

Abstract
Novel protein chimeras constituted of ‘silk’ and a silica-binding peptide (KSLSRHDHIHHH) were synthesized by genetic or chemical approaches and their influence on silica-silk based chimera composite formation evaluated. Genetic chimeras were constructed from 6 or 15 repeats of the 32 amino acid consensus sequence of Nephila clavipes spider silk ([SGRGGLGGQG AGAAAAAGGA GQGGYGGLGSQG]n) to which one silica binding peptide was fused at the N terminus. For the chemical chimera, 25 equivalents of the silica binding peptide were chemically coupled to natural Bombyx mori silk after modification of tyrosine groups by diazonium coupling and EDC/NHS activation of all acid groups. After silica formation under mild, biomaterial compatible conditions the effect of peptide addition on the properties of the silk and chimeric silk-silica composite materials was explored. The composite biomaterial properties could be related to the extent of silica condensation and to the higher number of silica binding sites in the chemical chimera as compared to the genetically derived variants. In all cases, the structure of the protein / chimera in solution dictated the type of composite structure that formed with the silica deposition process having little effect on the secondary structural composition of the silk based materials. Similarly to our study of genetic silk based chimeras containing the R5 peptide (SSKKSGSYSGSKGSKRRIL), the role of the chimeras (genetic and chemical) used in the present study resided more in aggregation and scaffolding than in the catalysis of condensation. The variables of peptide identity, silk construct (number of consensus repeats or silk source) and approach to synthesis (genetic or chemical) can be used to ‘tune’ the properties of the composite materials formed and is a general approach which can be used to prepare a range of materials for biomedical and sensor based applications.
Keywords: fusion protein, silk, silica, peptide, chemical chimera, genetic chimera
Introduction
In the living kingdom, many organisms produce siliceous structures such as frustules in diatoms, spicules in sponges, and silica phytoliths in higher plants, which confer advantages, including support and protection.1 Natural siliceous structures synthesized in living organisms are formed from an environment undersaturated in monosilicic acid (levels below 100 ppm). Soft interactions with various biomolecules trigger and template the formation of silica structures under mild conditions compatible with life, such as circumneutral pH and low temperature, e.g. 4-40°C.2 Hence soft, bio-inspired silicification processes would be of significant interest in the production of new materials through the use of structural biomolecules as templates for mineralization (reviewed by Fan et al3). Many applications could be considered in the biomedical sciences, especially in the field of biocompatible medical prostheses (i.e. bone and enamel synthesis). To reach this goal and to generate materials whose properties can be tuned on the nano to microscopic scales, an understanding of how these silicified composite materials are formed is needed. Exemplar biomolecules involved in silica formation in nature include silicatein from sponges and heavily post-translationally modified proteins and polyamines in some species of diatoms.4 From model studies performed to understand the roles that these molecules play it is evident that involvement in nucleation via catalysis and structure direction (templating/ scaffolding) may both be important although the process of biosilicification is still not fully understood.4–11 An example of a structural protein that is being used in the development of silica based biopolymer composites is silk. The mechanical properties of silks vary according to specie of spider and silkworm, type of silk (primary sequence) and mode of processing. The ability to correlate sequence and structure [mix of beta sheet crystals (hard segments) with less crystalline (soft segments)] provides significant input to the mechanical features. Recent molecular dynamics simulations have highlighted the importance of the confinement of the β domains in the form of nanocrystals (1-2nm by 2-4nm) and by further confinement of the crystallites in 50±30nm fibrils. The result of this confinement optimizes hydrogen bond interactions to maximize the mechanical toughness of the silk over the macroscopic scale.12-18 The strength, toughness and a resistance to mechanical compression, of silks can rival those of synthetic high performance fibers such as Kevlar.19 Due to the mechanical and biocompatibility properties, silk proteins, both silkworm and spider, are central to applications20, 21 such as controlled release, biomaterials22, 23 and tissue engineering.24, 25 In principle, silk-silica biocomposites could be tailored using the bio-inspired silicification process performed in vitro at circum-neutral pH and ambient temperature.6 In our earlier studies, we have shown that silk-silica binding peptide chimeras can be generated using genetic engineering and demonstrated the positive effect of the binding peptide on the silicifying properties of silk.26 We have further shown that these materials promote osteoblast development with the upregulation of key markers associated with bone formation27. In the genetically engineered chimera, the Si-peptide was coupled to one end of the silk protein. An alternative approach to introduce further functionalization into silk is to use chemical coupling techniques that are a rapid and a viable alternative to genetic engineering for the modification of biomolecules. Such methods could be used to graft target peptides to the side-chains of the protein, in some cases with a higher degree of substitution than the bioengineered variants.
In this study, we compare the synthesis and silicifying properties of both chemically and genetically engineered chimeric silk proteins. The silica-binding peptide sequence chosen for study (KSLSRHDHIHHH) has been previously determined by phage display28, 29 where it was shown to bind to preformed silica particles and affect the process of silica formation de novo. The chimeric proteins were silicified and their materials properties compared, with the final aim of producing biomaterials combining the elasticity and toughness of silk with the hardness of silica. The mechanism of silica condensation was further investigated for the chemical chimera due to the higher capacity for silica-binding.
Materials and Methods
Si-Peptide-Silk Protein Chimera Synthesis
Chemically Produced Chimera
Peptide Synthesis and Purity Determination
Silica-binding peptide (Si-peptide) designated as Pep 1 (KSLSRHDHIHHH from N to C terminal) and an additional peptide R-Pep1 (RKSLSRHDHIHHH), designed to enable more ready identification of the successful coupling of the peptides to the silk fibroin using mass spectrometry, were synthesised by solid phase peptide synthesis (SPPS) using Fmoc chemistry. The purity of the peptide was assessed by RP-HPLC (LC20 chromatography enclosure, Dionex, Sunnyvale, California, USA) and MALDI ToF Mass Spectrometry (Brucker Daltonics Ultraflex 3 Matrix-Assisted Laser Desorption Ionisation).
