

Abstract
Increased IFN-α signaling is a heritable risk factor for systemic lupus erythematosus (SLE). IFN induced with helicase C domain 1 (IFIH1) is a cytoplasmic dsRNA sensor that activates IFN-α pathway signaling. We studied the impact of the autoimmune-disease–associated IFIH1 rs1990760 (A946T) single nucleotide polymorphism upon IFN-α signaling in SLE patients in vivo. We studied 563 SLE patients (278 African-American, 179 European-American, and 106 Hispanic-American). Logistic regression models were used to detect genetic associations with autoantibody traits, and multiple linear regression was used to analyze IFN-α–induced gene expression in PBMCs in the context of serum IFN-α in the same blood sample. We found that the rs1990760 T allele was associated with anti-dsDNA Abs across all of the studied ancestral backgrounds (meta-analysis odds ratio = 1.34, p = 0.026). This allele also was associated with lower serum IFN-α levels in subjects who had anti-dsDNA Abs (p = 0.0026). When we studied simultaneous serum and PBMC samples from SLE patients, we found that the IFIH1 rs1990760 T allele was associated with increased IFN-induced gene expression in PBMCs in response to a given amount of serum IFN-α in anti-dsDNA–positive patients. This effect was independent of the STAT4 genotype, which modulates sensitivity to IFN-α in a similar way. Thus, the IFIH1 rs1990760 Tallele was associated with dsDNA Abs, and in patients with anti-dsDNA Abs this risk allele increased sensitivity to IFN-α signaling. These studies suggest a role for the IFIH1 risk allele in SLE in vivo.
Systemic lupus erythematosus (SLE) is a complex multi-system autoimmune disease resulting from both genetic and environmental factors (1). The type I IFN system of anti-viral immunity is pathologically overactive in many SLE patients, and multiple lines of evidence support the idea that increased type I IFN signaling is a primary pathogenic event in human lupus (2, 3). In some cases, SLE has been induced by rIFN-α that was given as a treatment for chronic viral infections and malignancy (4–6). Serum IFN-α activity is elevated in many SLE patients (7), and this abnormality also is found in some unaffected relatives of SLE patients, supporting the idea that high serum IFN-α level is a heri-table risk factor for SLE (8, 9). Many SLE risk genetic variants that function in the IFN-α pathway result in increased IFN-α pathway signaling in patients in vivo, providing further support for this idea (3, 10–15).
IFN induced with helicase C domain 1 (IFIH1, also known as MDA5) is a DEAD box helicase that senses viral RNA and helps to induce transcription of type I IFN and IFN-induced genes when activated (16). IFIH1 is localized in the cytoplasm and shares significant similarities with retinoic acid inducible gene I (RIG-I), another cytoplasmic RNA sensor (16). Genetic variants of IFIH1 have been associated with type I diabetes (17), autoimmune thyroid disease (18), psoriasis (19), and recently SLE (20, 21). A common coding-change variant in IFIH1 (rs1990760, A946T) has been associated with these autoimmune conditions, and rare variants in IFIH1 also have been associated with protection from type I diabetes (22). The autoimmune-disease-associated allele of IFIH1 (rs1990760 T, 946T) is not predicted to disrupt the protein structure by the PolyPhen database. In fact, the rs1990760 T risk allele is likely a gain-of-function variation, resulting in increased expression of IFIH1 (23), although this finding has not been replicated uniformly (24). The rare variants in IFIH1 that are associated with protection from type I diabetes result in decreased expression of IFIH1 (23). These data taken together suggest that increased expression or gain-of-function in IFIH1 predisposes to human autoimmunity.
Given the importance of this protein in type I IFN responses and the pathogenic importance of IFN-α in human SLE, we investigated the impact of the IFIH1 rs1990760 polymorphism on the IFN-α pathway in SLE patients in vivo. Autoantibodies against dsDNA and small nuclear RNA-binding proteins such as Ro, La, Sm, and ribonucleoprotein are strongly associated with serum IFN-α levels in SLE patients (7), and frequently SLE-associated loci demonstrate associations with particular autoantibodies (13, 25, 26). Therefore, we also investigated potential associations between IFIH1 rs1990760 T and SLE-associated autoantibodies.
