

Abstract
There has been no extensive characterization of the effects of Ginsenoside Rg1, a pharmacological active component purified from the nature product ginseng, in an Alzheimer's disease mouse model. The well-characterized transgenic Alzheimer disease (AD) mice over expressing amyloid precursor protein (APP)/Aβ (Tg mAPP) and nontransgenic (nonTg) littermates at age of 6 and 9 month were treated with Rg 1 for three months via intraperitoneal injection. Mice were then evaluated for changes in amyloid pathology, neuropathology and behavior. Tg mAPP treated with Rg1 showed a significant reduction of cerebral Aβ levels, reversal of certain neuropathological changes, and preservation of spatial learning and memory, as compared to vehicle-treated mice. Rg1 treatment inhibited activity of γ-secretases in both Tg mAPP mice and B103-APP cells, indicating the involvement of Rg1 in APP regulation pathway. Furthermore, administration of Rg1 enhanced PKA/CREB pathway activation in mAPP mice and in cultured cortical neurons exposed to Aβ or glutamate –mediated synaptic stress. Most importantly, the beneficial effects on attenuation of cerebral Aβ accumulation, improvement in neuropathological and behavioral changes can be extended to the aged mAPP mice, even to 12-13 months old mice that had extensive amyloid pathology and severe neuropathological and cognitive malfunction. These studies indicate that Rg1 has profound multi-faced and neuroprotective effects in an AD mouse model. Rg1 induces neuroprotection through ameliorating amyloid pathology, modulating APP process, improving cognition, and activating PKA/CREB signaling. These findings provide a new perspective for the treatment of AD and demonstrate potential for a new class of drugs for AD treatment.
Keywords: Rg1, AD mouse model, neuroprotection, gamma secretase
1. Introduction
Alzheimer's disease (AD) is defined by the progressive impairment in memory and cognition and by the presence of two major pathological hallmarks, accumulation of β-amyloid protein (Aβ) and formation of neurofibrillary tangles composed of hyperphosphorylated tau. Aβ, a key constituent of amyloid plaque, is a neurotoxic peptide that contributes to neuronal, synaptic and cognitive malfunction in an Aβ rich environment found in human AD and the AD mouse model. Thus, strategies that reduce Aβ levels in the brain and improve cognitive function are critical for prevention and/or halting of AD progression.
Ginseng, a drug used in traditional Chinese medicine that has a wide range of pharmacological effects, has been empirically used to alleviate many ailments, particularly those associated with aging and memory deterioration [1-3]. Rg1, one of the bioactive compounds in ginseng crosses the blood-brain barrier [4]. Several studies provide evidence of its neurotrophic, neuroprotective and anti-aging properties [5-8]. Pretreatment with Rg1 improved viability in cells injured by Aβ, reduced the levels of intracellular Aβ 1-42, and attenuated the activation of caspase-3 and apoptosis [9-11]. Furthermore, recent studies demonstrated that administration of ginsenosides including Rg 1 reduced cerebral Aβ in Tg 2576 mice [12], an AD mouse model as well as aging mice [13]. However, the mechanisms underlying Rg1-involved attenuation of Aβ accumulation and improvement in learning and memory in an Aβ-rich environment remain unclear.
Aβ1-40 and Aβ1-42 amino acid peptides generated by endoproteolysis of the Type I membrane protein, amyloid precursor protein(APP) [14]. Aβ formation involves the sequential cleavage of amyloid precursor protein (APP) by two proteases, the β- and γ-secretases [15]. β secretase cleaves APP at the N-terminus to form APP-CTF-β (C99), which is subsequently cleaved by a multi-protein γ-secretase complex that results in generation of the Aβ peptide and a smaller APP-CTF-γ. It follows then that γ secretase is a potential prime drug target for the treatment of Alzheimer's disease. However, it is unclear whether Rg1 is involved in regulation of the APP process via γ secretase.
Tg mAPP mice have been well characterized with respect to mitochondrial function and neuropathological, behavioral and electrophysiological endpoints [16-23]. The strengths of this model include the rapid accumulation of Aß (started at 2-4 months; plaque formation at 6 months), decreased density of presynaptic terminals, abnormalities in synaptic, electrophysiological and behavioral changes. Importantly, there is no report on studying Rg1 in this model with overexpression of human Aβ including APP process and neuropathological changes. Thus, it would be essential and logical for choosing an aggressive AD model to determine the impact of Rg1 on neuronal and cognitive function.
The goals of the present study are: 1) to fully assess the effects of Rg 1 on cerebral Aβ accumulation, amyloid pathology, neuronal function, and learning/memory capacity in a well-characterized mouse model for AD; and 2) to elucidate the pathways affected by Rg1 that suppress Aβ production and improve neuronal and cognitive function. Results generated from the study would provide substantial evidence for preclinical translation study of Rg1 as a new therapeutic agent for prevention of AD and slowing of the disease process.
2. Material and Methods
2.1 Tg mice and cell culture
All animal experiments were approved by the Institutional Animal Care and Use Committee of Columbia University and conformed to the guidelines outlined in the National Institutes of Health Guide for Care and Use of Laboratory Animals (NIH Pub. No. 85-23, 1996). Transgenic (Tg) mice with neuronal overexpression of a mutant human form of amyloid precursor protein (Tg mAPP, J-20 line, originally provided by Dr. Lennart Mucke, University of California, San Francisco) driven by the platelet-derived growth factor (PDGF) B-chain promoter were used in this study. These Tg mice are a well-established mouse model for the Alzheimer disease and exhibit many features of AD neuropathology [16, 18, 24], and have been used in previously published studies [17-21, 25]. Tg mice were divided into four groups: mAPP and nonTg littermate control mice treated with vehicle (PBS) or Rg1 in PBS), 5-7 mice per group. Rg1 (10 mg/kg based on the previous study [13]) was intraperitoneally injected daily, starting at age of 6 or 9-10 months for a period of three months.
For cellular studies, cortical neurons were prepared from mouse embryos at day 16 of gestation. Rat neuroblastoma B103 cells stably expressing human wild-type (wt) APP (B103-wtAPP) were kindly provided by Tony Wyss-Coray [26].
2.2. Reagents
Ginseng Rg1 (molecular weight 800, purity 98%) was provided by the Department of Biochemistry of Jilin University, China.