Silk Protein Purification
Silk protein was purified from Bombyx mori silkworm silk (Forest Fibers, cut silk cocoon, UK) similarly to the procedure reported30 and lyophilised prior to peptide chemical coupling.
Diazonium Coupling Reaction
The tyrosine residues of silk were modified with a diazonium salt 27 with detail provided in the supplementary data. Under these conditions, all the tyrosine residues, representing 5.26% of amino-acid residues in the silk proteins31, should be modified.
Surface Modification of Silk with Si-Peptides
The synthesised peptide was chemically coupled to N-(3-Dimethylaminopropyl) N’-EthylCarbo-imide (EDC; Sigma-Aldrich, Steinheim, Germany) and N-HydroxySuccinimide (NHS, 98%; Sigma-Aldrich, St Louis, MO, USA) activated silk as described in the supplementary information. The potentially reactive amino-acid residues in silk fibroin (6.7%) were calculated considering a 1:1 molar ratio of heavy chain:light chain as reported by Inoue et al.32, the diazo-coupled tyrosine residues (5.26% of total residues) and the acidic amino-acid residues (glutamic and aspartic acids: 1.41% of total residues; Table 1).
Table 1.
Theoretical numbers of amino-acid residues potentially involved in silica binding through diazo-coupling (Tyrosine, Y), EDC/NHS activation (diazo-Y, Aspartic and Glutamic acid residues, D and E) and unmodified basic amino-acids residues (Arginine, R, Histidine, H and Lysine, K) in silk.
The Si-peptides were added at a molar ratio of 28:1 and 25:1 respectively for Pep1: silk and R-Pep1: silk respectively. The spatial distribution of amino-acid residues potentially involved in silica binding on the chimeric silk proteins is presented in Figure 1. These can be either unmodified basic amino-acids residues (Arginine, R, Histidine, H and Lysine, K) or chemically grafted Si-binding peptides rich in such basic amino-acids. The modification of silk upon chemical coupling with Si-peptides was followed by UV/VIS spectrophotometry from 190 to 600 nm (UNICAM UV-1 spectrophotometer). No residual peptide was left in solution following the coupling procedure as measured using a fluorescamine assay developed specifically for the peptide used. In a typical assay, 20 μL of fluorescamine (5mg/ml in acetone) was added to a 180 μL aliquot of the supernatant in a 96-well plate and the fluorescence intensity was measured using a Tecan Spectrafluor XFLUOR4 plate reader equipped with a 360 nm excitation filter and a 465 nm absorption filter. The concentration of each specific peptide was calculated using a peptide specific calibration curve and the amount of peptide reacted was calculated by difference from the initial peptide concentration. All assays were repeated three times in order to guarantee their repeatability.
Figure 1.

Spatial distribution of amino-acid residues potentially involved in silica binding through diazo-coupling (Tyrosine, Y), EDC/NHS activation (diazo-Y, Aspartic and Glutamic acid residues, D and E) and unmodified basic amino-acids residues (Arginine, R, Histidine, H and Lysine, K) of silk block and Si-peptide in chemically generated protein chimeras. The silk block drawing is taken from Wong Po Foo, C.; Patwardhan, S. V.; Belton, D. J.; Kitchel, B.; Anastasiades, D.; Huang, J.; Naik, R. R.; Perry, C. C.; Kaplan, D. L. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9428–9433 and is “Copyright (2006) National Academy of Sciences, U.S.A.”.
The coupling of Si-peptides to silk was additionally checked via trypsin proteolysis of the Si-peptide-silk chimeras followed by Mass spectrometric analysis. For Mass spectrometry analysis, each sample was combined with ammonium bicarbonate (16.6 μL of 100 mM), water (7.6 μL), and trypsin (0.7 μL of 0.5 μg/μL, Promega Gold dissolved in ammonium bicarbonate), incubated at 37°C overnight and the reaction terminated by addition of 0.5 μL of 1% TFA. Digested samples were C18 ZipTipped to desalt and concentrate prior to MALDI target spotting which involved 3 cycles (aspirate and dispense) of 10 μL 80% acetonitrile, followed by 3 cycles of 10 μL 0.1% TFA. Sample binding consisted of 15 binding cycles of 10 μL, followed by 3 wash cycles of 10 μL 0.1% TFA and 15 elution cycles of 8 μL of 80 % acetonitrile. 1 μL of the eluate was mixed with 1 μL of CHCA matrix and spotted directly onto a Bruker 384 spot ground steel MALDI target for analysis.
MS analysis was carried on a Bruker Ultraflex III MALDI-TOFTOF instrument in positive reflectron mode. MSMS analysis was carried out on the same instrument in LIFT mode using a laser power boost of 45% and a detector gain boost of 110% following precursor ion selection of the MS ion. Spectra were processed in FlexAnalysis software (Bruker Daltonics, Germany, version 1.3) for baseline correction and smoothing prior to deriving sequence information using Biotools software (Bruker Daltonics, Germany).