Materials and Methods
Patients and methods
We studied serum and genomic DNA samples from 563 SLE patients from the University of Chicago Translational Research in the Department of Medicine registry, the Hospital for Special Surgery Lupus Registries, Rush University Medical Center, and the NorthShore University Health System. The SLE cohort consisted of 278 African-American, 179 European-American, and 106 Hispanic-American SLE patients. All of the patients met the revised 1982 American College of Rheumatology criteria for the diagnosis of SLE (27). PBMCs were obtained from 80 anti-dsDNA–positive SLE patients and 24 anti-dsDNA–negative SLE patients selected from the subjects above. The subjects in this study were not related to each other. Informed consent was obtained from all of the subjects at each site, and the study was approved by the Institutional Review Board at each institution.
Single nucleotide polymorphism genotyping
SLE patients were genotyped at IFIH1 rs1990760 and STAT4 rs7574865 using Applied Biosystems Taqman Assays-by-Design primers and probes on an Applied Biosystems 7900HT PCR machine with >98% genotyping success. All of the scatter plots were reviewed individually for quality, and genotype frequencies did not deviate significantly from the expected Hardy-Weinberg proportions (p > 0.01 in all of the ancestral backgrounds).
Reporter cell assay for IFN-α
The reporter cell assay for IFN-α has been described in detail elsewhere (8, 28). In this assay, reporter cells were used to measure the ability of patient sera to cause IFN-induced gene expression. The reporter cells (WISH cells, American Type Culture Collection CCL-25) were cultured with 50% patient sera for 6 h and then lysed. cDNA was made from total cellular mRNA, and cDNA then was quantified using real-time PCR. Forward and reverse primers for the genes IFN-induced protein with tetratricopeptide repeats 1 (IFIT1), myxovirus resistance 1 (MX1), and dsRNA-activated protein kinase, which are known to be highly and specifically induced by IFN-α, were used in the reaction (8).
PBMC processing and transcript analysis
PBMCs were processed immediately after phlebotomy. Cells were lysed, and cDNA was made from total cellular RNA. cDNA transcripts then were quantified using real-time PCR with the same primers for the IFIT1 and MX1 genes used above (8) (see Ref. 29 for further details regarding PBMC transcript analysis).
Real-time PCR data analysis
PCR data from both WISH cells and PBMCs were analyzed in the same way. The relative expression of each of the tested IFN-induced genes was calculated as a fold increase compared with its expression in either WISH cells cultured with media alone or healthy donor PBMCs, respectively. The PCR data were log-normally distributed, and we performed log10 transformation to normalize the relative expression data. We also used an algorithm to generate a single serum IFN-α activity score from the three genes tested in the WISH cells (8). Healthy unrelated donor sera (n = 141) were tested in the WISH assay. The mean and SD of the log-transformed IFN-α–induced gene relative expression in healthy individuals were calculated. The number of SDs above the mean of healthy donors was calculated for each gene tested in patient samples. The number of SDs above healthy donors then was summed for the three genes, and this number was reported as a score representing serum IFN-α activity (8). This assay has been highly informative when applied to SLE as well as other human autoimmune disease populations (8, 30, 31).
Measurement of autoantibodies
The anti-Ro, anti-La, anti-Sm, and anti-ribonucleoprotein Abs were measured in all of the samples by ELISA methods using kits from INOVA Diagnostics (San Diego, CA), and anti-dsDNA Abs were measured using Crithidia luciliae immunofluorescence, with detectable fluorescence considered positive. All of the samples were assayed in the University of Chicago clinical laboratory. Standard cutoff points for a positive test designated by the manufacturer were used to categorize samples as positive or negative.
