces are mounted on a nickel specimen puck (Ted Pella, Inc., Redding, CA) and incubated with 50 μL peptide under experimental conditions in a modified scintillation vial. The vial contains a centered hollow glass column which elevates the sample to minimize contact with warm elements during the freeze-dry process. When incubation is finished, the vial is immediately placed in a −80C deep freezer which freezes within 10–15 seconds. Samples are then immediately placed inside a glass jar and surrounded with crushed ice. The jar is placed in a liquid N2 bath and transferred to a standard freeze-drier (Virtis Benchtop K, SP Industries, Inc., Warminster, PA) and immediately placed in a vacuum with a −80C condensing plate to sublime frozen incubation solutions. The drying rate was ~ 6 μL/hr. All images in this study, except for Fig. 1a, were prepared using this method. Surfaces in Fig. 1a were prepared by wicking incubation solution with a KimWipe. For verification, we performed extensive time and concentration experiments using our 5 mutants and found (see Figs. 2,3 and SI Fig. 2) agreement in coverage, height and density trends. In all, ~60 samples were prepared in this study for reproducibility.
Atomic force microscopy
A Digital Instruments (Veeco, Santa Barbara, CA) Multimode Nanoscope IIIa scanning probe microscope equipped with high frequency NanoSensors PPP-NCHR (NanoandMore USA, Lady’s Island, SC, USA) non-contact probes, with a 42 N/m spring constant, at a 4V amplitude set point.
AFM Image Processing
Large ‘wavy’ topographic features coming from HOPG surfaces observed by AFM were removed by image subtraction to allow large area coverage analysis of peptides (e.g., see SI, Fig. S5A). Specifically, GWYDDION (Czech Metrology Institute, Czech Republic) image filters and simple image operations were applied to the raw AFM data. First, images were corrected for tilt by a first order plane subtraction while fast scan lines were normalized by aligning their median z-offset as seen in SI Fig. S5A. Next, an erosion filter was applied to AFM images using a neighborhood of 10–15 pixels, where peptide features are removed to create a secondary image containing only the topography of the bare surface, as in SI Fig. S5B. This secondary image is then directly subtracted from the original AFM image, yielding a background subtracted image for surface coverage analysis (SI Fig. S5C). To quantify surface coverage values, IMAGEJ (NIH, Bethesda, MD, USA) is used to determine the threshold depth in the image where full lateral surface features are measured. This threshold is then used to form a binary image.
To calculate coverage values for mixed images comprised of both disordered and ordered peptides (e.g., Fig. 2e), a particle analyzer was used to distinguish peptide features via SPIP (Image Metrology A/S, Denmark) as shown in the left-hand images of SI Fig. S4. Ordered peptides generally occupy higher area coverage over disordered peptides and can be quantified separately for independent coverage values based on particle size.
Height measurements of discretely bound, amorphous and ordered phases of peptide in Figure 5 were measured by AFM height image histogram analysis. Peaks in the histogram arise from a predominant number of pixels at certain heights. For peptides on HOPG, bare surface pixels form a prominent peak due to the atomically flat nature of graphite. Particles observed on the surface, likely clustered or monomeric peptides, are tip convoluted and fail to reflect the true maximum height of interest in the histogram so image filtering is necessary. To address this, dilation filtering was used, as seen in SI Fig. S6 insets, to enhance the total number of pixels representing the maximum heights of bound particles. This is reflected by an upward shift in peptide peak height on the histogram, where overall height is measured with respect to the bare graphite peak from the original image. These values agree with individual cross sectional measurements. In confluent films, no filtering was used since pixels mainly come from the flat film and form a dominant peak. For images with two phases present, height is measured from each phase independently as shown in SI Fig. S6.