Genetically Produced Chimera
The plasmid vector pET30 expression was modified with 6mer or 15mer silk repeats of the native spidroin 1 sequence of Nephila clavipes 34. The modified plasmid vector, pET30-silk (Novagen) was used for the cloning and expression of the fusion Si-peptide-silk protein in E. coli. The nucleotide sequence, coding for the Si-peptide designated Pep1 (amino-acid sequence from N to C terminal: KSLSRHDHIHHH) with N-terminal Nhe1 and C terminal SpeI cohesive ends, was designed using codon usage optimised for E. coli and obtained from Invitrogen (Carlsbad, CA). The synthesis of the genetically produced chimera and its purification procedure are described in detail in the supplementary information.
Bio-Inspired Silicification
Silicification Procedure
Tetraethoxysilane (TEOS, Sigma Aldrich, St. Louis, MO, USA) was used as the silica precursor. TEOS (1.115 mL) was hydrolysed for 15 min in a water: ethanol mixture (1:1 mix solution; 3.835 mL) acidified with HCl 1M (50μL; pHfinal = 2). For each sample, 870 μL of a 1 mg.mL−1 biomolecule solution (silk, peptide, or silk-peptide chimera) were mixed with 27 μL of citric acid 1M (Fisher Scientific, Loughborough, Leicester, UK) and 73 μL of bis-tris propane 1M (Sigma Aldrich, St Louis, MO, USA), to obtain pH 7 in the condensing system. After 15 min the pre-hydrolysed TEOS solution was added to give a final silica concentration of 30 mM. After pre-determined reaction times, samples were lyophilised and kept for further analysis.
Kinetics of Silica Condensation
The kinetics of silica condensation in the presence of the biomolecules was followed for 24 hours using the molybdenum blue test (adapted from Mullin and Riley.35). The molybdic acid stock solution and the reducing agent solution were prepared as described in Belton et al.36 At various times, aliquots were transferred to the molybdic acid solution (1.5 mL molybdic acid stock solution in 15 mL distilled water). The silicomolybdic complexation reaction was stopped after 15 min (± 5 sec) with 8 mL of reducing agent. The absorbance was read at 810 nm after two hours of reduction time and compared to a blank of distilled water. Calibration was performed using a 1,000 ppm silica standard solution (BDH laboratories supplies, Poole, UK) diluted to a range of concentrations varying between 0 and 50 μM.
Biomaterials Characterization
Scanning Electron Microscopy and EDX analysis
Freeze-dried samples were attached to double-sided adhesive tape attached to SEM stubs and coated with carbon (Edwards, Sputter coater S150B). The samples were then examined with a SEM JEOL JSM-840A instrument operated at 20 kV. Measurements were carried out under secondary vacuum (about 10−5 torr) by retro diffusion of secondary electrons. Energy Dispersive X-ray analysis (Oxford Inca system with light element detector) was carried out during SEM experiments to determine the local composition of the samples (mapping of Si, O, C, N) over a depth of a few μm.
Infra-Red Spectroscopy
ATR-FTIR spectroscopy was carried out using a Perkin Elmer Spectrum 100-FTIR spectrometer on freeze-dried samples (blank, silk, peptide, Si-peptide-silk chimeras), silicified or not. For each sample, 32 scans were acquired from 4000 to 380 cm−1, and the background previously acquired, was subtracted. The resolution was set at 4 cm−1. Data were treated with Nicolet OMNIC software. For each spectrum, the baseline was manually corrected and the various proportions of beta turn, beta sheet and alpha helix/ random conformation ascertained Curve fitting was carried out using the Grams A1 spectroscopy software (v8.0) Spectra were multipoint baseline corrected with the levelling mode set to level and zero. Where necessary the baseline corrected peaks were then binomially smoothed with a maximum of 10 points. 6 peaks were selected across the amide I absorption band (1720-1580cm−1) corresponding to β turns (1666-1688cm−1) α helix (1654-1658cm−1) random coil (1643-1654cm−1) and β sheet (1624-1642cm−1)37-43.
The 2 additional peaks were to compensate for side chains (1720-1690cm−1) and non-baseline resolution from the amide II band (1580-1620cm−1) the drift limits for each component listed in brackets. Peak width at half height was limited to between 8 and 40cm−1 for all peaks and the peak height limited to positive values only. Initially a single iteration was carried out to force all peaks positive and then the baseline function was set to linear. A total of 1000 iterations were then carried out to attain the best fit. The area under the random coil and α helix peaks were summed as overlap in this region is common and individual areas can be unreliable. Component compositions were then expressed as a percentage of the total assigned conformers. The errors associated with this type of analysis are illustrated in Figure S2(a and b) which shows the results of an example fitting and the errors associated with the analysis of α lactalbumin, 5 repeat samplings and peak fittings.
Nitrogen Gas Adsorption/Desorption Analysis
Samples were degassed (15 h, 50°C) under vacuum and analyses were carried out on freezedried samples using a Quantachrome nova 3200e surface area and pore size analyser. Surface areas were determined by the BET method, where nitrogen is assumed to have a crosssectional area of 0.16 nm2, over the range of relative pressures 0.05-0.3 at which point the monolayer is assumed to assemble. Particle radii were calculated from the BET surface area measurements on the assumption that all surface area was generated by spherical particles 1. Pore radii were determined by the BJH method.40 using the desorption branch of the isotherm.
Results and Discussion
Comparison of Silica-Based materials
Morphology and composition of the materials
SEM/EDX analyses were performed on the silk proteins and on the chemically and genetically produced protein chimeras, before and after silicification. From SEM data, silica sphere formation was observed upon silica condensation onto the biomolecule templates and the materials prepared in the presence of silica showed a tendency to aggregate into clumps (Figure 2).
Figure 2.

