

Abstract
(−)-Epigallocatechin gallate [(−)-EGCG] has been implicated in cancer chemoprevention and has been shown as an inhibitor of tumor proteasomal chymotrypsin-like activity in vitro and in vivo. However, EGCG is subjected to rapid biotransforming modifications such as methylation by catechol-O-methyltransferase (COMT) that limits its action. We recently reported that structure 7, an EGCG analog which should be resistant to COMT-mediated methylation and inactivation in cells, was able to inhibit the activity of purified 20S proteasome and cellular 26S proteasome. However, the involved molecular mechanism is unknown. Herein, we applied computational solution to understand the possible interaction between EGCG analogs including structure 7 and the proteasome β5 subunit which is responsible for the chymotrypsin-like activity. We report that the ester carbonyls at C2 and C3 carbon atoms may be the active sites for nucleophilic attack in structure 7 and 5. Equally spaced carbon atoms in COMT-resistant structure 7 give more stable conformation and lower docked free energy than other EGCG analogs. The absence of a second gallate group in structure 16 and 21 significantly decreases the ability to inhibit the proteasome.
Keywords: catechol-O-methyltransferase, methylation, proteasome inhibitor, (−)-epigallocatechin gallate, computational docking, cancer
Introduction
To facilitate protein binding, a ligand must exhibit correct shape and interaction properties complementary to the residues exposed towards the binding pocket of a target protein (1). Performing molecular docking of these ligands, which interact with active site of large macromolecules, has become a valuable strategy for predicting the preferred orientation of a ligand to a macromolecule to form a stable complex (2–6). Herein, we use the computational modeling application to analyze the interaction between synthetic analogs of naturally occurring green tea polyphenols and the 20S proteasome.
Proteasomes are large protein complexes whose main function is to degrade unneeded or damaged proteins by proteolysis, a chemical reaction that breaks peptide bonds. The cellular 26S proteasome contains one 20S core particle structure and two 19S regulatory caps that dock onto ends of the 20S proteasome (7). Although the three catalytic β subunits have a common mechanism, they have different substrate specificities, which are considered chymotrypsin-like (CT), trypsin-like (T), and peptidyl-glutamyl peptide-hydrolyzing (PHGH)-like (8). These variations in specificity are the result of interatomic contacts with local residues near the active site of each subunit. The mechanism of proteolysis by the β subunits of the 20S core particle is through a threonine-dependent nucleophilic attack. The S1 pocket of proteasomal β5 subunit is defined by the hydrophobic residues, Ala 20, Ala 49, Val 31, Ile 35, Met 45, Gln 53, which plays a significant role for binding of several types of inhibitors (8).
Polyphenols constitute one of the most ubiquitous natural compounds which are most commonly found in normal dietary intake (9). Polyphenols are present in tea, fruits and vegetables and have been reported to possess anti-cancer (10,11), anti-inflammatory (12,13) antiulcer, antidiarrheal and anti-oxidant (14) activities.
Many epidemiological studies have suggested that tea consumption confers cancer-preventive effects (15–18). Green tea is an aqueous infusion obtained from the dried green tea leaves Camellia sinensis. The major components of green tea include epicatechin (EC), epigallocatechin (EGC), epicatechin gallate (ECG), and epigallocatechin-3-gallate (EGCG). Other components include three other flavonoids, known as kaempferol, quercetin, and myricetin (15). Moreover, studies indicate that EGCG is the most abundant and biological active catechin with respect to anticancer activity in several human cancers. The effect of EGCG has been tested on various diseases including the topical treatment of genital warts (19).
We reported that the carbonyl carbon of green tea poly-phenols, EGCG, is the site of nucleophilic attack in the inhibition of the proteasome (20). Most recently we also reported that structure 7 (Fig. 1), an EGCG analog which should be resistant to COMT-mediated methylation and inactivation in cells, was able to inhibit the activity of purified 20S proteasome and cellular 26S proteasome (21). However, the involved molecular mechanism is unknown. Herein, we applied computational solution to understand the possible interaction between EGCG analogs including structure 7 and the proteasome β5 subunit which is responsible for the chymotrypsin-like activity. We performed silico modeling experiments using these EGCG analogs and found that carbonyl carbons are indeed highly susceptible for nucleophilic attack and may be involved in inhibiting the proteasomal chymotrypsin-like activity presumably by formation of a covalent bond at the N-terminal threonine. Importantly, equally spaced carbon atoms in COMT-resistant structure 7 gives it a more stable conformation and lower docked free energy than other EGCG analogs.