Contact Angle Study
For coverage normalized θCA values, 50 mm2 HOPG samples were freshly cleaved and immediately incubated with 80 μL of appropriate peptide solution in water ranging from 10 mins to 7 hrs. The drop was then wicked off using a tissue and the sample was dried under a gentle stream of N2. Samples were equilibrated in air for 30 min prior to contact angle measurements. Static sessile contact angles were measured by an FTA1000B Goniometer (First Ten Angstroms, Inc., Portsmouth, VA) with a digital camera and auto-capture software system by the vendor after 2 μL of peptide solution is dropped, performed in duplicate for each surface. The peptide solutions were placed on graphite with the same concentrations as used for their original assembly to prevent desorption. The samples were then dried with nitrogen and measured for coverage by AFM. AFM images were obtained from at least four different locations (4 μm x 0.5 μm sized scans each) on all samples by the methods described above. (See SI, Supplementary Methods S11)
Supplementary Material
Acknowledgments
The research was supported by NSF-MRSEC program (DMR-0520567) at GEMSEC, Genetically Engineered Materials Science and Engineering Center, University of Washington. CS and YH were supported by the NCI Training Grant T32CA138312 (NIH) and JST PRESTO programs (Japan), respectively, and also by NSF Biomat program. HY, CG, DK, CT and MS were supported by MRSEC. The work was carried out at GEMSEC-SECF, a member of Materials Facilities Network of MRSEC.
Footnotes
Author Contributions. C.R.S., Y.H., and M.S. designed the study; C.R.S., H.Y. and D.K. performed experiments; C.R.S., Y.H. and M.S. analyzed data; H.Y., C.T., and C.G. generated sequences and synthesized peptides; C.R.S., Y.H. and M.S. wrote the manuscript; M.S. supervised the research.
Supporting Information Available. A supporting information section with extensive figures, lengthy experimental procedures, and expanded discussions of peripheral findings is available. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lowenstam H, Weiner S. On Biomineralization. Oxford University Press; Oxford: 1989. [Google Scholar]
- 2.Sarikaya M, Aksay I. Biomimetics: Design & Processing of Materials. AIP; College Park: 1996. [Google Scholar]
- 3.Mann S. Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry. Oxford University Press; Oxford: 2001. [Google Scholar]
- 4.Addadi L, Weiner S. Interactions between Acidic Proteins and Crystals - Stereochemical Requirements in Biomineralization. Proc Natl Acad Sci USA. 1985;82:4110–4114. doi: 10.1073/pnas.82.12.4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mann S. Molecular Recognition in Biomineralization. Nature. 1988;332:119–124. [Google Scholar]
- 6.Cha JN, Shimizu K, Zhou Y, Christiansen SC, Chmelka BF, Stucky GD, Morse DE. Silicatein Filaments and Subunits From a Marine Sponge Direct the Polymerization of Silica and Silicones in vitro. Proc Natl Acad Sci USA. 1999;96:361–365. doi: 10.1073/pnas.96.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ratner BD, Hoffman AS, Schoen FJ, Lemons JE. Biomaterials Science, Second Edition: An Introduction to Materials in Medicine. 2. Elsevier Academic Press; San Diego: 2004. [Google Scholar]
- 8.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric Control of Cell Life and Death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 9.Templin MF, Stoll D, Schrenk M, Traub PC, Vohringer CF, Joos TO. Protein Microarray Technology. Trends Biotechnol. 2002;20:160–167. doi: 10.1016/s0167-7799(01)01910-2. [DOI] [PubMed] [Google Scholar]