2.3. Immunohistochemistry
The acetycholinesterase (AChE) activity was determined by the change in AChE density (area occupied) in brain region (entorhinal cortex). After 6 min of perfusion with saline, mice brains were divided sagittally in half; one half was immersed in paraformaldehyde (4%) for 26 hr. Serial vibratome sections (20 μm, two sections from each mouse) were incubated with the mixture of the two mediums: (A) 5 mg of acetylthiocholine iodide, 6.5 mL of 0.1-mmol/L acetate buffer (pH 6.0), 0.5 ml of 30-mmol/L copper sulphate, 1.0 ml of distilled water, and 0.2 mL of 4-mmol/L iso octamethyl pyrophosphoramide (iso OMPA); and (B) 1.0 ml of 5-mmol/L potassium ferricyanide at 37°C for 5 minutes and rinse with tap water for 10 second. Following this, they were developed with freshly prepared 3-3′ diamino benzidine tetrahydrochloride (25 mg in 100 mL of 0.05-mol/L phosphate buffer [pH 7.0]), containing 0.015% H202 for 5 minutes at room temperature. Afterward, wash with tap water, dehydrate and mount. Acetylcholine esterase (AChE) activity was analyzed histochemically by the area occupied by AChE-positive staining neuritis in the entorhinal cortex (the area in which cholinergic neuritis is prominent) in the brain using MetaMorph software as previously described [18, 27]. Sections were also reacted with anti-Aß antibody 3D6 (dilution 1:1000) to identify Aß deposits. Areas with Aß plaques were analyzed using MetaMorph software evaluating multiple sections of the same level in each animal group. To ensure objective assessments and reliability of the results, brain sections from mice in any given experiment were blind coded and processed in parallel. Codes were revealed only after the analysis was completed.
2.4. Aβ ELISA
The brain tissue was extracted with 8 volumes of guanidine-Tris buffer (5.0M Guanidine HCl /50mM Tris-HCl, pH 8.0). The homogenates were mixed for 3-4 hours at room temperature. The specific human Aβ1-40 and human Aβ1-42 levels in brain tissue and cultured medium of B103-wtAPP cells derived from Rg1 (5μM or 10 μM) or vehicle treatment for 48h were measured by ELISA kit following the manufacture's instructions (Invitrogen, CA).
2.5. γ-Secretase activity
γ-secretase activity was measured as previously described [28]. Freshly dissected mouse cerebral cortex was homogenized in homogenization buffer (10mM MOPS, pH 7.0, 10mM KCL, 1× complete protease inhibitor cocktail) and centrifuged at 2500 rpm for 15 minutes at 4 °C. The supernatant was collected and centrifuged at 14000 rpm for 20 minutes at 4 °C. The pellet was rinsed once in homogenization buffer and then resuspended in assay buffer (150mM sodium citrate, pH 6.4, 1× complete protease inhibitor cocktail, protein concentration 4ug/ul). Samples were incubated at 37 °C for 2 hours, and then subjected to Western blot. The APP-CTF antibody was used for APP-CTF-γ (6kDa) identification (Calbiochem, USA). B103-wtAPP cells treated with Rg1 or vehicle were processed similar to tissue samples.
2.6. PKA activity and Phosphor-CREB (p-CREB) and total-CREB (T-CREB)
Cultured cortical neurons were treated with Rg1 and/or oligomeric Aβ1-42, and then harvested in lysis buffer (20mM MOPS, 50mM β-glycerolphosphate, 50mM sodium fluoride, 1mM sodium vanadate, 5mM EGTA, 2mM EDTA, 1% NP40, 1mM dithiothreitol (DTT), 1mM benzamidine, 1mM phenylmethane- sulphonylfluoride (PMSF) and 10μg/mL leupeptin and aprotinin). PKA activity was measured using a PKA Kinase Activity Assay Kit (Stressgen, Ann Arbor, Michigan, USA), according to the manufacturer's instructions.
Levels of p-CREB and T-CREB in cultured cortical neurons were analyzed using previous described methods [29]. Briefly, cortical neurons were treated with Rg1 and/or oligomer Aβ1-42, and then exposed to 50 μM glutamate (from Sigma) in a salt medium (containing 119 mM NaCl, 5 mM KCl, 2 mM CaCl2 20 mM Hepes, 1 μM glycine, 300 mM glucose) for 15min (ginsenoside Rg1 and/or Aβ1-42 were added to the culture medium to maintain stable concentration throughout the experiments). Lysates were subjected to immunoblotting with specific antibodies to p-CREB or T-CREB (1:1,000 dilution, Cell Signaling technology, Danvers, MA, USA). Levels of p-CREB/T-CREB in the brains were measured using p-CREB and T-CREB ELISA kits (Invitrogen).
2.7. Behavioral studies
Spatial learning and memory was evaluated using the radial arm water maze as previously described [18, 19, 21, 25]. Briefly, the mouse started the task from a different randomly chosen arm for each trial. The mouse could not use its long-term memory of the location of the platform on previous days, but had to rely on the short-term memory of its location on the day in question based on spatial clues that were present in the room. Each trial lasted one minute and errors were counted each time the mouse entered the wrong arm or needed more than 20 sec to reach the platform. After each error, the mouse was gently pulled back to the start arm for that trial. At the end of the trial, the mouse remained on the platform for 30sec. After four consecutive trials, the mouse was placed in its home cage for 30 min, then returned to the maze and administered a retention trial. Testing was considered complete when wild-type mice reached an asymptotic performance (below 2 errors on trials 4 and 5). The generally took about 10-14 training days. The scores for each mouse on the last three days of testing were averaged and used for statistical analysis. The investigators were blinded to mouse genotypes and experimental conditions.
2.7. Statistical analysis
One-way ANOVA was used for repeated measure analysis followed by Fisher's Protected Least Significant Difference for post-hoc comparisons. STATVIEW computer software was utilized. Results were expressed as mean ± Standard Error of the Mean (SEM).
3. Results
3.1. Effect of Rg1 on Aβ accumulation
Tg mAPP mice and nonTg littermates at age of 6 months (beginning Aβ accumulation in the brain) and 9-10 months (severe amyloid pathology, late stage) were treated with Rg1 for 3 months to determine the effect of Rg1 on early and late stage AD pathology and to examine whether Rg1 is able to reverse existing AD pathology. After 3 month of Rg1 treatment, levels of Aβ1-40 and Aβ1-42 including soluble and insoluble Aβ were significantly lower in hippocampus of Rg1-treated mAPP mice than in vehicle-treated mice (Fig 1A). Intriguingly, similar results were obtained from mAPP mice treated with Rg1 from 9-10 to 12-13 months of age (Fig. 1B). Consistent with this result, Aβ plaque loads were significantly reduced in cerebral cortex including hippocampus of Rg1-treated mAPP mice, whereas vehicle-treated mAPP mice displayed expected plentiful Aβ deposits in the brains. Quantification of Aβ plaque load revealed that the area covered by Aβ deposits in the brains of Rg1-treated mAPP mice was significantly reduced compared to those of vehicle-treated mAPP mice (Fig 1C-D; P<0.01).