Statistical analysis
Associations between the presence of autoantibodies and genotype at IFIH1 rs1990760 were detected using logistic regression models as implemented in the PLINK program, version 1.07 (http://pngu.mgh.harvard.edu/~purcell/plink/) (32). Each ancestral background was analyzed separately initially. When similar tendencies were observed across ancestral backgrounds, a meta-analysis was performed using a fixed-effects model, and testing for homogeneity between data sets was performed using Cochrane's Q statistic. The serum IFN-α activity score data were distributed nonnormally, and nonparametric Mann–Whitney U test was used to compare quantitative serum activity IFN-α data between two genotype subgroups. Relative expression data for each individual IFN-α–induced gene was log-normally distributed, and when data were compared for individual gene expression these data first were log10-transformed. The individual gene relative expression data then passed statistical testing for normality by the D'Agostino and Pearson omnibus normality test (p > 0.05), and parametric t testing was used to compare differences between columns of individual gene relative expression data. Linear regression also was used to analyze the relative expression of individual IFN-α–induced genes in the reporter cell line versus the relative expression of the same transcript in PBMCs from the same blood sample. p values for this analysis were calculated using the extra sum-of-squares F test for a difference in slope or y-intercept of the best-fit line.
The p values shown in the article are uncorrected for multiple comparisons. When testing differences in the serum IFN-α activity score between different genotype groups, a p value of 0.016 is sufficient to control the family-wise type I error rate at 0.05. In the t tests comparing the differences in IFIT1 and MX1 expression between reporter cells and PBMCs comparing CC versus CT or TT genotypes, p values of 0.025 or less would control the type I error rate at 0.05. For the serum IFN-α versus IFN-α–induced gene expression in PBMC regressions, df are taken into account in the determination of the p value, and further statistical correction for the number of lines plotted on the graph is not necessary.
Results
IFIH1 rs1990760 T allele is associated with serologic profile in SLE patients
We used logistic regression to test for associations between IFIH1 genotype and serum autoantibodies in the SLE cohort. There was a similar trend toward association of rs1990760 T with anti-dsDNA autoantibodies in each ancestral background, which was statistically significant in the meta-analysis (Table I, meta-analysis using fixed effects model odds ratio = 1.34, p = 0.026, Cochrane's Q = 0.99, indicating no evidence for heterogeneity in the association between groups). We did not detect any other significant consistent associations between IFIH1 rs1990760 T and autoantibodies in our cohort.
Table I.
Association of IFIH1 rs1990760 T with anti-dsDNA Abs across multiple ancestral backgrounds
| Ancestral Background | T Allele Frequency dsDNA− | T Allele Frequency dsDNA+ | Odds Ratio |
|---|---|---|---|
| African-Americans (n = 268) | 0.198 | 0.252 | 1.36 |
| European-Americans (n = 179) | 0.583 | 0.646 | 1.31 |
| Hispanic-Americans (n = 106) | 0.400 | 0.473 | 1.35 |
For the T allele frequency, dsDNA−/dsDNA+ is the frequency of the T allele in SLE patients lacking anti-dsDNA Abs (dsDNA−) or those with a positive anti-dsDNA Ab test (dsDNA+). Meta-analysis: odds ratio = 1.34, p = 0.026; Cochrane's Q = 0.99.
rs1990760 T is associated with lower serum IFN-α levels in anti-dsDNA–positive SLE patients
In patients stratified by IFIH1 genotype, serum IFN-α levels were higher in CC subjects than those in CT subjects (p = 0.034, Fig. 1A). Due to the association that we had detected with anti-dsDNA Abs and the large impact that these autoantibodies have upon serum IFN-α levels, we performed an analysis stratified by dsDNA Abs (Fig. 1B). In this analysis, subjects with anti-dsDNA Abs clearly show a dose-response decrease in serum IFN-α in the presence of the IFIH1 rs1990760 T allele (p = 0.0026 for CC versus TT). In contrast, subjects lacking anti-dsDNA Abs showed no relationship between IFIH1 genotype and serum IFN-α level. This pattern was consistent across all of the studied ancestral backgrounds.
FIGURE 1.

Serum IFN-α activity in SLE patients stratified by rs1990760 genotype and anti-dsDNA autoantibodies. A, All patients in aggregate. B, Patients stratified by presence or absence of anti-dsDNA Abs. The line indicating the central tendency represents the median, the error bars show the interquartile range, and the p value was calculated using the Mann–Whitney U test.