SEM images of biomolecules (+ SiO2, 30 mM in the condensing system). (A) Blank + SiO2. (B) Silk + SiO2. (C) Pep1 + SiO2. (D) Chimera silk-Pep1 + SiO2. (E) 6 mers + SiO2 (F) 6 mers-Pep1 + SiO2. (G) 15 mers + SiO2. (H) 15 mers-Pep1 + SiO2. After 24h silica condensation and direct lyophilisation.
In the absence of biomolecules, the silica sample (Figure 2A) showed a granular structure with spherical particles approximately 50 nm in diameter. Non-silicified silk samples (natural reconstituted silk, 6 mers and 15 mers) exhibited a network of fibers of the protein (results shown in supplementary information, Figure S2). Upon silicification of the silk samples (Figure 2B), spherical silica particles similar to those present in the blank sample appear to be deposited on silk, giving rise to a rough granular appearance superimposed on the biotemplate network morphology. The silicification of peptide Pep1 (Figure 2C) led to the formation of larger particles (≈100 nm in diameter) than found for silica condensation on its own. This phenomenon appears to be amplified by the coupling of Pep1 to silk, demonstrating that the silicification properties of the peptide were conserved upon grafting, with an added templating effect induced by the reconstituted silk network (Figure 2D).
In contrast, the genetic coupling of Si-binding peptide Pep1 to 15 mer silk did not significantly change the size of the silica spheres produced upon silicification (1.1 μm standard deviation 0.3 μm with or without pep 1) and the granular structure was less noticeable (Figures 2 G/H). The addition to the 6 mer changed both the size (average aggregate size 820nm without pep 1, 690 nm with pep 1) with more control over monodispersity (standard deviations of 240 nm and 130 nm respectively) and appearance of the silica spheres, compared to materials formed in the presence of the 15 mers, whereby the granular structure was no longer visible at these magnifications (Figures 2 E/F). These observations are in accordance with the lower ratio of Si-binding peptide coupled to silk mers (1:1). Yet, as compared to the chemical protein chimera, the genetically produced protein chimera showed larger silica particles upon silicification (Figure 2, panel F, H and D), possibly indicating a higher efficiency of the silica condensation process, which was not anticipated with regard to the lower number of potential binding sites for silica in the genetic chimera (Table 1). An alternative interpretation is that the chemical protein chimeras have structures that are mainly stabilized through intramolecular attractions and therefore remain as isolated molecules which collapse into a globular form on silicification whereas the genetic chimeras may be able to form large aggregates through intermolecular attractions with their size limited by surface charge density.
In summary, each biomolecule used had a different effect on silicification; natural reconstituted silk serving as a non-specific nucleation site or simple support for silica nuclei deposition whereas Pep1 acted as a specific trigger for the growth and aggregation of such nuclei. The chemical coupling of Pep1 to silk led to a molecule able to act both as a specific nucleation site and as a template for the directed assembly of silica nuclei and larger structures.
In support of the structural evidence, EDX elemental analysis of Silicon (Si), Oxygen (O), and Nitrogen (N) performed on silicified samples showed an external and visible silicon partition, whereas the Nitrogen (N) map arising from the silk protein and peptides was hidden inside the silicified structure (See supplementary data, Figure S3).
Fourier-Transform Infra-Red Spectroscopy
FTIR analysis was used to identify the presence of silica and the biomolecules within the materials formed after silica condensation. The silicified blank sample gave three distinct bands at 810, 1000 and 1050 cm−1, corresponding to Si-O-Si bending, Si-OH and Si-O-Si stretching vibrations respectively (representative results shown in the supplementary information, Figure S4). The materials prepared in the presence of peptides and/or chimeras contained additional bands between 1400 and 1800 cm−1, characteristic of protein amide bands (i.e. amide I: 1620 cm−1, amide II: 1520 cm−1, amide III: 1240 cm−1), see supplementary information, Figure S4. These results are in accordance with the EDS elemental analysis showing biomolecule occlusion inside the silica formed. Deconvolution of the data provided in the amide bands was used to assess the effect of silica forming on the structure of the silk based biomolecules, Figure 3.
Figure 3.

Percentage of conformers in 6 mers, 15 mers, silk and respective chimeras, silicified (30 mM in the condensing system) or not.
The ‘control’ silks (6 mer, 15 mer and Bombyx mori silk) all showed the expected tendency to form beta sheet and beta turn structures in solution. The 15 mer which has the most uniformly repetitive structure shows a higher ratio of beta sheet to beta turns which would suggest more intermolecular associations than for the 6 mer or B. silks. The ratio of β turn to β sheet observed is higher for the natural silk than observed in the unmodified genetic proteins suggesting that intramolecular associations are comparably favoured and this may represent the formation of the confined nanocrystalline states which are reported to be crucial to the mechanical properties of silk13-15. Addition of the pep 1 moiety at multiple locations through the primary sequence disrupts this resulting in shorter β sheet domains replaced with an increased random or helical nature. Compared to the changes in conformation observed during chimera formation the conservation of structure during the silicification process is relatively high but were the proteins to be intimately associated with the siliceous part of the composite a combination of steric and electrostatic disruptions would have been expected to be very apparent. The fact that there is no significant conformational change during silica condensation experiments provides evidence that in all cases the observed siliceous materials have been templated onto an already existing solution structure which itself remains largely unaltered at the core of the materials formed.
Nitrogen Adsorption
The surface area of the materials was determined using the BET method, with the pore volume and pore radius being calculated from the desorption branch of the isotherm using the BJH method (Table 2). The surface areas of the silks alone were below the limit of detection of the apparatus. All materials were mesoporous with the peptide, Pep1 having little effect on the nature of the silica produced. The observed reduction in the surface areas of the silica condensed in the presence of the genetically produced silk chimeras indicated a change in primary particle size (Table 2).