Fig. 1.
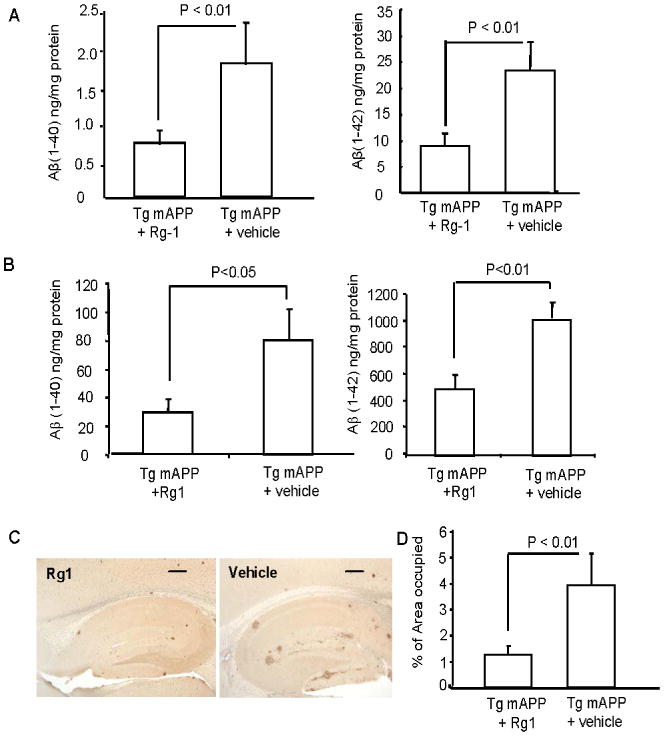
Effect of Rg1 on Aβ accumulation. Panels A and B. ELISA used for the measurement of Aβ 40 (left) and Aβ 42 (right) in the hippocampus of 9 (A) and 12-13 (B) month old Tg mAPP mice injected intraperitoneally with Rg1 (10mg/kg) daily continuously for 3 month or with vehicle. Aβ 40 and Aβ 42 levels were significantly decreased in Rg1 injection group as compared to vehicle. Panel C. Immunostaining for Aβ plaque using Aβ antibody (3D6) in 9 month old Tg mAPP mice injected with Rg1 or vehicle. Representative sections showed Aβ deposits from Tg mAPP mice injected with Rg1(left) or with vehicle (right). Panel D. Quantification of area (percentage) occupied by Aβ plaques in the indicated Tg mice. N = 5-6 mice per group. Scale bar 30 μm.
3.2. Effect of Rg1 on γ secretase activity
To probe the mechanism by which Rg1 reduces Aβ accumulation, we first measured γ-secretase activity in brain tissue using an established enzymatic assay based on carboxyl terminal fragment detection APP-CTF-γ (6 kDa) formation from membrane bound APP [28], which is an index of γ secretase activity. Measurement of APP in the cortex of the mice showed no differences in full length APP in mAPP mice with and without Rg1 treatment. However, the intensity of immunoreactive bands corresponding to 6kDa APP-CTF-γ was significantly reduced in Rg1-treated mAPP brains as compared with those from vehicle-treated mAPP brains, indicating that γ secretase activity was significantly diminished (Fig 2A-B; P<0.05). No significant differences were found in full length APP among Rg1-treated and vehicle-treated nonTg mice or mAPP mice (Fig. 2). Immunoblotting for β-actin verified an equal amount protein loading in each groups of mice (Fig. 2B).
Fig. 2.
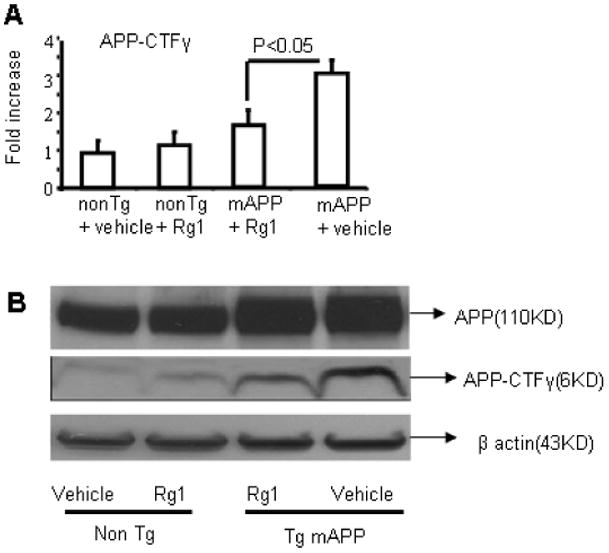
Effect of Rg1 on γ-secretase activity in the brain of Tg mAPP mice and nonTg mice. Panel A. Densitometry of APP C-terminal fragment cleavage by gamma secretase (APP-CTF-γ) from indicated Tg mice at age of 12-13 m. Decreased γ-secretase activity in the cerebral cortex assessed by APP-CTF- γ from membrane-associated APP in Tg mAPP mice injected with Rg1 as compared to Tg mAPP mice injected with vehicle. Panel B. Representative Western blots of brain extracts for full length APP, APP-CTF-γ and β-actin in the indicated mice. Rg1 did not affect total APP expression, whereas the cleavage products of APP-CTF-γ generated from membrane-bound APP were diminished in Rg1-treated mAPP mice. N = 3-5 mice per group.
