

Abstract
A 59-year-old woman presented with the clinical symptoms and radiologic investigations of a liver lesion suspect of metastasis. However, postoperative histopathology revealed a primary hepatic lymphoma (PHL). The case of a patient with a solitary PHL, which was treated by resection and subsequent chemotherapy, will be discussed with a short overview of the literature.
Keywords: Primary, Hepatic, Lymphoma, Liver, Diagnosis, Computed tomography, Magnetic resonance imaging
INTRODUCTION
Primary tumors of the liver are difficult to characterize and are frequently associated with a poor prognosis. Primary hepatic lymphoma (PHL) is a rare primary liver tumor. Due to its clinical and radiological resemblance to liver metastases of adenocarcinoma, PHL is frequently diagnosed intra- or post-operatively. Since chemotherapy is the treatment of first choice for lymphoma, adjuvant chemotherapy should be given to patients for optimal treatment[1]. The existing literature on PHL reveals the difficulties involved in diagnosis and treatment. Here we present a case of an immunocompetent patient with a large primary lymphoma of the liver. This case study can provide references for the diagnosis and treatment of patients suspected of having PHL. Due to the rarity of the disease controlled studies are lacking, thus the recommendations made are based almost completely on case reports.
CASE REPORT
A 59-year-old woman without prior medical history was referred to our clinic with a computed tomography (CT) confirmed 10 cm liver lesion. During the previous year she complained of fatigue and weight loss of 13 kg. Physical examination only indicated paleness. Lymph nodes were not enlarged. Blood analysis revealed iron deficiency anemia and biliary obstruction. The tumor markers α fetoprotein, carcinoembryonic antigen, carbohydrate antigen and 5-hydroxyindoleacetic acid were not elevated.
No positive antigens to human immunodeficiency virus (HIV), Ebstein-Barr virus (EBV), cytomegalovirus, toxoplasmosis or hepatitis A, B, or C (hep A, B, C) were observed. Chest X-ray, gastric and colon endoscopy did not show any abnormalities. A 3-phase (arterial, portal/venous and equilibrium phase) iodinated contrast enhanced CT-scan (Siemens Somatom Sensation 10) revealed a 10 cm × 8 cm × 7.5 cm hypovascular, inhomogeneous lesion in segments V and VI of the liver without calcifications (Figure 1). Supplemental magnetic resonance imaging (MRI) (Philips Achieva 1.5 T), performed at our hospital, using gadolinium-EOB-DTPA (Primovist®; Bayer Schering) (Figure 2) confirmed the presence of a hypovascular, inhomogeneous solitary lesion, without contrast uptake in the hepatobiliary phase. The periphery of the lesion was accompanied by hypoattenuated regions in the arterial phase, some of which show diminished contrast uptake in the hepatobiliary phase. These findings suggest the radiological differential diagnosis of metastases of adeno- or squamous tumors or cholangiocarcinoma.
Figure 1.
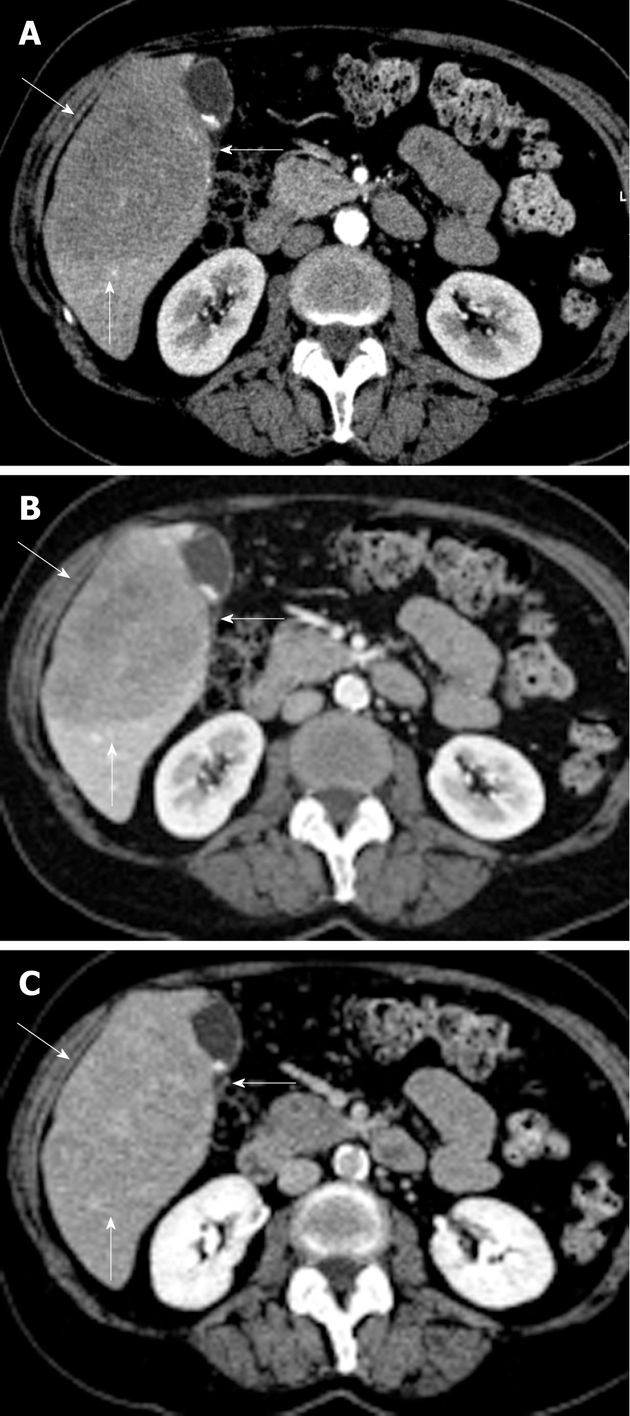
Radiological depiction of the liver lesion. Arterial (A), portal (B) and equilibrium phase (C) computed tomography scan with a large (10 cm × 8 cm × 7.5 cm) hypovascular lesion in segments V and VI of the liver with some inhomogeneity and without calcifications (lesion indicated by 3 white arrows).