IFIH1 rs1990760 T is associated with increased IFN-α–induced gene expression for a given amount of serum IFN-α activity in SLE patients with dsDNA Abs
Finding a decrease in serum IFN-α levels in subjects with the risk allele of IFIH1 seemed somewhat paradoxical, given the role for increased IFN-α signaling in SLE pathogenesis. We have noted this pattern once previously with the STAT4 autoimmune disease risk allele (STAT4 rs7574865) (13). In this case, the STAT4 autoimmune disease risk allele was associated with increased IFN-α–induced gene expression in PBMCs for a given amount of serum IFN-α in SLE patients in vivo. This suggested that the STAT4 allele increased sensitivity to IFN-α, because risk allele carriers had more robust IFN-α–induced gene expression at lower levels of serum IFN-α. This could explain lower serum IFN-α levels in patients carrying this allele, if IFN-α signaling results in risk of lupus in a dose-effect manner.
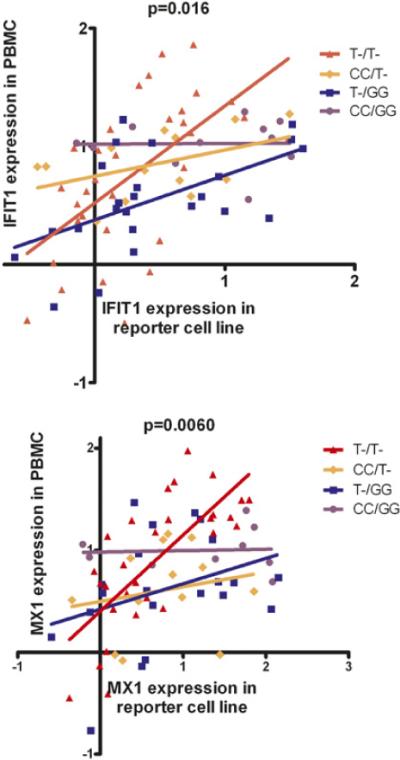
We looked for a similar sensitivity effect in relation to the IFIH1 allele in our SLE patients. Simultaneous serum and PBMC samples were available for 80 anti-dsDNA–positive patients, and we compared the relative expression of the IFN-α–induced IFIT1 and MX1 genes in these samples. The relative expression of IFIT1 and MX1 induced by patient serum in the WISH reporter cell line was subtracted from the relative expression of the same gene in PBMCs, and the results were analyzed in the context of the IFIH1 rs1990760 genotype. As shown in Fig. 2, after controlling for serum IFN-α activity in this way, greater IFN-α–induced gene expression was observed in the presence of the IFIH1 T allele. In anti-dsDNA–negative patients, no pattern was observed in IFN-α–induced gene expression related to IFIH1 genotype (data not shown).
FIGURE 2.

Difference in IFIT1 and MX1 expression in reporter cells versus PBMCs stratified by IFIH1 genotype. Relative expression values were log10-transformed, and the relative expression of each transcript in reporter cells exposed to patient sera was subtracted from the relative expression of the same transcript in PBMCs from the same blood sample. A, IFIT1. B, MX1. Lines indicate the mean, and error bars show the SDs. Data were distributed normally, and the p value was calculated using an unpaired t test for the difference between CC and CT or TT genotypes.
Impact of IFIH1rs1990760 T upon sensitivity to IFN-α is independent of STAT4 genotype
Given that we had observed previously a similar phenomenon in relation to the STAT4 rs7574865 genotype, we performed a stratified regression analysis to control for the STAT4 genotype. The difference in serum-induced versus PBMC gene expression was similar in the CT and TT genotypes in Fig. 3, so these were combined into one group to decrease the number of subgroups. Similarly, the rare STAT4 homozygous risk allele (TT) genotype (7 of the 80 subjects) was combined with the heterozygous risk allele carriers (GT genotype). Thus, both IFIH1 and STAT4 genotypes were binned into risk allele carrier versus nonrisk allele carriers, resulting in four genotype categories. Linear regression analysis was performed on these four subgroups of anti-dsDNA–positive SLE patients to determine the relationship between the capacity of serum to induce IFN-α–induced gene expression versus the actual IFN-α–induced gene expression observed in PBMCs from the same sample. As shown in Fig. 3, the effects of IFIH1 and STAT4 were additive, and the influence of IFIH1 was statistically independent of STAT4 genotype (in STAT4 risk allele carriers, carriage of the rs1990760 T allele resulted in a statistically significant increase in the slope of the regression line in Fig. 3, p < 0.05).