Second, to investigate the direct effect of Rg1 on γ secretases, rat neuroblastoma cells (B103) stably transfected with wild-type APP (wtAPP) were incubated with Rg1 (5μM and10μM) for 48 hrs, followed by analysis of APP-CTF-γ levels in the cell lysate by immunoblotting and measurement of Aβ levels in cell-free culture medium. Aβ levels were significantly decreased in cultured medium of Rg1-treated cells compared to those in the vehicle-treated group (Fig. 3A; *P < 0.01; and #P < 0.05 compared to the vehicle). Importantly, the intensities of APP-CTF-γ immunoreactive bands in the cell lysate were significantly decreased in Rg1-treated B103-wtAPP cells compared to those without Rg1 treatment (Fig. 3B and D, APP-CTF-γ: P<0.01). Remarkably, Rg1 treatment almost abolished γ-secretase activity as shown by prominently diminished 6kDa APP-CTF-γ immunoreactive bands by 3-4 folds. There are no significant differences in levels of APP-CTF-γ in cells exposed to 10 μM Rg1 compared to cells exposed to 5 μM (P=0.33) though Rg1 5uM appears lower than 10uM in panel D. Similarly, levels of Abeta 40 in cells treated with 10 μM Rg1 were comparable to the cells treated with 5 μM Rg1 (P=0.298). Fig. 3B shows the representative immunoreactive bands of the various groups. Since B103-wtAPP cells used in our experiments are over-expressed wild-type APP gene, whereas nonTg mice express endogenous wtAPP, the gamma secretase activity was higher in B103-wtAPP cells than that in nonTg mice. This could explain that the activity of gamma-secretase appears to be profoundly suppressed in B103-wt APP cells as compared to that in nonTg mice. There were no differences in full length APP for Tg mice or B103-wtAPP cells with and without Rg1 treatment (Fig. 2B and 3B-C), suggesting that Rg1 does not interfere with APP generation/production. These in vitro results support the observations made for in vivo Rg1-treated mAPP mice both of which indicate that Rg1 treatment decreased γ secretases, which are responsible for Aβ production.
Fig. 3. Effect of Rg1 on γ-secretase activity in neuronal cells expressing wt APP.
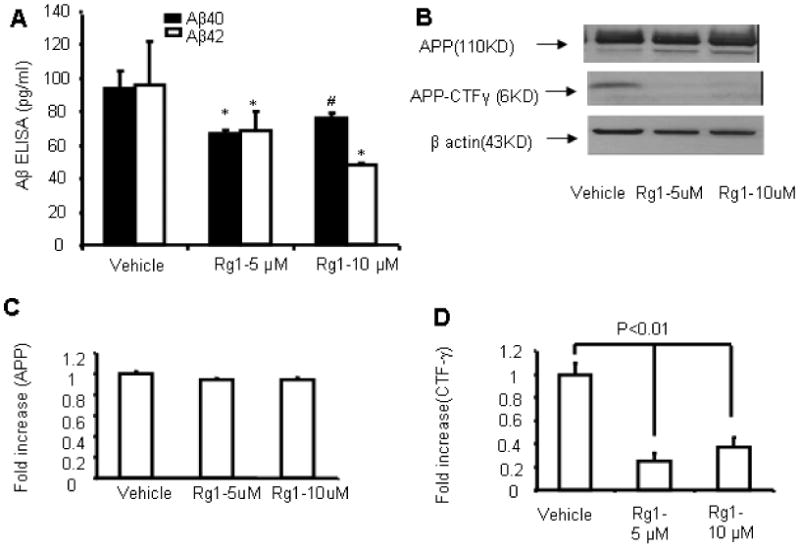
Panel A. Aβ ELISA used for measurement of Aβ1-40 and Aβ1-42 levels in the supernatant of cultured neuronal cells (B103) stably expressed wild-type APP (wtAPP) in the presence of Rg1 (5 and 10μM) or vehicle for 48 hours. Panel B. Representative Western blots of cultured B103-wtAPP cells for APP and APP-CTF-γ, showing decreased APP-CTF-γ generated from membrane associated APP because of reduced γ-secretase activity in cultured B103-wtAPP cells treated with 5μM or 10μM Rg1 for 48 hours. Panels C-D. Densitometry of APP (C) and APP-CTF-γ (D) immunoreactive bands in cells with and without treatment of Rg1. (*P<0.01, #P<0.05 compared to those from vehicle-treated B103-wtAPP cells).
3.3. Effect of Rg1 on neuropathologic development in Tg mAPP mice
In view of decreased Aβ accumulation in Rg1-treated mAPP brain, we next determined whether Rg1 treatment could reverse neuropathological changes in mAPP brain. Since diminished density of cholinergic fibers and synapses is associated with AD-like pathology [30, 31] and because density of acetylcholinesterase (AChE)-positive neuritis was significantly decreased in the AD-affected brain regions of mAPP mice [18, 27], we examined AChE activity. AChE activity was determined histochemically and reported as the area occupied by positively staining neurites in the entorhinal cortex (the area in which cholinergic neuritis is predominant) in brain. As previously reported [18, 27], at the 12-13 months of age, mAPP mice exhibited decreased levels of AChE-positive neurites in the entorhinal cortex compared to nonTg mice (Fig. 4; P<0.01). Such decreases in AChE-positive fibers in mAPP brain were significantly reversed by the administration of Rg1. A representative example of AChE histochemically stained sections of entorhinal cortex from each group of mice is shown in the lower panel of Fig. 4. There were no differences between Rg1-treated and vehicle-treated nonTg mice, indicating no effect of Rg1 on neuronal structure and function. These data indicate a protective effect of Rg1 on Aβ-induced neuropathologic alterations in mAPP mice.
Fig. 4.
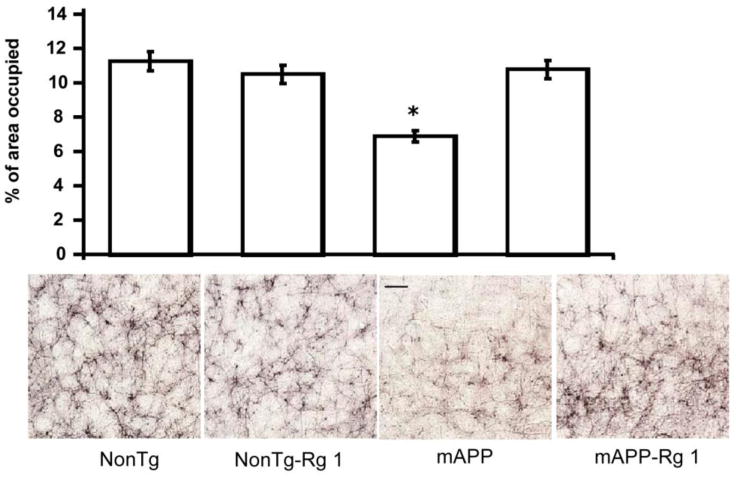
Effect of Rg1 on neuropathological findings in Tg mAPP mice. AChE activity was determined histochemically in the entorhinal cortex of the indicated mice groups at 12-13 months of age. Quantification of density of AChE-positive neurites in the entorhinal cortex of hippocampus in the indicated Tg mice (* P < 0.01 vs. other groups of mice, n = 4-6 mice/group). Representative images for AChE staining in the entorhinal cortex of the indicated mice are shown in the lower panel. Scale bar=5 μm.