Table 2.
Surface area (m2/g), calculated particle radius, pore volume (cm3/g) and pore radius (nm) in silicified materials.
| BET | BJH | ||||
|---|---|---|---|---|---|
| Sample | Surface Area (m2/g) |
Calculated particle radius nm1 | Pore volume (cc/g) |
Pore Radius (Dv(r), nm) |
|
| Blank - SiO2 | 353.9 | 4 | 0.72 | 2.08 | |
|
| |||||
| Chemical chimera | Silk - SiO2 | 392.3 | 4 | 1.87 | 1.85 |
| Pep1 - SiO2 | 368.9 | 4 | 1.48 | 3.18 | |
| Chimera Pep1 - SiO2 | 167.9 | 7 | 0.96 | 1.64 | |
|
| |||||
| Genetic chimera | 6 mer SiO2 | 238.7 | 6 | 1.05 | 5.95 |
| 6 mer - pep 1 SiO2 | 78.0 | 18 | 0.61 | 18.39 | |
| 15 mer SiO2 | 110.2 | 12 | 0.35 | 18.44 | |
| 15 mer - pep 1 SiO2 | 61.7 | 20 | 0.21 | 30.18 | |
Particle radii were calculated from surface area measurements on the assumption that the area found arose from interconnected spherical particles. At low radii (< 4nm) the connectivity (coordination number) of the particles in relation to particle packing pattern becomes significant in reducing the area available to nitrogen molecules, but at 4nm and above an error of ca. 1nm in the radius calculated is observed 1. The results of the surface analysis indicate that the silk and Pep1 alone have little influence on primary particle size (ca. 4nm) when compared with the blank. The chimeras increasingly appear to control the particle size (chemical chimera, 7nm) with the genetic chimeras showing most effect (6mer-Pep1-silica 18nm, 15mer-Pep1-silica 20nm). These values largely correlate with the pore radii found in that they appear to be voids between the interconnected particles. A second explanation for the observed surface areas and porosity is that there could be greater affinity of the modified and genetic chimeras for the silica as it forms resulting in more irreversible entrainment of the organic phase in the voids between the interconnected particles. The data indicate that the smallest features observed by SEM (∼50nm for non chimeric and >400nm for chimeric silica precipitates) were aggregated structures and in order to understand the activity of the chemical chimera the efficiency of the Si-peptide grafting onto silk and the mechanisms of silica condensation were further investigated.
Efficiency of surface modification of silk with Si-Peptide
Si-Peptide Purity
Pep1 (KSLRSHDHIHHH) and R-Pep1 (RKSLRSHDHIHHH) prepared to enable an easy quantification of bound peptides using mass spectrometry following trypsinisation had purities as determined by RP-HPLC of 90% and 92%, respectively. The mass determined for Pep1 by mass spectrometry (1503.817 g.mol−1) was close to the calculated one (1503.643 g.mol−1). For R-pep 1, two main peaks were detected at 1659.914 and 1796.978 g.mol−1. The first peak was close to the expected mass (1659.832 g.mol−1), while the second corresponded to the additional coupling of one histidine residue (137.142 g.mol−1) to R-Pep1 in the His-tail, i.e. the RPep1 sample contained both RPep1 and RPep1H. This was not an issue as the latter peptides were only used to help confirm coupling of the desired peptide to the silk fibroin components.
Diazonium and EDC/NHS Coupling evaluation
Carboxylic acid groups of aspartic and glutamic acid are the main targets for the surface modification of proteins.45 Such modifications have already been applied to silk,46 however only 1.41% of the amino-acid residues of silkworm silk fibroin comprise carboxylic groups, limiting the extent of functionalization. In order to increase the number of potential grafting sites, silk was modified with a diazonium salt containing a carboxylic acid functionality. Such modification targets tyrosine and histidine residues (5.30% for Tyr and less than 0.1% for His).31, 46 With diazonium coupling the total number of carboxylic side groups on silk available for grafting can be theoretically increased up to 6.7%. Upon diazo-coupling and EDC/NHS activation, 365 moles of amino-acid residues can be potentially activated per mole of silk protein (Table 1). In this study, 25-28 moles of Si-peptide were added to react with these activated residues and may be coupled with silk. The excess of potential grafting sites compared to the number of Si-peptides was expected to favour peptide coupling onto silk while preventing steric hindrance issues. Figure 1 and Table 1 present the spatial distribution and the theoretical quantification of amino-acid residues potentially involved in silica binding on the chimeric silk proteins.
The diazo-coupling reaction and the modification of silk proteins with Si-peptides were followed by UV-VIS scanning spectrophotometry. Purified silk absorbs at 280 nm due to the presence of aromatic rings of the free tyrosine residues. Upon diazonium coupling, this band was completely replaced by a peak assigned to the azobenzene groups at 325 nm with a shoulder at 390 nm (Figure 4 LHS, horizontal arrow showing change in peak position).30 Unbound diazonium salts were removed using a disposable PD-10 desalting column, as can be demonstrated by a decrease in absorbance at 325 nm (Figure 4, vertical arrows).
Figure 4.