FIGURE 3.
IFN-β–induced gene expression in PBMCs versus serum IFN-β activity in anti-dsDNA–positive SLE patients. Patients were stratified by IFIH1 rs1990760 and STAT4 rs7574865 genotypes. T indicates carriage of the risk allele in each case (includes both heterozygous and homozygous risk allele genotypes). Data were log-transformed, and the lines shown are the result of linear regression in each genotype category. The p values were calculated by the sum-of-squares F test for a difference in the slope of the regression line between genotype categories.
Discussion
We provide evidence that the autoimmune disease susceptibility allele IFIH1 rs1990760 T is associated with anti-dsDNA Abs in SLE patients. In this anti-dsDNA–positive subgroup of SLE patients, we demonstrate a biological impact of the autoimmune disease risk allele rs1990760 T within the IFN-α pathway in vivo, suggesting that this allele is particularly important in this patient group. The rs1990760 T allele is associated with a modest increase in risk of SLE (odds ratio = 1.16–1.17) in overall case-control genetic studies (20, 21), and it is possible that these modest results reflect the heterogeneity of the SLE cohorts studied with respect to anti-dsDNA Abs. Incorporating serologic data in case-control studies of this locus may result in more robust findings, because it is quite possible that this allele is not of equal importance to all of the SLE patients.
Members of the TLR-independent pathway of viral defense and type I IFN generation have been implicated in SLE, including IFIH1 (20, 21) and more recently mitochondrial antiviral signaling protein (MAVS) (33). MAVS is an adaptor protein that facilitates signaling via RIG-I, which is a cytosolic DEAD box helicase nucleic acid sensor similar to IFIH1. Interestingly, the MAVS allele that was associated with SLE was loss-of-function and also was associated with a lack of autoantibodies against RNA-binding proteins and lower serum IFN-α levels in SLE patients (33). Although the MAVS allele is clearly a loss-of-function variant (33), existing data suggest that IFIH1 may be a gain-of-function variant (23). The MAVS and IFIH1 alleles associated with SLE both are associated with decreased serum IFN-α levels, but the MAVS allele is loss-of-function and is associated with a lack of autoantibodies of a particular specificity, whereas the IFIH1 allele is gain-of-function and is associated with the presence of particular autoantibodies. We do not suspect that the MAVS allele is associated with an increased sensitivity to IFN-α signaling and instead propose that this allele results in some dysfunction of the RIG-I pathway that results in SLE susceptibility via some other mechanism that is not known at present. Interestingly, a recent study demonstrated decreased IFN-β production in PBMCs from type I diabetes patients with the rare loss-of-function IFIH1 alleles that are associated with protection from type I diabetes (34). It would be of high interest to examine these polymorphisms in SLE patients as we have done with IFIH1 rs1990760, because we would predict that these variations would be associated with lower serum IFN-α levels and decreased sensitivity to IFN-α signaling.
Viruses have been proposed as potential triggers of SLE, with intriguing evidence implicating EBV (35). EBV nucleic acids have been shown to stimulate the TLR-independent system of IFN-α production (36). EBV also has been implicated in autoantibody formation in SLE, possibly via molecular mimicry (37). It is possible that viral infection could interact with gain-of-function genetic polymorphisms in cytosolic viral sensors such as IFIH1 to result in overactive IFN-α responses and risk of autoimmunity. Virus-like endogenous retroelements such as long interspersed nuclear element 1 also could provide a stimulus to the TLR-independent pathway, and these retroelements also have been implicated in SLE pathogenesis (38). Recent studies of the cytosolic RNA sensors suggest that cytosolic dsDNA may be transcribed to RNA by RNA polymerase III and that this RNA can be recognized by RIG-I (39). Circulating cell-free DNA is present in SLE patients, and levels are elevated as compared with those in healthy individuals (40). Free circulating dsDNA could be taken up into cells via endocytosis and subsequently stimulate the cytosolic RNA sensors after transcription of the DNA to RNA.