3.4. Administration of Rg1 improves learning/memory in Tg mAPP mice
Based on our observations that Rg1 treatment reduced AD-type amyloidosis and neuropathological changes, we then explored the possibility that Rg1 could improve cognitive ability even in aged mAPP mice, whose behaviors were severely impaired by the age of 9-13 months. Using the radial arm water maze, known to detect hippocampal-dependent learning and memory deficits, we tested whether Rg1 treatment improves learning and memory. At 9-13 months of age, vehicle-treated mAPP mice displayed impaired spatial learning memory for platform location between trials (average of about 4-6 errors by trial 4 and retention test). In contrast, Rg 1-treated mAPP mice demonstrated good spatial memory, finding the hidden platform more rapidly with increasing trials as compared to vehicle (Fig 5; P<0.01 at age 9-10m, P<0.05 at age12-13 m). Remarkably, Rg1 treatment for three months (from age of 6 months) completely reversed or largely improved spatial learning/memory as shown by ∼2-3 errors on the maze compared to vehicle-treated mAPP mice (∼ 4-6 errors by trials 4 or 5) (Fig. 5A-B). Further, Rg1 treatment markedly improved spatial learning/memory even in aged mAPP mice with severely impaired behavior and neuropathological impairment (Fig. 5B). Rg1 did not affect normal behavior as demonstrated by a good performance in Rg 1 treated nonTg mice. Four groups of Tg mice showed no difference in their speed of swimming or in the time required to reach the platform in the visible platform test (data not shown). Taken together, these results indicate that learning and memory improvement is a consequence of Rg1 treatment in mAPP mice.
Fig. 5.

Effect of Rg1 on spatial learning/memory in Tg mAPP mice. Spatial/learning memory was tested with the radial arm water maze at 9-10 months (left) and 12-13 months (right) of age in the indicated groups of mice (n=5-8 male mice /group). Trials 1-4 denote acquisition trials and trial 5 denotes retention trial. Tg mAPP mice injected with Rg1 showed improved spatial learning memory as compared to Tg mAPP mice injected with vehicle. *P < 0.01, # P < 0.05 vs. Tg mAPP+ vehicle.
3.5. Rg1 activation of PKA and phosphorylation of CREB
We next explored the possible mechanisms underlying improved neuronal and cognitive function and apparent protection in Rg1-treated mAPP mice, compared to findings in vehicle-treated mAPP mice. cAMP-dependent protein kinase (PKA) is critical for hippocampal-dependent long-term memory and for long-lasting potentiation (LTP) [29, 32-38]. Activation of the PKA intracellular signaling pathway has been implicated in synaptic facilitation and enhanced memory of age-related [33, 36, 37, 39, 40] and Aß-induced deficits [29, 32-34, 38]. Aβ is known to perturb signal transduction pathways, including that of PKA/CREB, leading to changes in synaptic plasticity and memory [29, 33, 34]. We therefore measured PKA activity and its downstream target, phosphorylated CREB (pCREB) in brain extracts. As shown in Fig. 6A, 12-13 month-old mAPP mice displayed significantly decreased PKA activity as compared to nonTg littermates (P < 0.01 vs. nonTg mice). However, Rg1 treatment for 3 months (from age 9-10 months to 12-13 month) completely restored PKA activity. Because CREB phosphorylation depends on PKA activity, we next measured pCREB levels in cortex extracts by ELISA. mAPP mice showed significantly decreased CREB phosphorylation at Ser 133 compared to nonTg littermates, whereas levels of CREB phosphorylation at serine 133 were significantly greater in Rg1-treated Tg mAPP mice than vehicle-treated Tg mAPP mice (Fig. 6B; *P < 0.01 vs. nonTg + vehicle, # P<0.05 vs. mAPP+vehicle). Total CREB levels were similar in Rg1-treated and vehicle-treated mAPP mice.
Fig. 6.
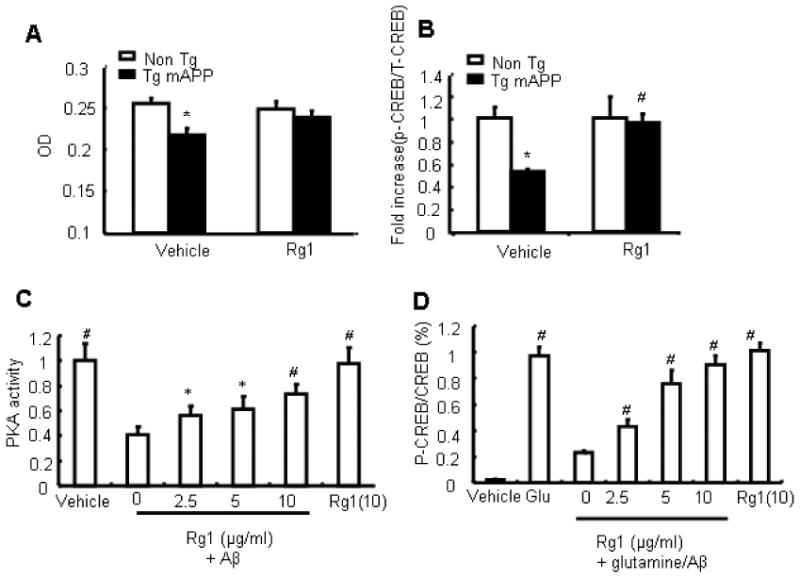
Rg1 activation of PKA and p-CREB pathway. Panel A. PKA activity was determined in brain extracts of the indicated Tg mice treated with Rg1 or vehicle for three months (started at 9 months old), respectively. Panel B. ELISA was performed to measure levels of phosphor- and total-CREB in the cortex of the indicated Tg mice. The bar graphs of Figure 6B represented fold-increased phosphor-CREB normalized to total of CREB. There was no significant change in the total of CREB. * p<0.05 vs. vehicle-treated nonTg mice and #P<0.05 vs. vehicle-treated Tg mAPP mice. Panel C. Ginsenoside Rg1 attenuated the decrease in PKA activity induced by oligomeric Aβ1-42. The cultured cortical neurons were preincubated with ginsenoside Rg1 (0, 2.5, 5, 10 μM) for 1 hour, and then oligomeric Aβ1-42 (5 μM) or vehicle was added to the cells. Two hours later, PKA activity in cell lysates was measured and data were expressed as fold-increased compared to vehicle-treated cells. Results are shown as the mean ± SEM. *P < 0.05, #P < 0.01 vs. neurons with 5.0 μM Aβ1-42 treatment. Panel D. Modulation of glutamate-stimulated CREB phosphorylation by oligomeric Aβ1-42. Phosphorylation is stimulated strongly by treatment with 50 μM glutamate (Glut) for 15 min. Pretreatment with 5 μM Aβ for 2 h results in a decrease in pCREB /T-CREB. This effect is opposed by the preincubation of 2.5/5/10 μM ginsenoside Rg1 (Rg1-Aβ-Glut). Data are shown as the mean ± SEM. #P < 0.01 vs. neurons with 5.0μM Aβ1-42 treatment. Studies were repeated at least three times.