- 10.Pauling L. Molecular architecture and Biological Reactions. Chem Eng News. 1946;24:1375–1377. [Google Scholar]
- 11.Bradley DE. Ultrastructure of Bacteriophages and Bacteriocins. Bacteriol Rev. 1967;31:230–314. doi: 10.1128/br.31.4.230-314.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gyorvary ES, Stein O, Pum D, Sleytr UB. Self-assembly and Recrystallization of Bacterial S-layer Proteins at Silicon Supports Imaged in Real Time by Atomic Force Microscopy. J Microsc (Oxf) 2003;212:300–306. doi: 10.1111/j.1365-2818.2003.01270.x. [DOI] [PubMed] [Google Scholar]
- 13.Sneer R, Weygand MJ, Kjaer K, Tirrell DA, Rapaport H. Parallel β,-Sheet Assemblies at Interfaces. Chemphyschem. 2004;5:747–750. doi: 10.1002/cphc.200301046. [DOI] [PubMed] [Google Scholar]
- 14.Zhang F, Du HN, Zhang ZX, Ji LN, Li HT, Tang L, Wang HB, Fan CH, Xu HJ, Zhang Y, et al. Epitaxial Growth of Peptide Nanofilaments on Inorganic Surfaces: Effects of Interfacial Hydrophobicity/Hydrophilicity. Angew Chem Int Edit. 2006;45:3611–3613. doi: 10.1002/anie.200503636. [DOI] [PubMed] [Google Scholar]
- 15.Harper JD, Wong SS, Lieber CM, Lansbury PT. Observation of Metastable A-β, Amyloid Protofibrils by Atomic Force Microscopy. Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 16.Brown CL, Aksay IA, Saville DA, Hecht MH. Template-Directed Assembly of a de novo Designed Protein. J Am Chem Soc. 2002;124:6846–6848. doi: 10.1021/ja0261271. [DOI] [PubMed] [Google Scholar]
- 17.Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, Willis L, Drndic M, Kikkawa JM, DeGrado WF. Computational Design of Virus-Like Protein Assemblies on Carbon Nanotube Surfaces. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarikaya M, Tamerler C, Jen AKY, Schulten K, Baneyx F. Molecular Biomimetics: Nanotechnology through Biology. Nat Mater. 2003;2:577–585. doi: 10.1038/nmat964. [DOI] [PubMed] [Google Scholar]
- 19.Whaley SR, English DS, Hu EL, Barbara PF, Belcher AM. Selection of Peptides with Semiconductor Binding Specificity for Directed Nanocrystal Assembly. Nature. 2000;405:665–668. doi: 10.1038/35015043. [DOI] [PubMed] [Google Scholar]
- 20.Sano KI, Shiba K. A Hexapeptide Motif that Electrostatically Binds to the Surface of Titanium. J Am Chem Soc. 2003;125:14234–14235. doi: 10.1021/ja038414q. [DOI] [PubMed] [Google Scholar]
- 21.So CR, Kulp JL, Oren EE, Zareie H, Tamerler C, Evans JS, Sarikaya M. Molecular Recognition and Supramolecular Self-Assembly of a Genetically Engineered Gold Binding Peptide on Au{111} ACS Nano. 2009;3:1525–1531. doi: 10.1021/nn900171s. [DOI] [PubMed] [Google Scholar]
- 22.Wang SQ, Humphreys ES, Chung SY, Delduco DF, Lustig SR, Wang H, Parker KN, Rizzo NW, Subramoney S, Chiang YM, et al. Peptides with Selective Affinity for Carbon Nanotubes. Nat Mater. 2003;2:196–200. doi: 10.1038/nmat833. [DOI] [PubMed] [Google Scholar]
- 23.Naik RR, Brott LL, Clarson SJ, Stone MO. Silica-Precipitating Peptides Isolated From a Combinatorial Phage Display Peptide Library. J Nanosci Nanotechnol. 2002;2:95–100. doi: 10.1166/jnn.2002.074. [DOI] [PubMed] [Google Scholar]
- 24.Brown S. Metal-Recognition by Repeating Polypeptides. Nat Biotechnol. 1997;15:269–272. doi: 10.1038/nbt0397-269. [DOI] [PubMed] [Google Scholar]
- 25.So CR, Tamerler C, Sarikaya M. Adsorption, Diffusion, and Self-Assembly of an Engineered Gold-Binding Peptide on Au(111) Investigated by Atomic Force Microscopy. Angew Chem Int Edit. 2009;48:5174–5177. doi: 10.1002/anie.200805259. [DOI] [PubMed] [Google Scholar]