It is currently unclear why a risk allele in an RNA-sensing viral defense protein would be associated with Abs directed at dsDNA. It is possible that overactivity in the IFN-α pathway conferred in part by IFIH1 rs1990760 T could increase the chance that tolerance is broken toward DNA and that the polymorphism results in risk of anti-dsDNA Abs. In humans, there is evidence to support the idea that IFN-α could facilitate a break in tolerance to nuclear Ags in SLE (7, 41), although this concept is not uniformly supported across all of the autoimmune disease states (42). Alternately, it is also possible that anti-dsDNA Abs enhance the gain-of-function signaling tendency of IFIH1 rs1990760 T, resulting in pathogenic overactivity of the IFN-α pathway and subsequent risk of SLE. This could happen in an indirect way, because anti-dsDNA Ab immune complexes could stimulate TLR9, resulting in greater IFN-α production. This increase in IFN-α then could result in greater IFIH1 transcription that is enhanced by the rs1990760 T risk variant. In this way, the formation of anti-dsDNA Abs in SLE patients could exacerbate an underlying genetic tendency toward greater IFN-α signaling. The increased IFN-α sensitivity that we observe in the setting of the risk variant may be a result of this type of “priming,” presuming that increased expression of IFIH1 then results in increased downstream IFN-α-induced gene expression.
Our results indicate that the IFIH1 autoimmune disease risk allele modulates IFN-α-induced gene expression in SLE patients in vivo. Given the relevance of IFN-α pathway signaling in a number of different autoimmune diseases (31, 43, 44), we expect that these results will inform studies of this allele in other auto-immune disease populations. Additionally, the autoimmune diseases that have been associated with IFIH1 rs1990760 to date are characterized frequently by autoantibody formation, and we hypothesize that the IFIH1 risk variant may play a role in humoral tolerance in these conditions as well.
Acknowledgments
This work was supported by the Howard Hughes Medical Institute Gilliam Fellowship for Advanced Study (to S.N.K.), the Lupus Clinical Trials Consortium (to T.O.U.), National Institutes of Health Grants K08 AI083790 and P30 DK42086, National Institute of Allergy and Infectious Diseases Clinical Research Loan Repayment AI071651, National Institutes of Health Clinical and Translational Science Awards Core Subsidy Grant and Clinical and Translational Science Awards Pilot Grant UL1 RR024999, a Lupus Research Institute Novel Research grant, an Alliance for Lupus Research Target Identification in Lupus grant, and an Arthritis National Research Foundation Eng Tan Scholar award (to T.B.N.).
Abbreviations used in this article
- IFIH1
IFN induced with helicase C domain 1
- IFIT1
IFN-induced protein with tetratricopeptide repeats 1
- MAVS
mitochondrial antiviral signaling protein
- MX1
myxovirus resistance 1
- RIG-I
retinoic acid inducible gene I
- SLE
systemic lupus erythematosus.
Footnotes
Disclosures The authors have no financial conflicts of interest.