Figure 2.

Magnetic resonance imaging showing a large, sharply demarcated lesion measuring 11 cm in the right liver lobe. The lesion is hypointense on T1 (A) and hyperintense on T2 (B) weighed images with slight inhomogeneity. Arterial (C), portal (D), equilibrium (E) and hepato-biliary (20 min) (F) phase magnetic resonance imaging after Gd-EOB-DTPA contrast enhancement reveal a hypovasular lesion without uptake in the hepato-biliary phase (lesion indicated by white arrows).
Treatment consisted of an en-bloc resection of tumor, gallbladder, and hepatoduodenal lymphadenectomy (Figure 3). Postoperative recovery was uncomplicated.
Figure 3.

Intraoperative aspect of lesion. A: Tumor presentation intra-operatively; B: Tumor in segment V/VI of the liver with en bloc in the resection specimen the gall bladder (black arrow indicates gall bladder); C: Resection plane of the liver; D: Resection specimen with centrally white/yellow shiny tumor (small white arrow indicates resection plane, long white arrow indicates tumor).
Histopathology revealed a diffuse, large B-cell, non-Hodgkin lymphoma (NHL) with negative surgical margins (Figure 4). Immunostaining of the tumor showed reactivity for CD45, CD20, CD79a, BCL6, MIB1 and BCl2. Postoperative investigations for disseminated NHL by CT scan, FDG-PET scan and bone marrow biopsy were negative. Adjuvant chemotherapy consisted of 6 cycles of cyclophosphamide, hydroxydaunorubicin (Adriamycin), oncovin (Vincristine), prednisone (CHOP) and rituximab.
Figure 4.

Histopathological results. A: Hematoxylin eosin staining (150 ×) shows a large cell malignancy with mostly loose tumor cells with nuclear polymorphism. Furthermore, frequent giant nuclear bodies with macro-nucleoli and numerous cell mitoses. No central necrosis is observed; B: A photomicrograph (400 ×) showing lymphocytic tumor cells which are positive for CD20 staining around the plasma membrane, indicating non-Hodgkin lymphoma of B-cell origin.
During 24-mo follow-up our patient showed no symptoms or signs of recurrent disease.
DISCUSSION
PHL was first described in 1965 by Ata el al[2]. In 1986 Caccamo et al[3] defined PHL as a lymphoma localized and limited to the liver without extrahepatic involvement[3]. Symptoms should be explainable by involvement of the liver. Furthermore, superficial lymphadenopathy, splenomegaly, abnormal hematological parameters, spleen or bone marrow localization cannot be present for at least 6 mo after appearance of the hepatic lesion[3].
Primary hepatic NHL is very rare, only 0.016% of all NHL. Of all primary extranodal NHL only 0.4% arise in the liver[4]. 1.1% of all primary hepatic tumors in 30 years in the Johns Hopkins tumor registry consisted of PHL[5]. The incidence of hepatic involvement in NHL is described between 16% and 22%, stressing the importance of careful investigation to disseminated disease outside of the liver[6]. Associations in the literature have been made with HIV, hep B and C, EBV, liver cirrhosis, primary biliary cirrhosis, immunosuppressive therapy, and autoimmune disease. However, until now the pathogenesis of PHL is still unclear[7].