Figure 1.

Structure of four EGCG analogs.
Materials and methods
Molecule building and nucleophilic susceptibility analysis
CAChe workstation (Fujitsu, Inc.) was used for the construction of chemical structures. After being constructed, the molecules were then subjected to geometry optimization using PM5 geometry in water as the parameter. Nucleophilic susceptibility analysis was determined by using PM5 geometry and PM5 wave function in water and is saved as PDB format using CAChe conversion filters. Surface analysis was also performed. A colored ‘bull's-eye’ with a white center denotes atoms that are highly susceptible to nucleophilic attack. The PDB files generated in CAChe were imported to AutoDock for molecular docking (20).
Molecular docking of synthetic EGCG analogs to proteasome β5 subunit
In silico docking was performed on a Linux Red Hat 9.0 based platform using AutoDock 3.0. The AutoDock suite of programs, which was used for the docking calculation, employs an automated docking approach, allowing ligand flexibility as described to a full extent elsewhere (22). AutoDock has been compared with various other docking programs and has been found to be able to locate docking modes that are consistent with X-ray crystal structure (23–26).
The Eukaryotic yeast 20S proteasome used in this study were selected from the protein databank (27) (ref. no. 1JD2) and used for all docking studies presented herein. The yeast 20S proteasome is structurally similar to mammalian 20S proteasome, and the chymotrypsin active site between the two species is highly conserved (28,29). The default parameters were used to prepare 20S proteasome and the ligand, except where noted. The energy scoring grid was prepared as a 40x40x40 Å box centered on the β5 catalytic Thr1, and the ligand was limited in search space during docking. Atomic solvation parameters were assigned to the proteasome using default parameters. Among the different search algorithm offered by AutoDock 3.0., we chose Lamarckian genetic algorithm which search both globally and locally. Genetic algorithms allow the exploration of a large conformational space (which is basically spanned by the protein and ligand jointly), by representing each spatial arrangement of the protein and ligand with a particular energy. The number of the genetic algorithm runs, which was set to 100, and the maximum number of energy evaluations, which was set to 5 million (the population size was retained at 50). Genetic algorithm was quite suitable for solving docking problems because of their usefulness in solving complex optimization problem.
For mutation, crossover and elitism the default parameters were used. Even for local search method, the pseudo-Solis and wets was included by using the default parameters. AutoDock is designed to predict how small molecules, such as substrates or drug candidates, bind to a receptor of known 3D structure. AutoDock reports the docked energy that we have referred as final docked free energy. It includes the ligand internal energy, or the intermolecular interaction energy of the ligand.
The emphasis was given to the docked free energies since the number of rotatable bonds in our inhibitor is relatively constant and we also believe that the internal energy of ligand should not be neglected. In Table I, we indicate the number of multiple clusters, cluster rank of the docking mode selected, their intermolecular energy, internal energy of ligand along with the torsional energies and the docked free energies of the docked mode selected. Dockings were chosen by fulfilling two criteria we used for resolving the docking of (−)-EGCG and related compounds to the β5 subunit. Briefly, the carbonyl carbon of the ester function attached to C-ring should lie within 4 Å of the N-terminal threonine (a distance suitable for nucleophilic attack). The probability of adopting the inhibitory conformation was the number of genetic runs (out of 100) in which the molecule docked into the active site and fulfilled the above criteria.
Table I.
Docking of EGCG analogs to the proteasomal β5 subunit.
| Compounds | No. of multiple-conformation clustersa | No. of conformationsb | Final energy intermolecular (kcal/mol) | Final internal energy (kcal/mol) | Torsional free energy (kcal/mol) | Free energy of binding (kcal/mol) | Ki | Final docked energy (kcal/mol)c | Distance between carbonyl, carbon and N terminal Thr |
|---|---|---|---|---|---|---|---|---|---|
| Structure 5 | 23 (7) | 3 | −11.12 | −0.99 | +1.87 | −9.25 | +1.66e-07 | −12.11 | 2.99, 3.08 |
| Structure 16 | 6 (5) | 13 | −9.58 | +1.51 | +0.93 | −8.65 | +4.59e-07 | −8.07 | 4.01 |
| Structure 7 | 20 (1) | 10 | −11.54 | −0.76 | +1.87 | −9.67 | +8.19e-08 | −12.29 | 3.09, 2.98 |
| Structure 21 | 7 (9) | 5 | −8.23 | +2.56 | +0.93 | −7.30 | +4.48e-06 | −5.68 | 3.46 |
Dock structure selected based on effective nucleophilic attack distance between the carbonyl carbon and N terminal Thr.