References
- 1.Harley JB, Kelly JA, Kaufman KM. Unraveling the genetics of systemic lupus erythematosus. Springer Semin. Immunopathol. 2006;28:119–130. doi: 10.1007/s00281-006-0040-5. [DOI] [PubMed] [Google Scholar]
- 2.Niewold TB, Clark DN, Salloum R, Poole BD. Interferon alpha in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010;2010:948364. doi: 10.1155/2010/948364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kariuki SN, Niewold TB. Genetic regulation of serum cytokines in systemic lupus erythematosus. Transl. Res. 2010;155:109–117. doi: 10.1016/j.trsl.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rönnblom LE, Alm GV, Oberg KE. Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J. Intern. Med. 1990;227:207–210. doi: 10.1111/j.1365-2796.1990.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 5.Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin. Rheumatol. 2005;24:178–181. doi: 10.1007/s10067-004-1024-2. [DOI] [PubMed] [Google Scholar]
- 6.Niewold TB. Interferon alpha-induced lupus: proof of principle. J. Clin. Rheumatol. 2008;14:131–132. doi: 10.1097/RHU.0b013e318177627d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weckerle CE, Franek BS, Kelly JA, Kumabe M, Mikolaitis RA, Green SL, Utset TO, Jolly M, James JA, Harley JB, Niewold TB. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 2011;63:1044–1053. doi: 10.1002/art.30187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8:492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niewold TB, Adler JE, Glenn SB, Lehman TJ, Harley JB, Crow MK. Age- and sex-related patterns of serum interferon-alpha activity in lupus families. Arthritis Rheum. 2008;58:2113–2119. doi: 10.1002/art.23619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kariuki SN, Moore JG, Kirou KA, Crow MK, Utset TO, Niewold TB. Age- and gender-specific modulation of serum osteopontin and interferon-alpha by osteopontin genotype in systemic lupus erythematosus. Genes Immun. 2009;10:487–494. doi: 10.1038/gene.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salloum R, Franek BS, Kariuki SN, Rhee L, Mikolaitis RA, Jolly M, Utset TO, Niewold TB. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-alpha activity in lupus patients. Arthritis Rheum. 2010;62:553–561. doi: 10.1002/art.27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum. 2008;58:2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, Niewold TB. Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-alpha in lupus patients in vivo. J. Immunol. 2009;182:34–38. doi: 10.4049/jimmunol.182.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harley ITW, Niewold TB, Stormont RM, Kaufman KM, Glenn SB, Franek BS, Kelly JA, Kilpatrick JR, Hutchings D, Divers J, et al. The role of genetic variation near interferon-kappa in systemic lupus erythematosus. J. Biomed. Biotechnol. 2010;2010:706825. doi: 10.1155/2010/706825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kariuki SN, Crow MK, Niewold TB. The PTPN22 C1858T polymorphism is associated with skewing of cytokine profiles toward high interferon-alpha activity and low tumor necrosis factor alpha levels in patients with lupus. Arthritis Rheum. 2008;58:2818–2823. doi: 10.1002/art.23728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilkins C, Gale M., Jr Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, Guja C, Ionescu-Tirgoviste C, Widmer B, Dunger DB, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat. Genet. 2006;38:617–619. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- 18.Surtherland A, Davies J, Owen CJ, Vaikkakara S, Walker C, Cheetham TD, James RA, Perros P, Donaldson PT, Cordell HJ, et al. Genomic polymorphism at the interferon-induced helicase (IFIH1) locus contributes to Graves' disease susceptibility. J. Clin. Endocrinol. Metab. 2007;92:3338–3341. doi: 10.1210/jc.2007-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, Barton A, Band G, Bellenguez C, Bergboer JG, et al. Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2. 2010. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, et al. International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN). 2008. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, Ortmann W, Kosoy R, Ferreira RC, Nordmark G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Downes K, Pekalski M, Angus KL, Hardy M, Nutland S, Smyth DJ, Walker NM, Wallace C, Todd JA. Reduced expression of IFIH1 is protective for type 1 diabetes. PLoS One. 2010;5:e12646. doi: 10.1371/journal.pone.0012646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zouk H, Marchand L, Polychronakos C. Study of transcriptional effects in Cis at the IFIH1 locus. PLoS One. 2010;5:e11564. doi: 10.1371/journal.pone.0011564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kariuki SN, Franek BS, Mikolaitis RA, Utset TO, Jolly M, Skol AD, Niewold TB. Promoter variant of PIK3C3 is associated with autoimmunity against Ro and Sm epitopes in African-American lupus patients. J. Biomed. Biotechnol. 