Clinical presentation of PHL is nonspecific. Most often fever, loss of weight and night sweats (also known as ‘B’ symptoms) occur. Alternative symptoms described are: right upper abdominal pain, epigastric pain, abdominal distension, nausea, vomiting, asthenia or itch. No specific physical complaints are typical for PHL. Abdominal pain, jaundice and hepatomegaly are the only physical findings described for various patients. Blood count can show abnormal aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, total and direct bilirubin and LDH[7]. Hypercalcemia and Bence Jones protein peak are rare but have been described[7].
At initial presentation a third of patients present with a solitary liver nodule while another third have multiple lesions, and the remaining cases have diffuse infiltration of the liver[7]. Radiological investigation consists of an ultrasound of the liver, on which the tumor is hypo-attentuating or iso-attentuating[8]. On tri-phasic liver CT scan PHL usually presents itself as a hypodense lesion, with possible areas of inhomogeneity. Occasionally local areas of rim enhancement or calcifications may be seen[8]. On MRI, lesions tend to be hypointense compared to healthy liver parenchyma at T1, and have slight enhanced signal intensity on T2 weighed images. Hepato-biliary specific contrast does not show any enhancement of PHL either in the early dynamic or late hepato-biliary phase. This is similar for gadobenate and gadopentate dimeglumine[9].
The majority of PHL consist of B-cell NHL (63%) and T-cell lymphoma (25%)[10]. Diagnosis is often made upon histopathological investigation of the resection specimen. This is due to the hesitance in obtaining tissue biopsies of suspect liver lesions and risking needle track metastases[11,12]. Further differentiation can be done by immunohistochemical investigation[7,10]. The tumor has a nodular or diffuse growth pattern, in which the lymphoma cells expand into the liver parenchyma[13]. Tumor tissue consists of atypical cells with little basophilic cytoplasm, large vesicular nucleus, irregular nuclear membrane and often multiple prominent nucleoli[14].
Additional diagnostic methods could be flow cytometry, gene rearrangement and cytogenetic studies. However, the histopathology can vary considerably, complicating the diagnosis.
Chemotherapy is the recommended therapeutic treatment for all extranodal diffuse large B-cell lymphoma and T-cell lymphoma, making it the treatment of choice when PHL is diagnosed preoperatively[1]. Indications for surgical treatment are localized disease, which can be resected completely, or surgical debulking[10]. However, due to the radiological resemblance to liver metastases of adeno- or squamous carcinoma and the accompanying risk of needle track metastases from biopsies, most patients will be diagnosed after resection and will receive adjuvant chemotherapy[11,12].
Reports in literature discuss one or a combination of treatments. The lack of controlled studies, due to the low incidence of PHL, make well supported treatment recommendations based on available literature difficult.
Survival rates for PHL vary considerably among reported cases, largely depending on co-morbidity[7]. Page et al[15] discuss the 20-year experience of The University of Texas MD Anderson Cancer Center. Twenty-four cases with varying co-morbidity and treated only with chemotherapeutics resulted in an overall 5-year survival of 83%[15].
PHL is associated with a poor prognosis due to its aggressive nature and frequent severe co-morbidity. Of 72 PHL patients described in literature the median survival is only 15.3 mo. The co-morbidity, especially immunoincompetence, causes a large variation in survival of 3 to 123.6 mo[10]. Previous reports suggest an association between survival and histopathological subtype of the tumor, based upon analysis of case reports. Emile et al[16] show a significant difference between 1- and 3-year survival (70% and 57%) for nodular PHL and 1- and 3-year survival (38% and 18%) for diffuse PHL (P = 0.0033).
In conclusion, PHL is rare, occurring often in immunoincompetent patients. The presented case of PHL in an immunocompetent patient emphasizes the difficulties of diagnosing PHL and shows that PHL should be included in the differential diagnosis of solid, hypovascular liver lesions. Although the primary treatment should be chemotherapy, the current consensus not to take pre-operative biopsies from solitary liver lesions, usually results in resection followed by chemotherapy as the most frequently performed treatment strategy.