The various numbers of conformations present in each of the selected docked structures.
Final docking energy = Final intermolecular energy of the ligand + Final internal energy of the ligand.
The resultant structure files were visualized and analyzed by using PyMOLv0.99 (30) (DeLano Scientific LLC, San Carlos, CA) visualization program.
Results
We recently reported that structure 7 (Fig. 1), which should be resistant to COMT-mediated methylation and inactivation in cells, was able to inhibit the activity of purified 20S proteasome and cellular 26S proteasome (21). However, the involved molecular mechanism is unknown. To build upon our understanding of how structure 7 inhibits the proteasome, we did automated docking to explore the computational interaction between this EGCG analog and β5 subunit of proteasome. We also examined 3 other structures: 5, 16 and 21. Each of the four analogs (Fig. 1) was first examined for sites of nucleophilic susceptibility. Analysis revealed that structure 5 and 7 possessed two sites with similar susceptibility whereas structure 16 and 21 possessed a single site (Fig. 2), suggesting that these sites could be attacked, and subsequently covalently bound, by the OH group of N-Thr of proteaosmal β5 subunit (22). For better understanding of the possible chemical nature of these four analogs to inhibit the chymotrypsin-like activity of the proteasome, each was docked to the active site of the proteasome β5-subunit, which is responsible for the chymo-trypsin-like activity (22).
Figure 2.
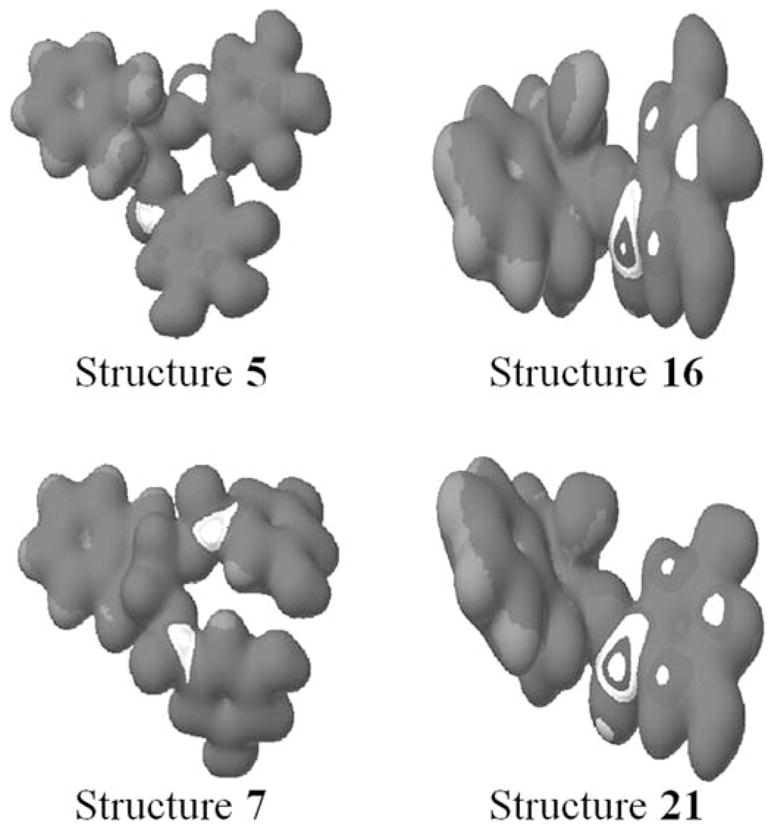
Structures 5, 7, 16 and 21 were drawn in Quantam CAChe software. After that nucleophilic susceptibility analysis was performed using PM5 Geometry and wave function in water. The bull's eye indicated the nucleophilic susceptibilty region in the structures. Absence of one gallate group decreases the nucleophilic susceptibilty in structures 16 and 21, as there is only one susceptibile region as compared to two in structures 5 and 7.