To determine the direct effect of Rg1 on PKA activation and CREB phosphorylation, we measured PKA activity and CREB phosphorylation in cultured cortical neurons exposed to various concentrations of Rg1 (2.5, 5, and 10 μM) in the presence of Aβ. As previously reported, treatment of cultured neurons with oligomer Aβ depressed PKA activity and inactivated CREB phosphorylation in response to glutamate stimulation. PKA activity was significantly decreased in neurons treated with Aβ compared to those from vehicle-treated neurons. Notably, Rg1 abolished Aβ-induced suppression of PKA activity in a dose-dependent manner. Similarly, in the presence of Aβ, pCREB decreased in response to glutamate stimulation; the addition of Rg1 suppressed Aβ-induced inhibitory effects on pCREB in a dose-dependent manner (Fig 6 C-D; * P<0.05, # P<0.01). These results strongly suggest that inhibition of the PKA/CREB pathway underlies the effects of Aβ on synaptic function; further Rg1 enhances PKA activity and CREB phosphorylation, and may be responsible for protection against Aβ-induced neuronal dysfunction and cognitive decline in mAPP mice.
4. Discussion
In the present study, we established that Rg1 treatment protects against Aβ-mediated abnormalities in a well-characterized AD mouse model over expressing Aβ. Rg1 treatment reversed deleterious effects in mAPP mice, and even in aged mAPP mice that had severe neuropathological changes and impaired learning/memory.
First, we demonstrated a role of Rg-1 on regulation of γ-secretase, a potential mechanism of Rg1-mediated reduction in Aβ accumulation in brain both in vitro Aβ-rich neuronal culture and in vivo mAPP mice over expressing Aβ. Second, we showed a protective effect of Rg1 on Aβ-induced neuropathological changes in mAPP mice as shown by increased AChE-positive fibers in mAPP brain upon Rg1 treatment. Third, using different transgenic AD mouse model, an aggressive model of beta-amyloid pathology, we were able to observe the protective effects of Rg1 as previously described in the non-AD mouse model (SAMP8 mice) [12, 13, 41]. These reproducible results in particular from a different transgenic AD mouse model provide the additional evidence of the protective effects of Rg1 on Aβ-induced amyloid pathology and impaired cognitive function, which will significantly strengthen and support the significance of Rg1 on the clinical translation application in the future.
We investigated the possible mechanisms underlying the Rg1 protection noted above. Rg1 treatment significantly inhibited γ-secretases in studies of in vitro cultured neuronal cells and in vivo mAPP mice overexpressing Aβ. It was previously reported that oral Rg1 treatment decreased Aβ levels in the brain of Tg 2576 mice [12], but the underlying mechanism of lower cerebral amyloid accumulation was unclear. Our results suggest the involvement of Rg1 in modulation of APP cleavage. APP is cleaved by three secretases to generate various distinct products. β -secretase cleaves APP at the N-terminus to form CTF-β, which is subsequently cleaved by γ-secretase to form Aβ 40 and 42 residues [42]. Therapeutic inhibition of β- and γ-secretase decreased all forms of Aβ, including the pathogenic Aβ42. Given that inhibition of γ-secretase decreases Aβ production, development of compounds that inhibit and/or modulate γ-secretase activity will represent a rational therapeutic approach for the treatment of AD. Several groups have shown that γ-secretase inhibitor treatment results in therapeutic reduction of neurotoxic Aβ peptides [43, 44]. Rg1-mediated reduction of γ secretase activity would therefore provide additive protection against Aβ amyloidosis. Rg1 might also be a promising therapeutic candidate for AD due to its strong protection and low incidence of adverse effects.
The cAMP-respond element-binding protein (CREB) is important for learning and memory and may be a core component of the molecular switch that converts memory from short-term to long-term [45]. The major regulator of CREB activity is cAMP-dependent protein kinase (PKA). Increases in intracellular cAMP activated PKA act to disassociate the regulatory subunits from the catalytic subunits. Activated PKA moves into the cell nucleus, where it phosphorylates CREB. Interestingly, recent studies have shown that PKA/CREB-dependent signaling, which is required for long-term potentiation, is inhibited by sublethal concentrations of Aβ in cultured hippocampal neurons in mouse models of AD or in human AD brain [29, 33, 46-48]. Rg1 treatment increases cAMP levels in adipocytes and prevents memory loss in aged SAMP8 mice through increased hippocampal antioxidation and up-regulation of plasticity-related proteins including phosphor-CREB [41]. Taken together, these studies suggest that Rg1 activates the PKA/CREB signal transduction pathway thereby contributing to the improved or the retained cognitive function. To date, there is no report on the effects of Rg1 on activation of the PKA/CREB pathway in an accelerating AD mouse model.
The data presented herein clearly shows that spatial learning/memory impairment is significantly improved in Tg mAPP mice treated with Rg1. Further, our results demonstrate that Rg1 enhances the PKA/CREB signaling pathway in an Aβ-rich environment in mAPP mice as well as in cultured cells. PKA activity and CREB phosphorylation play a key role in synaptic function and memory. CREB is a key molecule in initiation of transcriptional activation of other genes encoding proteins. It has been proposed that the effect of Aβ on long term potentiation (a form of synaptic function) and memory is mediated by the inhibition of phosphorylation of CREB, a transcription factor that is activated by cAMP-dependent protein kinase (PKA) activity[29, 33]. Acute alterations in dendritic spines induced by Aβ as well as chronic loss of spine density seen in mAPP transgenic mice are both reversible by treatments that restore the cAMP/PKA/CREB signaling pathway [49]. Puzzo et al. demonstrated that CREB activation ameliorates synaptic function and memory in a mouse model of amyloid deposition [50]. Therefore, Rg1-mediated activation of PKA/CREB signal transduction mechanisms may be responsible for alleviating deficits of learning and memory in mAPP mice. Aβ can also disrupt multiple signal transduction pathways including Ca2+-regulated signaling pathways and oxidative stress-mediated kinase system. The potential effects of Rg1 on these signaling pathways require for the further investigation.