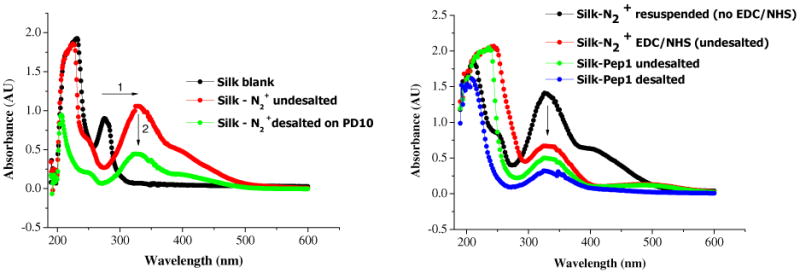
UV-Vis absorption spectra of silk solutions after different stages of diazocoupling. UV-Vis absorption spectra of silk solutions, LHS- after diazo-coupling, RHS-EDC/NHS activation, and Pep1 coupling (before and after desalting step).
The Si-peptides were then grafted through EDC/NHS coupling to the carboxylic acid groups of diazo-tyrosine, aspartic and glutamic acid residues of the modified silk protein. Upon EDC/NHS coupling, the absorbance at 325 nm progressively decreased, demonstrating the efficiency of the grafting to azobenzene groups (mechanism in suppl. information). However, the peak attributed to the azobenzene ring did not completely disappear due to an excess of potential coupling sites on silk compared to the number of Si-binding peptides introduced, steric hindrance or incomplete EDC/NHS grafting. The extent of the absorbance peak decrease at 325 nm would indicate that almost 80% of the diazo-modified residues have been coupled to Pep1 which is not possible given the molar ratio of peptide: coupling sites used. This over-estimation could be explained by an absorbance quenching of diazo-modified tyrosine residues in the vicinity of the grafted peptides. Hence, although reduction in band at 325 nm can be used to evidence Si-peptide coupling onto silk, it is not an appropriate method to quantify grafting.
The chemical coupling of Si-peptide onto silk protein was further probed by mass spectrometry carried out after trypsin proteolysis of the silk-peptide protein chimera and the corresponding controls (silk and Si-peptides). Trypsin cleaves peptide bonds at the carboxyl side of the amino-acids lysine (K) or arginine (R) except when either is followed by proline (P). Fragments released from Si-peptide-silk chimera trypsinisation can either be linear (fragment constituted of silk or peptide solely) or ramified (silk-peptide fragment) as explained in Figure 5.
Figure 5.

Potential cleavage sites upon trypsinisation of Si-peptide-silk chimera. The peptide can be cleaved at 1, 2 or 3 locations in the meantime (inset figure). Silk block taken from Wong Po Foo, C.; Patwardhan, S. V.; Belton, D. J.; Kitchel, B.; Anastasiades, D.; Huang, J.; Naik, R. R.; Perry, C. C.; Kaplan, D. L. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9428–9433 and is “Copyright (2006) National Academy of Sciences, U.S.A.”.
Evidence for the coupling of the silica binding peptides to both the heavy and light chain components of silk fibroin was obtained with coupling to both modified tyrosine and acidic amino acids being seen, Table 3. Additional mass spectrometric evidence for the presence of the peptide and silk fragments is given in the supplementary information, Tables S1 and S2.
Table 3.
Molecular weight of various silk fibroin and ramified Si-peptide fragments obtained from trypsin proteolysis of Si-peptide-silk chimera with peptide ions sequenced by MS/MS.
| Ramified fragment | ||||
|---|---|---|---|---|
| Silk fibroin fragment | Si-peptide fragment | Theoretical MW (g.mol−1) | Experimental MW (g.mol−1) | |
| Light chain | YIAQAASQVHV | K | 1444.795 | 1444.737 |
| AWDYVDDTDKSIAILNVQEILK | K | 2807.458 | 2807.276 | |
| RLYNSR | RK | 1092.27 | 1091.466 | |
|
| ||||
| Heavy chain | GYGQGAGSAASSVSSASSR | KSLSRHDHIHHH | 3324.529 | 3323.638 |
| DASGAVIEEQITTK | R | 1617.67 | 1618.7 | |
| EGYEYAWSSK | R | 1375.46 | 1375.674 | |
Silica Condensation on Chemically Produced Chimera
Kinetic Study of Silica Condensation
Silica condensation in the presence of the biomolecules (silk, peptides, silk-peptide chimera) was followed using the molybdenum blue method.1 Kinetic constants (figure 6) were determined between 1-4 min (apparent third order rate constant) and 4.5-20 min of silica condensation (reversible first order rate constants)47, with Figure S6 showing the relationship between speciation and rate constants and raw data being included in the supplementary information, Figure S7.
Figure 6.

Relative third order (Top) and relative first order constants (Bottom) corresponding to the condensation of silica dimers into trimers and to the transition between trimers and oligomers, respectively (30 mM Si).
Silica condensation occurred in the presence of silk, Pep1, and their respective chimera Silk-Pep1. No significant differences could be detected between the systems in the first minutes of condensation, where up to 85% of monosilicic acid condensation occurred. Small differences were detected for condensation times greater than 300 minutes where the condensing trend evolved in the following order: Silk < Chimera Silk-Pep1 < Pep 1< Blank in line with the Si-binding peptide concentration, Figure S7. The small differences in the rate constants for reaction between ca. 4-20 minutes of reaction do not relate to presence of Pep1. Although differences in materials properties were observed, it is clear that for the biomolecules investigated these did not have a catalytic effect, rather a templating effect was found as evidenced from the structures observed.
Concerning the relative forward first order constants (k+), the condensation of trimers into oligomers was slower for Chimera Silk-Pep1 as compared to Pep1, probably due to a concentration effect. A similar trend was observed for chimera Silk-Pep1 concerning the reverse first order constant (k−), indicating that the transition between trimers and oligomers is slowed down.