After docking these EGCG analogs to the β5 chymotrypsin active site using 50 genetic algorithms runs, AutoDock reports the best docking outputs (lowest docked free energy) for each GA run and also performs a cluster analysis in which the total number of clusters represents the reliability of docking results based on similarity of final docked conformation. The final docked energy was the sum of the internal energy of the ligand and the intermolecular energy. We analyzed different multiple clusters out of which we chose a specific cluster rank. The criteria of choosing these ranks were based upon i) the distance from the carbonyl carbon to N terminal Thr must be within 4 Å; ii) the calculated docked free energy values that are favorable for binding of (−)-EGCG analogs to the proteasome. The lower the docking energy is and the larger the cluster is, the greater the possibility is predicted (22).
Docking of EGCG analog structure 7
In structure 7, we analyzed 20 different multiple conformation clusters along with single clusters. Preference was given to multiple conformation clusters, from which we chose cluster rank 1 having 10 conformations. This was found consistent with our criteria of selection. As obtained from the AutoDock report, the final docked energy was found to be minimum in our selected cluster (−12.29 kcal/mol, Table I). This places the ester bonds of structure 7 oriented directly over the N terminal Thr at a distance of 3.09 and 2.88 Å from the oxygen atom of Thr OH, respectively (Fig. 3). This orientation and conformation of structure 7 is well suited for nucleophilic attack. The distance of the hydrophobic portion of aromatic ring to the hydrophobic residues Val 31, Ala 49, Ala 20, Met 45, Gln 53, Ile 35 in the S1 pocket of β5-subunit were within favorable distances.
Figure 3.

Binding modes of compounds into the β5-subunit of proteasome. Docking was performed by using AutoDock 3.0 software. The resultant files were analyzed and visualized by using PyMol v. 0.99. (A) The carbonyl carbons of structure 5 were at distance of 3.08 and 2.99 Å having energy of −12.11 kcal/mol, that is suitable for nucleophillic attack. (B) In structure 16 the distance was 4.01 Å in the cluster having 13 conformations. The lowest docked energy was found to be −8.07 kcal/mol. (C) The distance of the two carbonyl carbons of structure 7 and hydroxyl of N-terminal Thr was 3.09 and 2.88 Å in cluster having ten conformations with docking energy −12.29 kcal/mol. (D) The distance was 3.46 Å in structure 21 having docked energy of −5.68 kcal/mol in the cluster having 5 conformation.
In structure 7, B- and G-rings are symmetrical to each other having the two hydroxyl groups in meta-substitution at 3′ and 5′ carbon atoms. The presence of two OH groups on each ring gives more stable conformation and there exists a good probability that these two OH moieties or groups in any of these rings may interact with the amino acids in the active site to form hydrogen bonds. These interactions further help in getting the carbonyl atoms very close to the N-terminal of active site of the proteasome. Since identical B- or G-rings having equally spaced OH atoms and carbonyl groups, structure 7 has higher probability of adopting a more stable conformation. To confirm the favorable binding mode of structure 7 to the proteasomal chymotrypsin active site, we analyzed hydrogen bond (H-bond) formation. There are four polar hydrogen atoms and two carbonyl oxygen atoms on structure 7 that are available for H-bonding in which only 2 are actively participating in H-bond formation (to Lys 33 4.19 Å and to Ser 96 2.72 Å).
Predicted values obtained from the AutoDock were further compared with the observed experimental free energies values. From experimental data we found that structure 7 inhibited the proteasomal chymotrypsin-like activity with an IC50 value of 29 μM (21).
Docking of other EGCG analogs
Like structure 7, structure 5 also consists of A, C, B and G rings (Fig. 1), suggesting that it may act as a more potent inhibitor of the proteasome. The only difference between the two structures is the presence of three (instead of two) OH groups on both B and G rings in structure 5. In our docking studies, structure 5 was oriented in the similar mode as structure 7; we analyzed 29 single conformation clusters and 20 multiple conformation clusters for structure 5 (Table I). Out of all the multiple clusters, the carbonyl carbons of cluster rank 7 were at the closest proximity of 2.99 and 3.08 Å to hydroxyl oxygen atom of N terminal Thr (Table I), favoring an effective nucleophilic attack of the proteasome (Fig. 3).