- 26.Hnilova M, Oren EE, Seker UOS, Wilson BR, Collino S, Evans JS, Tamerler C, Sarikaya M. Effect of Molecular Conformations on the Adsorption Behavior of Gold-Binding Peptides. Langmuir. 2008;24:12440–12445. doi: 10.1021/la801468c. [DOI] [PubMed] [Google Scholar]
- 27.Kowalewski T, Holtzman DM. In situ Atomic Force Microscopy Study of Alzheimer's β,-Amyloid Peptide on Different Substrates: New Insights into Mechanism of β,-Sheet Formation. Proc Natl Acad Sci USA. 1999;96:3688–3693. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie H, Becraft EJ, Baughman RH, Dalton AB, Dieckmann GR. Ranking the Affinity of Aromatic Residues for Carbon Nanotubes by Using Designed Surfactant Peptides. J Pept Sci. 2008;14:139–151. doi: 10.1002/psc.978. [DOI] [PubMed] [Google Scholar]
- 29.Tomasio SM, Walsh TR. Modeling the Binding Affinity of Peptides for Graphitic Surfaces. Influences of Aromatic Content and Interfacial Shape. J Phys Chem C. 2009;113:8778–8785. [Google Scholar]
- 30.Whitehouse C, Fang JY, Aggeli A, Bell M, Brydson R, Fishwick CWG, Henderson JR, Knobler CM, Owens RW, Thomson NH, et al. Adsorption and Self-Assembly of Peptides on Mica Substrates. Angew Chem Int Edit. 2005;44:1965–1968. doi: 10.1002/anie.200462160. [DOI] [PubMed] [Google Scholar]
- 31.Sowerby SJ, Petersen GB, Holm NG. Primordial Coding of Amino Acids by Adsorbed Purine Bases. Origins Life Evol B. 2002;32:35–46. doi: 10.1023/a:1013957812213. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz DK. Mechanisms and Kinetics of Self-Assembled Monolayer Formation. Annu Rev Phys Chem. 2001;52:107–137. doi: 10.1146/annurev.physchem.52.1.107. [DOI] [PubMed] [Google Scholar]
- 33.Barth JV, Costantini G, Kern K. Engineering Atomic and Molecular Nanostructures at Surfaces. Nature. 2005;437:671–679. doi: 10.1038/nature04166. [DOI] [PubMed] [Google Scholar]
- 34.Jensen P, Barabasi AL, Larralde H, Havlin S, Stanley HE. Deposition, Diffusion and Aggregation of Atoms on Surfaces - a Model for Nanostructure Growth. Phys Rev B. 1994;50:15316–15329. doi: 10.1103/physrevb.50.15316. [DOI] [PubMed] [Google Scholar]
- 35.Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, Lan N, Jansen R, Bidlingmaier S, Houfek T, et al. Global Analysis of Protein Activities using Proteome Chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]
- 36.Langer R, Peppas NA. Advances in Biomaterials, Drug Delivery, and Bionanotechnology. Aiche J. 2003;49:2990–3006. [Google Scholar]
- 37.Joachim C, Gimzewski JK, Aviram A. Electronics using Hybrid-Molecular and MonoMolecular Devices. Nature. 2000;408:541–548. doi: 10.1038/35046000. [DOI] [PubMed] [Google Scholar]
- 38.Baneyx F, Mujacic M. Recombinant Protein Folding and Misfolding in Escherichia Coli. Nat Biotechnol. 2004;22:1399–1408. doi: 10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- 39.Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, Clair JLS, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, et al. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science. 2010;329:309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gross H. Cryotechniques in Biological Electron Microscopy. 1. Springer; New York: 1987. [Google Scholar]
- 41.Engel A. Biological Applications of Scanning Probe Microscopes. Annu Rev Biophys Bio. 1991;20:79–108. doi: 10.1146/annurev.bb.20.060191.000455. [DOI] [PubMed] [Google Scholar]
- 42.Kyte J, Doolittle RF. A Simple Method for Displaying the Hydropathic Character of a Protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 43.Cassie ABD, Baxter S. Wettability of Porous Surfaces. T Faraday Soc. 1944;40:0546–0550. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.