To summarize the impact of silk-based biomolecules on the kinetics of silicification observed using the molybdenum blue assay, the main effect appeared to be a decreased rate of trimerisation and oligomer formation, and a promotion of oligomer dissolution, both effects being amplified at increasing molecular weight of the biomolecule. This can be explained by a complexation of charged silicate monomers and small oligomers along the chains of the biomolecules, with the larger chains leading to a higher probability for strong cooperative binding sites to exist.
Conclusions
The impact of the silica binding peptide and novel chimeras (genetic and chemical) containing this peptide on protein structure and silica composite formation were compared. In all cases, the structure of the protein / chimera in solution dictates the type of composite structure that forms with the silica deposition process having little effect on the secondary structural composition of the silk based materials. In our previous work, the presence of both charged and uncharged amine groups within a number of polyamines was demonstrated as being a key factor for the catalysis of silica condensation leading to large silica structures6 but in this study we have shown that silica-binding peptides that do not contain such groups can also be used to generate distinct particulate structures. Similarly to our study of genetic silk based chimeras containing the R5 peptide (SSKKSGSYSGSKGSKRRIL),26 the role of the chimeras (genetic and chemical) used in this present study resides more in aggregation and scaffolding than in the catalysis of condensation. A comparison of the early stages of the silica formation process for the genetic R5-silk chimera and the chimeras in this study using a peptide that carries much less positive charge also shows that by choice of peptide, the rate of silica formation can be varied. Changing the silk based component from 6mer to 15mer to natural silk together with the addition of the chosen peptide at different levels can be used to tune the morphology and physical properties of the materials formed.
The variables of peptide identity, silk construct (number of consensus repeats or silk source) and approach to synthesis (genetic or chemical) can be used to ‘tune’ the properties of the composite materials formed and is a general approach which can be used to prepare a range of materials for biomedical and sensor based applications.
Supplementary Material
Acknowledgments
The authors would like to thank Dr David Boocock for MALDI ToF MS analysis. Financial support from EPSRC (EP/E048439/1), the AFOSR and the NIH (5RO1DE017207-05) made the work possible and their support is gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental procedures for the diazonium coupling reaction; EDC/NHS activation procedure; generation of the genetically produced chimeras; Figures S1 (mechanism of EDC/NHS amine coupling), S2 (SEM of non-silicified biomolecules), S3 (EDX elemental analysis of silicified biomolecules), S4 (ATR-FTIR spectra for non-silicified and silicified biomolecules), S5 (example of amide I band curve fitting), S6 (kinetic constants determination upon silica condensation), S7 (silicic acid concentration %) for silica condensation on biomolecules); Tables S1 (molecular weight of trypsin fragments obtained from trypsin autolysis), S2 (Molecular weight of various Si-peptide fragments and silk fragments following trypsin proteolysis of Si-peptide-silk chimera).
This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.Iler RK. The Chemistry of Silica. Plenum Press; New York: 1979. [Google Scholar]
- 2.Perry CC. Prog Mol Subcell Biol. 2009;47:295–313. doi: 10.1007/978-3-540-88552-8_13. [DOI] [PubMed] [Google Scholar]
- 3.Fan TX, Chow SK, Zhang D. Prog Mater Sci. 2009;54:542–659. [Google Scholar]
- 4.Poulsen N, Sumper M, Kröger N. Proc Natl Acad Sci USA. 2003;100:12075–12080. doi: 10.1073/pnas.2035131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patwardhan SV, Clarson SJ, Perry CC. Chem Commun. 2005:1113–1121. doi: 10.1039/b416926c. [DOI] [PubMed] [Google Scholar]
- 6.Belton DJ, Patwardhan SV, Annenkov VV, Danilovtseva EN, Perry CC. Proc Natl Acad Sci USA. 2008;105:5963–5968. doi: 10.1073/pnas.0710809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brzezinski MA. Proc Natl Acad Sci USA. 2008;105:1391–1392. doi: 10.1073/pnas.0711994105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li C, Kaplan DL. Curr Opin Solid State Mater Sci. 2003;7:265–271. [Google Scholar]
- 9.Patwardhan SV, Clarson S. J Mater Sci Eng C. 2003;23:495–499. [Google Scholar]
- 10.Pozzolini M, Sturla L, Cerrano C, Bavestrello G, Camardella L, Parodi AM, Raheli F, Benatti U, Müller WEG, Giovine M. Mar Biotechnol. 2004;6:594–603. doi: 10.1007/s10126-004-3036-y. [DOI] [PubMed] [Google Scholar]
- 11.Wong Po Foo C, Huang J, Kaplan DL. Trends Biotechnol. 2004;22(11):577–585. doi: 10.1016/j.tibtech.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Grubb DT, Jelinski LW. Macromolecules. 1997;30:2860–2867. [Google Scholar]