2010;2010:826434. doi: 10.1155/2010/826434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kariuki SN, Franek BS, Kumar AA, Arrington J, Mikolaitis RA, Utset TO, Jolly M, Crow MK, Skol AD, Niewold TB. Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res. Ther. 2010;12:R151. doi: 10.1186/ar3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 28.Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum. 2006;54:1906–1916. doi: 10.1002/art.21890. [DOI] [PubMed] [Google Scholar]
- 29.Kirou KA, Lee C, George S, Louca K, Papagiannis IG, Peterson MG, Ly N, Woodward RN, Fry KE, Lau AY, et al. Coordinate over-expression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum. 2004;50:3958–3967. doi: 10.1002/art.20798. [DOI] [PubMed] [Google Scholar]
- 30.Niewold TB, Rivera TL, Buyon JP, Crow MK. Serum type I interferon activity is dependent on maternal diagnosis in anti-SSA/Ro-positive mothers of children with neonatal lupus. Arthritis Rheum. 2008;58:541–546. doi: 10.1002/art.23191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niewold TB, Kariuki SN, Morgan GA, Shrestha S, Pachman LM. Elevated serum interferon-alpha activity in juvenile dermatomyositis: associations with disease activity at diagnosis and after thirty-six months of therapy. Arthritis Rheum. 2009;60:1815–1824. doi: 10.1002/art.24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pothlichet J, Niewold TB, Vitour D, Solhonne B, Crow MK, Si-Tahar M. A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO Mol. Med. 2011;3:142–152. doi: 10.1002/emmm.201000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chistiakov DA, Voronova NV, Savost'Anov KV, Turakulov RI. Loss-of-function mutations E6 27× and I923V of IFIH1 are associated with lower poly(I:C)-induced interferon-β production in peripheral blood mono-nuclear cells of type 1 diabetes patients. Hum. Immunol. 2010;71:1128–1134. doi: 10.1016/j.humimm.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 35.James JA, Kaufman KM, Farris AD, Taylor-Albert E, Lehman TJ, Harley JB. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J. Clin. Invest. 1997;100:3019–3026. doi: 10.1172/JCI119856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samanta M, Iwakiri D, Kanda T, Imaizumi T, Takada K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 2006;25:4207–4214. doi: 10.1038/sj.emboj.7601314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poole BD, Scofield RH, Harley JB, James JA. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity. 2006;39:63–70. doi: 10.1080/08916930500484849. [DOI] [PubMed] [Google Scholar]
- 38.Crow MK. Long interspersed nuclear elements (LINE-1): potential triggers of systemic autoimmune disease. Autoimmunity. 2010;43:7–16. doi: 10.3109/08916930903374865. [DOI] [PubMed] [Google Scholar]
- 39.Bauernfeind F, Ablasser A, Kim S, Bartok E, Hornung V. An unexpected role for RNA in the recognition of DNA by the innate immune system. RNA Biol. 2010;7:151–157. doi: 10.4161/rna.7.2.11058. [DOI] [PubMed] [Google Scholar]
- 40.Atamaniuk J, Hsiao YY, Mustak M, Bernhard D, Erlacher L, Fodinger M, Tiran B, Stuhlmeier KM. Analysing cell-free plasma DNA and SLE disease activity. Eur. J. Clin. Invest. 2011;41:579–583. doi: 10.1111/j.1365-2362.2010.02435.x. [DOI] [PubMed] [Google Scholar]
- 41.Ioannou Y, Isenberg DA. Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis Rheum. 2000;43:1431–1442. doi: 10.1002/1529-0131(200007)43:7<1431::AID-ANR3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 42.Cantaert T, De Rycke L, Mavragani CP, Wijbrandts CA, Niewold TB, Niers T, Vandooren B, Veys EM, Richel D, Tak PP. Exposure to nuclear antigens contributes to the induction of humoral autoimmunity during tumour necrosis factor alpha blockade. Ann. Rheum. Dis. 2009;68:1022–1029. doi: 10.1136/ard.2008.093724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Båve U, Nordmark G, Lövgren T, Rönnelid J, Cajander S, Eloranta ML, Alm GV, Rönnblom L. Activation of the type I interferon system in primary Sjögren's syndrome: a possible etiopathogenic mechanism. Arthritis Rheum. 2005;52:1185–1195. doi: 10.1002/art.20998. [DOI] [PubMed] [Google Scholar]
- 44.Devendra D, Eisenbarth GS. Interferon alpha—a potential link in the pathogenesis of viral-induced type 1 diabetes and autoimmunity. Clin. Immunol. 2004;111:225–233. doi: 10.1016/j.clim.2004.01.008. [DOI] [PubMed] [Google Scholar]