In summary, our results clearly demonstrate the protective effect of Rg1 treatment on cerebral Aβ accumulation, neuropathological change, and impaired learning/memory in a transgenic mouse model for AD. Notably, Rg1 treatment is able to reverse largely Aβ-induced deteriorated effects observed in an aged transgenic mouse model for Alzheimer disease, which exhibits the extensive amyloid pathology and cognitive decline. The mechanism through which Rg1 regulates APP process via decreased γ-secretase activity and enhances PKA/CREB signaling may explain its neuroprotective role. Given that lowering Abeta on its own so far (based on immunization or beta/gamma secretase inhibitors to suppress Abeta production) does not appear to be sufficient for attenuating neuronal dysfunction and improving cognitive function in AD patient, multiple targets such as PKA/CREB or combination with agonists of neuronal and cognitive function in addition to lowering Abeta would be therapeutic strategy for AD. Together with the previous studies, our data indicate that Rg1 is a potential therapeutic agent for preventing and halting AD progression.
Highlights.
>Rg1 treatment inhibits activity of γ-secretases in transgenic mAPP mice> Rg1 significantly reduced cerebral Aβ accumulation >Administration of Rg1 improves learning/memory and reverses neuropathological changes.
Acknowledgments
This work was supported by a grant from the USPHS (PO1AG17490 and AG037319).
Footnotes
Authors' contributions
F.F., X.C. and S.S.Y. designed research; F.F., X.C., T.H. and J.S.L. performed research and analyzed data; F.F. and S.S.Y. wrote the paper. All authors read and approved the final manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Attele AS, Wu JA, Yuan CS. Ginseng pharmacology: multiple constituents and multiple actions. Biochemical pharmacology. 1999;58:1685–1693. doi: 10.1016/s0006-2952(99)00212-9. [DOI] [PubMed] [Google Scholar]
- 2.Cheng Y, Shen LH, Zhang JT. Anti-amnestic and anti-aging effects of ginsenoside Rg1 and Rb1 and its mechanism of action. Acta pharmacologica Sinica. 2005;26:143–149. doi: 10.1111/j.1745-7254.2005.00034.x. [DOI] [PubMed] [Google Scholar]
- 3.Fu LM, Li JT. A systematic review of single Chinese herbs for Alzheimer's disease treatment. Evid Based Complement Alternat Med. 2009 doi: 10.1093/ecam/nep136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.C DH, Zhang JT, Chen Cf, Liu GZ. The chemistry, metabolism and biological activities of ginseng. Chemical Industry Press; Beijing: 2006. [Google Scholar]
- 5.Hu SQ, Yu HM, Liu TS, Yang DJ, Chen XZ, He CJ. Neuroprotective effects of water extracts of American Ginseng on SH-SY5Y cells apoptosis induced by Abeta25-35. Zhong yao cai = Zhongyaocai = Journal of Chinese medicinal materials. 2008;31:1373–1377. [PubMed] [Google Scholar]
- 6.Chang Y, Wang SJ. Ginsenoside Rg1 and Rb1 enhance glutamate exocytosis from rat cortical nerve terminals by affecting vesicle mobilization through the activation of protein kinase C. European journal of pharmacology. 2008;590:74–79. doi: 10.1016/j.ejphar.2008.05.032. [DOI] [PubMed] [Google Scholar]
- 7.Zhang YF, Fan XJ, Li X, Peng LL, Wang GH, Ke KF, Jiang ZL. Ginsenoside Rg1 protects neurons from hypoxic-ischemic injury possibly by inhibiting Ca2+ influx through NMDA receptors and L-type voltage-dependent Ca2+ channels. European journal of pharmacology. 2008;586:90–99. doi: 10.1016/j.ejphar.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 8.Xu L, Chen WF, Wong MS. Ginsenoside Rg1 protects dopaminergic neurons in a rat model of Parkinson's disease through the IGF-I receptor signalling pathway. British journal of pharmacology. 2009;158:738–748. doi: 10.1111/j.1476-5381.2009.00361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei C, Jia J, Liang P, Guan Y. Ginsenoside Rg1 attenuates beta-amyloid-induced apoptosis in mutant PS1 M146L cells. Neuroscience letters. 2008;443:145–149. doi: 10.1016/j.neulet.2008.07.089. [DOI] [PubMed] [Google Scholar]
- 10.Ji ZN, Dong TT, Ye WC, Choi RC, Lo CK, Tsim KW. Ginsenoside Re attenuate beta-amyloid and serum-free induced neurotoxicity in PC12 cells. Journal of ethnopharmacology. 2006;107:48–52. doi: 10.1016/j.jep.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Choi RC, Zhu JT, Leung KW, Chu GK, Xie HQ, Chen VP, Zheng KY, Lau DT, Dong TT, Chow PC, Han YF, Wang ZT, Tsim KW. A Flavonol Glycoside, Isolated From Roots of Panax notoginseng, Reduces Amyloid-beta-Induced Neurotoxicity in Cultured Neurons: Signaling Transduction and Drug Development for Alzheimer's Disease. J Alzheimers Dis. 2009 doi: 10.3233/JAD-2010-1293. [DOI] [PubMed] [Google Scholar]
- 12.Chen F, Eckman EA, Eckman CB. Reductions in levels of the Alzheimer's amyloid beta peptide after oral administration of ginsenosides. FASEB J. 2006;20:1269–1271. doi: 10.1096/fj.05-5530fje. [DOI] [PubMed] [Google Scholar]
- 13.Shi YQ, Huang TW, Chen LM, Pan XD, Zhang J, Zhu YG, Chen XC. Ginsenoside Rg1 Attenuates Amyloid-beta ontent, Regulates PKA/CREB Activity, and Improves Cognitive Performance in SAMP8 Mice. J Alzheimers Dis. 2009 doi: 10.3233/JAD-2010-1296. [DOI] [PubMed] [Google Scholar]
- 14.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 15.Vassar R, Citron M. Abeta-generating enzymes: recent advances in beta- and gamma-secretase research. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 16.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. Faseb J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 18.Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, Puzzo D, Liu S, Hegde A, Yan SF, Stern A, Luddy JS, Lue LF, Walker DG, Roher A, Buttini M, Mucke L, Li W, Schmidt AM, Kindy M, Hyslop PA, Stern DM, Du Yan SS. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. Embo J. 2004;23:4096–4105. doi: 10.1038/sj.emboj.7600415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 20.Takuma K, Yao J, Huang J, Xu H, Chen X, Luddy J, Trillat AC, Stern DM, Arancio O, Yan SS. ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. Faseb J. 2005;19:597–598. doi: 10.1096/fj.04-2582fje. [DOI] [PubMed] [Google Scholar]