- 13.Keten S, Xu Z, Ihle B, Buehler MJ. Nat Mat. 2010;9:359–367. doi: 10.1038/nmat2704. [DOI] [PubMed] [Google Scholar]
- 14.Giesa T, Arslan M, Pugno MM, Buehler MJ. Nano Lett. 2011;11:5038–5046. doi: 10.1021/nl203108t. [DOI] [PubMed] [Google Scholar]
- 15.Keten S, Buehler MJ. J R Soc, Interface. 2010;7:1709–1721. doi: 10.1098/rsif.2010.0149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simmons A, Michal C, Jelinski L. Science. 1996;271:84–87. doi: 10.1126/science.271.5245.84. [DOI] [PubMed] [Google Scholar]
- 17.Termonia Y. Macromolecules. 1994;27:7378–7381. [Google Scholar]
- 18.Porter D, Vollrath F, Shao Z. Eur Phys J E. 2005;16:199–206. doi: 10.1140/epje/e2005-00021-2. [DOI] [PubMed] [Google Scholar]
- 19.Gosline JM, Guerette PA, Ortlepp CS, Savage KN. J Exp Biol. 1999;202:3295–3303. doi: 10.1242/jeb.202.23.3295. [DOI] [PubMed] [Google Scholar]
- 20.Kluge JA, Rabotyagova O, Leisk GG, Kaplan DL. Trends Biotechnol. 2008;26:244–251. doi: 10.1016/j.tibtech.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Hardy JG, Romer LM, Scheibel TR. Polymer. 2008;49:4309–4327. [Google Scholar]
- 22.Cheng C, Shao Z, Vollrath F. Adv Funct Mater. 2008;18:2172–2179. [Google Scholar]
- 23.Feng XX, Zhang LL, Chen JY, Guo YH, Zhang HP, Jia CI. Int J Biol Macromol. 2007;40:105–111. doi: 10.1016/j.ijbiomac.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Wong Po Foo C, George A, Kaplan DL. Biomaterials. 2007;28:2358–2367. doi: 10.1016/j.biomaterials.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 25.Mieszawska AJ, Fourligas N, Georgakoudi I, Ouhib NM, Belton DJ, Perry CC, Kaplan DL. Biomaterials. 2010;31:8902–8910. doi: 10.1016/j.biomaterials.2010.07.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong Po Foo C, Patwardhan SV, Belton DJ, Kitchel B, Anastasiades D, Huang J, Naik RR, Perry CC, Kaplan DL. Proc Natl Acad Sci USA. 2006;103:9428–9433. doi: 10.1073/pnas.0601096103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mieszawska AJ, Nadkarni LD, Perry CC, Kaplan DL. Chem Mater. 2010;22:5780–5785. doi: 10.1021/cm101940u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Currie HA, Deschaume O, Naik RR, Perry CC, Kaplan DL. Adv Funct Mater. 2011;21:2889–2895. doi: 10.1002/adfm.201100249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patwardhan SV, Emami FS, Berry RJ, Jones SE, Naik RR, Deschaume O, Heinz H, Perry CC. J Am Chem Soc. doi: 10.1021/ja211307u. In submission. [DOI] [PubMed] [Google Scholar]
- 30.Murphy AR, St John P, Kaplan DL. Biomaterials. 2008;29:2829–2838. doi: 10.1016/j.biomaterials.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ha SW, Gracz HS, Tonelli AE, Hudson SM. Biomacromolecules. 2005;6:2563–2569. doi: 10.1021/bm050294m. [DOI] [PubMed] [Google Scholar]
- 32.Inoue S, Tanaka K, Arisaka F, Kimura S, Ohtomo K, Mizuno S. J Biol Chem Mol Biol. 2000;275(22):40517–40528. doi: 10.1074/jbc.M006897200. [DOI] [PubMed] [Google Scholar]
- 33.Kikuchi Y, Mori K, Suzuki S, Yamagushi K, Mizuno S. Gene. 1992;110:151–158. doi: 10.1016/0378-1119(92)90642-3. [DOI] [PubMed] [Google Scholar]
- 34.Bini E, Wong Po Foo C, Huang J, Karageorgiou V, Kitchel B, Kaplan DL. Biomacromolecules. 2006;7:3139–3145. doi: 10.1021/bm0607877. [DOI] [PubMed] [Google Scholar]
- 35.Mullin JB, Riley JP. Anal Chim Acta. 1955;12:162–176. [Google Scholar]
- 36.Belton DJ, Deschaume O, Patwardhan SV, Perry CC. J Phys Chem B. 2010;114:9947–9955. doi: 10.1021/jp101347q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goormaghtigh E, Cabiaux V, Ruysschaert JM. Eur J Biochem. 1990;193:409–20. doi: 10.1111/j.1432-1033.1990.tb19354.x. [DOI] [PubMed] [Google Scholar]
- 38.Byler DM, Susi H. Biopolymers. 1986;25:469–87. doi: 10.1002/bip.360250307. [DOI] [PubMed] [Google Scholar]
- 39.Susi H, Byler DM. Methods Enzymol. 1986;130:290–311. doi: 10.1016/0076-6879(86)30015-6. [DOI] [PubMed] [Google Scholar]
- 40.Kong J, Yu S. Acta Biochim Biophys Sin. 2007;39:549–559. doi: 10.1111/j.1745-7270.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- 41.Barth A. Prog Biophys Mol Biol. 2000;74:141–173. doi: 10.1016/s0079-6107(00)00021-3. [DOI] [PubMed] [Google Scholar]
- 42.Kauppinen JK, Moffatt DJ, Mantsch HH, Cameron DG. Appl Spectrosc. 1981;35:271–276. [Google Scholar]
- 43.Sofia S, Mc Carty MB, Gronowicz G, Kaplan DL. J Biomed Mater Res. 2001;54:139–148. doi: 10.1002/1097-4636(200101)54:1<139::aid-jbm17>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 44.Barret EP, Joyner LG, Halenda PP. J Am Chem Soc. 1951;73:373–380. [Google Scholar]
- 45.Vepari C, Kaplan DL. Prog Polym Sci. 2007;32:991–1007. doi: 10.1016/j.progpolymsci.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou CZ, Confalonieri F, Jacquet M, Perasso R, Li ZG, Janin J. Proteins. 2001;44:119–122. doi: 10.1002/prot.1078. [DOI] [PubMed] [Google Scholar]
- 47.Harrison CC, Loton N. J Chem Soc Faraday Trans. 1995;91:4287–4297. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.