Both structure 5 and 7 had almost a similar number of multiple conformation clusters and equal distances from N terminal threonine, but the docking energy for structure 5 was a little higher than in structure 7 (−12.11 kcal/mol vs. −12.29 kcal/mol; Table I). Though the orientation and conformation of both the structures were quite similar, the presence of three OH groups on B and G rings in structure 5 may render intramolecular steric hindrances, thus conferring a slightly higher free energy of docking in comparison with structure 7. There were six polar hydrogen atoms and two carbonyl oxygen atoms in structure 5. Although it is possible for all of them in structure 5 to form hydrogen bonds with β5 subunit of proteasome, only two have high potential to do so (Lys 33 4.11 Å and Ser 96 2.77 Å).
Structure 16 lacks one gallate group which is replaced with a hydroxyl group. This significantly decreases its potency against the 20S proteasome (Fig. 1). This was further substantiated when we investigated for final docked free energy of structure 16. In this case, we analyzed all nine (single and multiple) clusters and we found that there was only one cluster which meets the criteria. Additionally, the vast majority of clusters were not able to place carbonyl carbon within the 4 Å of the N-terminal threonine, a distance suitable for nucleophilic attack. AutoDock reports the final docked energy to be minimum in our selected cluster (−8.07 kcal/mol) (Table I) which meets the preset criteria. This places the ester carbon at an appropriate distance of 4.01 Å (Fig. 3).
Structure 16 allows the OH groups on its gallate ring to form two potential hydrogen bonds while the OH group on the C ring, farther out of the S1 pocket, has one OH group that might form one hydrogen bond. However, although this conformation might be good for hydrogen bonding, it places the G and A–C rings, out of the S1 pocket of the proteasome. The distance of hydrophobic portion of structure 16 to the hydrophobic amino acid residues on the S1 pocket was more than 4 Å, supporting the hypothesis that there was no hydrophobic interaction between structure 16 and the S1 pocket. Consistent with its high docked free energy, it inhibits the proteasome to a lesser extent in comparison with structures 7 and 5.
Structure 21 is similar to structure 16 as both lacks one gallate moiety, and this observation is highlighted by showing a dramatic change in docking results. Along with absence of one gallate group, structure 21 also lacks one hydroxyl group on its ring G (Fig. 1), suggesting that it might be a poor proteasome inhibitor. Loss of one gallate moiety and one hydroxyl group significantly decreases binding of this structure to the active site of proteasome. The distance of hydrophobic amino acid residues on the S1 pocket to the hydrophobic portion on structure 21 was found to be <4 Å and docked energy was found to be higher as compared to other structures (Table I). The carbonyl carbon is ~3.46 Å away from the hydroxyl of Thr (Fig. 3). The extent of inhibitory potencies was also evaluated on the basis of free energy of binding that equals to the sum of intermolecular energy and torsional free energy. Contrary to torsional free energy, intermolecular energy (−8.23 kcal/mol for structure 21 vs. −9.58 kcal/mol for structure 16; Table I) and ligand internal energy (+2.56 kcal/mol for structure 21 vs. +1.51 kcal/mol for structure 16) was found to be increased in structure 21. As a final effect free energy of binding was also increased from −8.65 kcal/mol in structure 16 to −7.30 kcal/mol in structure 21, thus giving a net increase in docked energy (−5.68 kcal/mol). Consistent with this finding, structure 21 was a very poor inhibitor of the proteasome in comparison with the other structures.
Discussion
Recently, proteasome inhibition has been developed as a viable favorable therapeutics strategy in the treatment of cancer. It has been shown that proteasome inhibition is associated with induction of apoptosis in tumor, but not in normal cells (31). Previously, we reported that green tea polyphenols with an ester bond, such as (−)-EGCG, possess proteasome-inhibitory properties (20). It is suggested that the lower the docking energy is and the larger the cluster is, the greater the inhibitory potency is predicted (22). The current docking data (Fig. 3) are consistent with the order of the potencies of these four analogs to inhibit the chymotrypsin-like activity of purified 20S proteasome. Since the analogs presented herein are structurally similar to (−)-EGCG and (−)-ECG, we hypothesized that two of these compounds may act as proteasome inhibitors which may contribute to their cancer-preventative properties (32).