- 21.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du H, Guo L, Zhang W, Rydzewska M, Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2011;32:398–406. doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du H, Guo L, Zhang W, Rydzewska M, Yan S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu X, Yang D, Wyss-Coray T, Yan J, Gan L, Sun Y, Mucke L. Wild-type but not Alzheimer-mutant amyloid precursor protein confers resistance against p53-mediated apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:7547–7552. doi: 10.1073/pnas.96.13.7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang F, Lue LF, Yan S, Xu H, Luddy JS, Chen D, Walker DG, Stern DM, Yan S, Schmidt AM, Chen JX, Yan SS. RAGE-dependent signaling in microglia contributes to neuroinflammation, A{beta} accumulation, and impaired learning/memory in a mouse model of Alzheimer's disease. Faseb J. 2009 doi: 10.1096/fj.09-139634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, Peng Y, Cambareri G, Rocher A, Mobbs CV, Hof PR, Pasinetti GM. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer's disease. Faseb J. 2004;18:902–904. doi: 10.1096/fj.03-0978fje. [DOI] [PubMed] [Google Scholar]
- 29.Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geula C. Abnormalities of neural circuitry in Alzheimer's disease: hippocampus and cortical cholinergic innervation. Neurology. 1998;51:S18–29. doi: 10.1212/wnl.51.1_suppl_1.s18. discussion S65-17. [DOI] [PubMed] [Google Scholar]
- 31.Masliah E, Terry RD, DeTeresa RM, Hansen LA. Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neuroscience letters. 1989;103:234–239. doi: 10.1016/0304-3940(89)90582-x. [DOI] [PubMed] [Google Scholar]
- 32.Yamamoto-Sasaki M, Ozawa H, Saito T, Rosler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300–303. doi: 10.1016/s0006-8993(99)01220-2. [DOI] [PubMed] [Google Scholar]
- 33.Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114:1624–1634. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin hydrolase Uch-L1 rescues beta-amyloid-induced decreases in synaptic function and contextual memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- 35.Arancio O, Chao MV. Neurotrophins, synaptic plasticity and dementia. Curr Opin Neurobiol. 2007;17:325–330. doi: 10.1016/j.conb.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 36.Duffy SN, Nguyen PV. Postsynaptic application of a peptide inhibitor of cAMP-dependent protein kinase blocks expression of long-lasting synaptic potentiation in hippocampal neurons. J Neurosci. 2003;23:1142–1150. doi: 10.1523/JNEUROSCI.23-04-01142.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen PV, Woo NH. Regulation of hippocampal synaptic plasticity by cyclic AMP-dependent protein kinases. Prog Neurobiol. 2003;71:401–437. doi: 10.1016/j.pneurobio.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bach ME, Barad M, Son H, Zhuo M, Lu YF, Shih R, Mansuy I, Hawkins RD, Kandel ER. Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc Natl Acad Sci U S A. 1999;96:5280–5285. doi: 10.1073/pnas.96.9.5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 41.Zhao H, Li Q, Zhang Z, Pei X, Wang J, Li Y. Long-term ginsenoside consumption prevents memory loss in aged SAMP8 mice by decreasing oxidative stress and up-regulating the plasticity-related proteins in hippocampus. Brain Res. 2009;1256:111–122. doi: 10.1016/j.brainres.2008.12.031. [DOI] [PubMed] [Google Scholar]
- 42.Lahiri DK, Kotwal GJ, Farlow MR, Sima A, Kupsky W, Sarkar FH, Sambamurti K. The role of the carboxyl-terminal fragments of amyloid precursor protein in Alzheimer's disease. Ann N Y Acad Sci. 2002;973:334–339. doi: 10.1111/j.1749-6632.2002.tb04661.x. [DOI] [PubMed] [Google Scholar]
- 43.Elvang AB, Volbracht C, Pedersen LO, Jensen KG, Karlsson JJ, Larsen SA, Mork A, Stensbol TB, Bastlund JF. Differential effects of gamma-secretase and BACE1 inhibition on brain Abeta levels in vitro and in vivo. Journal of neurochemistry. 2009;110:1377–1387. doi: 10.1111/j.1471-4159.2009.06215.x. [DOI] [PubMed] [Google Scholar]
- 44.Wolfe MS. Inhibition and modulation of gamma-secretase for Alzheimer's disease. Neurotherapeutics. 2008;5:391–398. doi: 10.1016/j.nurt.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barco A, Pittenger C, Kandel ER. CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert opinion on therapeutic targets. 2003;7:101–114. doi: 10.1517/14728222.7.1.101. [DOI] [PubMed] [Google Scholar]
- 46.Matsuzaki K, Yamakuni T, Hashimoto M, Haque AM, Shido O, Mimaki Y, Sashida Y, Ohizumi Y. Nobiletin restoring beta-amyloid-impaired CREB phosphorylation rescues memory deterioration in Alzheimer's disease model rats. Neuroscience letters. 2006;400:230–234. doi: 10.1016/j.neulet.2006.02.077. [DOI] [PubMed] [Google Scholar]
- 47.Liang Z, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer disease brain. Journal of neurochemistry. 2007;103:2462–2470. doi: 10.1111/j.1471-4159.2007.04942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O. Amyloid-beta peptide inhibit sactivation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J Neurosci. 2005;25:6887–6897. doi: 10.1523/JNEUROSCI.5291-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith DL, Pozueta J, Gong B, Arancio O, Shelanski M. Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16877–16882. doi: 10.1073/pnas.0908706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puzzo D, Staniszewski A, Deng SX, Privitera L, Leznik E, Liu S, Zhang H, Feng Y, Palmeri A, Landry DW, Arancio O. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer's disease mouse model. J Neurosci. 2009;29:8075–8086. doi: 10.1523/JNEUROSCI.0864-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]