Footnotes
Peer reviewers: Thomas J George, Jr., MD, FACP, Assistant Professor, Director, GI Oncology Program, Associate Director, HemOnc Fellowship Program, University of Florida, Division of Hematology and Oncology, 1600 SW Archer Road, PO Box 100277, Gainesville, FL 32610, United States; Wei Lu, MD, PhD, Associate Professor, Department of Interventional Radiology, Nanfang Hospital, Southern Medical University, Guangzhou 510515, Guangdong Province, China
S- Editor Cheng JX L- Editor O’Neill M E- Editor Zheng XM
References
- 1.Sehn LH, Donaldson J, Chhanabhai M, Fitzgerald C, Gill K, Klasa R, MacPherson N, O’Reilly S, Spinelli JJ, Sutherland J, et al. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B-cell lymphoma in British Columbia. J Clin Oncol. 2005;23:5027–5033. doi: 10.1200/JCO.2005.09.137. [DOI] [PubMed] [Google Scholar]
- 2.Ata AA, Kamel IA. Primary reticulum cell sarcoma of the liver. A case report. J Egypt Med Assoc. 1965;48:514–521. [PubMed] [Google Scholar]
- 3.Caccamo D, Pervez NK, Marchevsky A. Primary lymphoma of the liver in the acquired immunodeficiency syndrome. Arch Pathol Lab Med. 1986;110:553–555. [PubMed] [Google Scholar]
- 4.Freeman C, Berg JW, Cutler SJ. Occurrence and prognosis of extranodal lymphomas. Cancer. 1972;29:252–260. doi: 10.1002/1097-0142(197201)29:1<252::aid-cncr2820290138>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 5.Craig JR, Peters RL, Edmondson HA. Tumors of the liver and intrahepatic bile ducts. 2nd Series. Washington, DC: Armed Forces Institute of Pathology; 1989. pp. 244–246. [Google Scholar]
- 6.Civardi G, Vallisa D, Bertè R, Lazzaro A, Moroni CF, Cavanna L. Focal liver lesions in non-Hodgkin’s lymphoma: investigation of their prevalence, clinical significance and the role of Hepatitis C virus infection. Eur J Cancer. 2002;38:2382–2387. doi: 10.1016/s0959-8049(02)00481-1. [DOI] [PubMed] [Google Scholar]
- 7.Santos ES, Raez LE, Salvatierra J, Morgensztern D, Shanmugan N, Neff GW. Primary hepatic non-Hodgkin’s lymphomas: case report and review of the literature. Am J Gastroenterol. 2003;98:2789–2793. doi: 10.1111/j.1572-0241.2003.08766.x. [DOI] [PubMed] [Google Scholar]
- 8.Gazelle GS, Lee MJ, Hahn PF, Goldberg MA, Rafaat N, Mueller PR. US, CT, and MRI of primary and secondary liver lymphoma. J Comput Assist Tomogr. 1994;18:412–415. doi: 10.1097/00004728-199405000-00013. [DOI] [PubMed] [Google Scholar]
- 9.Maher MM, McDermott SR, Fenlon HM, Conroy D, O’Keane JC, Carney DN, Stack JP. Imaging of primary non-Hodgkin’s lymphoma of the liver. Clin Radiol. 2001;56:295–301. doi: 10.1053/crad.2000.0649. [DOI] [PubMed] [Google Scholar]
- 10.Avlonitis VS, Linos D. Primary hepatic lymphoma: a review. Eur J Surg. 1999;165:725–729. doi: 10.1080/11024159950189474. [DOI] [PubMed] [Google Scholar]
- 11.Durand F, Regimbeau JM, Belghiti J, Sauvanet A, Vilgrain V, Terris B, Moutardier V, Farges O, Valla D. Assessment of the benefits and risks of percutaneous biopsy before surgical resection of hepatocellular carcinoma. J Hepatol. 2001;35:254–258. doi: 10.1016/s0168-8278(01)00108-8. [DOI] [PubMed] [Google Scholar]
- 12.Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD. Liver biopsy. Hepatology. 2009;49:1017–1044. doi: 10.1002/hep.22742. [DOI] [PubMed] [Google Scholar]
- 13.Noronha V, Shafi NQ, Obando JA, Kummar S. Primary non-Hodgkin’s lymphoma of the liver. Crit Rev Oncol Hematol. 2005;53:199–207. doi: 10.1016/j.critrevonc.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Collins MH, Orazi A, Bauman M, Vik T, West K, Heerema NA, Klatte E, Neiman RS. Primary hepatic B-cell lymphoma in a child. Am J Surg Pathol. 1993;17:1182–1186. doi: 10.1097/00000478-199311000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Page RD, Romaguera JE, Osborne B, Medeiros LJ, Rodriguez J, North L, Sanz-Rodriguez C, Cabanillas F. Primary hepatic lymphoma: favorable outcome after combination chemotherapy. Cancer. 2001;92:2023–2029. doi: 10.1002/1097-0142(20011015)92:8<2023::aid-cncr1540>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 16.Emile JF, Azoulay D, Gornet JM, Lopes G, Delvart V, Samuel D, Reynès M, Bismuth H, Goldwasser F. Primary non-Hodgkin’s lymphomas of the liver with nodular and diffuse infiltration patterns have different prognoses. Ann Oncol. 2001;12:1005–1010. doi: 10.1023/a:1011131930409. [DOI] [PubMed] [Google Scholar]