In our previous studies on EGCG, we reported that EGCG is susceptible to nucleophilic attack and bind to hydroxyl group of N-terminal threonine of β5 subunit thus resulting in inhibition of the proteasomal activity (22). By analyzing the electron density (Fig. 2) we determined that the compounds possess a site susceptible for nucleophilic attack at the C-2 and C-3 positions in structure 5 and 7 and the C-3 position in structure 16 and 21 while C-2 OH may alter the ability of these structures to inhibit the proteasome (Fig. 1). Next, we docked the four analogs to the β5 subunit of the proteasome and found that these compounds are suitable for nucleophilic attack. The order of the docking energy is therefore: structure 7 < structure 5 < structure 16 < structure 21.
One of the key differences between structures 5 and 7 vs. structures 16 and 21 is the absence of a gallate group at the C2 position which is replaced by hydroxyl group, suggesting that removal of the gallate group decreases the probability of favorable binding to the active site of β5-subunit. This hypothesis is further supported by our previous study suggesting that absence of gallate group may cause decreased inhibition to the chymotrypsin-like activity of 20S proteasome (22). When compared with the experimental data, we found that structures 5 and 7 inhibited the proteasomal chymotrypsin-like activity with IC50 values of 19 and 29 μM, respectively (21). Although structure 7 was less potent to inhibit purified 20S proteasome, our previous results showed that it was more potent to inhibit cellular proteasome in cell lysate that contains a COMT activity compared with structure 5. In addition, a specific inhibitor of COMT could increase the action of structure 5, but not structure 7, further indicating that structure 7 was relatively resistant to COMT modification. These results suggest that structure 7 could overcome the disadvantage of EGCG which is easily subject to methylation by COMT (21).
By computational molecular modeling we estimated the internal energy of ligand, docked intermolecular energy and the torsional energy (Table I). Among them the intermolecular energy plays a significant role for the final free energy of binding and docked free energy. Herein, we found that the major intermolecular interactions may be due to the hydrophobic interaction and hydrogen bonding. Highly susceptible structures have more hydrophobic interactions when compared to other structures, with the hydrophobic amino acid residues in the β5-subunit of proteasome.
Collectively, consistent with prediction from the docking results, our data demonstrates that i) absence of one gallate group dramatically decreases the inhibitory potency of the ligand, ii) the change from 3,4,5-trihydroxy to 3,5-dihyroxy in the B and G gallate rings has only a small effect in the final docked energy, consistent with the small difference in the inhibition of purified proteasome. On the other hand, other processes such as metabolic methylation by COMT could substantially modify the proteasome-inhibitory ability of EGCG and its analogs. The current finding that structure 7 has more stable conformation and lower docked free energy than other analogs suggests that this COMT-resistant EGCG analog has a great potential to be developed into a novel anti-cancer agent for cancer prevention and therapy.
Acknowledgments
Funding support from the National Cancer Institute (Grant number: 1R01CA120009 and 3R01CA120009-04S1) and the Natural Sciences and Engineering Research Council of Canada (NSERC) is gratefully acknowledged.
References
- 1.Burgoyne NJ, Jackson RM. Predicting protein function from surface properties. In: Rigden DJ, editor. From Protein Structure to Function with Bioinformatics. Springer; Dordrecht, Netherlands: 2009. pp. 167–186. [Google Scholar]
- 2.Fernandez-Recio J, Totrov M, Skorodumov C, Abagyan R. Optimal docking area: a new method for predicting protein-protein interaction sites. Proteins. 2005;58:134–143. doi: 10.1002/prot.20285. [DOI] [PubMed] [Google Scholar]
- 3.Lensink MF, Mendez R, Wodak SJ. Docking and scoring protein complexes: CAPRI 3rd edition. Proteins. 2007;69:704–718. doi: 10.1002/prot.21804. [DOI] [PubMed] [Google Scholar]
- 4.Ruvinsky AM, Vakseer IA. Interaction cutoff effects on ruggedness of protein-protein energy landscape. Proteins. 2008;70:1498–1505. doi: 10.1002/prot.21644. [DOI] [PubMed] [Google Scholar]
- 5.Liu Z, Guo JT, Li T, Xu Y. Structure-based predicted of transcription factor binding sites using a protein-DNA docking approach. Proteins. 2008;72:1114–1124. doi: 10.1002/prot.22002. [DOI] [PubMed] [Google Scholar]
- 6.Sternberg MJ, Gabb HA, Jackson RM. Predictive docking of protein-protein and protein-DNA complexes. Curr Opin Struct Biol. 1998;8:250–256. doi: 10.1016/s0959-440x(98)80047-x. [DOI] [PubMed] [Google Scholar]
- 7.Goldberg AL. Functions of the proteasome: The lysis at the end of the tunnel. Science. 1995;268:522–523. doi: 10.1126/science.7725095. [DOI] [PubMed] [Google Scholar]
- 8.Seemuller E, Lupas A, Stock D, Lowe J, Huber R, Baumeister W. Proteasome from thermoplasma acidophilum: a threonine protease. Science. 1995;268:579–582. doi: 10.1126/science.7725107. [DOI] [PubMed] [Google Scholar]
- 9.Bravo L. Polyphenols: chemistry, dietary sources, metabolism and nutritional significance. Nutr Rev. 1998;56:317–333. doi: 10.1111/j.1753-4887.1998.tb01670.x. [DOI] [PubMed] [Google Scholar]
- 10.Miller AB. Diet and cancer. A review. Acta Oncol. 1990;29:87–95. doi: 10.3109/02841869009089996. [DOI] [PubMed] [Google Scholar]
- 11.Davis JN, Kucuk O, Sarkar FH. Geinstein inhibits NF-kappa B activation in prostate cancer cells. Nutr Cancer. 1999;35:167–174. doi: 10.1207/S15327914NC352_11. [DOI] [PubMed] [Google Scholar]
- 12.Sartor L, Pezzato E, Dell'Aica I, Caniato R, Biggin S, Garbisa S. Inhibition of matrix-proteases by phenols: chemical insights for anti-inflammatory and anti-invasion drug design. Biochem Pharmacol. 2002;64:229–237. doi: 10.1016/s0006-2952(02)01069-9. [DOI] [PubMed] [Google Scholar]
- 13.Tipoe GL, Leung TM, Hung MW, Fung ML. Green tea polyphenols as an anti-oxidant and anti-inflammatory agent for cardiovascular protection. Cardiovasc Hematol Disord Drug Targets. 2007;7:135–144. doi: 10.2174/187152907780830905. [DOI] [PubMed] [Google Scholar]
- 14.Paganga G, Miller N, Rice-Evans CA. The polyphenolic content of fruit and vegetables and their antioxidant activites. What does a serving constitute. Free Radic Res. 1999;30:153–162. doi: 10.1080/10715769900300161. [DOI] [PubMed] [Google Scholar]
- 15.Fujiki H. Cancer prevention with green tea polyphenols for the general population, and for patients following cancer treatment. J Cancer Res Clin Oncol. 1999;125:589–597. doi: 10.1007/s004320050321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuroda Y, Hara Y. Antimutagenic and anticarcinogenic activity of tea polyphenols. Mutat Res. 1999;436:69–97. doi: 10.1016/s1383-5742(98)00019-2. [DOI] [PubMed] [Google Scholar]
- 17.Yang CS. Effects of tea consumption on nutrition and health. Nutrition. 1999;15:946–949. doi: 10.1093/jn/130.10.2409. [DOI] [PubMed] [Google Scholar]
- 18.Ahmad N, Mukhtar H. Green tea polyphenols and cancer: biological mechanisms and practical implications. Nutr Rev. 1999;57:78–83. doi: 10.1111/j.1753-4887.1999.tb06927.x. [DOI] [PubMed] [Google Scholar]
- 19.Stockfleth E, Beti H, Orasan R, Grigorian F, Mescheder A, Tawfik H, Thielert C. Topical Polyphenon E in the treatment of external genital and perianal warts: a randomized controlled trial. Br J Dermatol. 2008;158:1329–1338. doi: 10.1111/j.1365-2133.2008.08520.x. [DOI] [PubMed] [Google Scholar]
- 20.Nam S, Smith DM, Dou QP. Ester bond-conatining tea poly-phenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem. 2001;276:13322–13330. doi: 10.1074/jbc.M004209200. [DOI] [PubMed] [Google Scholar]
- 21.Huo C, Yang H, Cui QC, Dou QP, Chan TH. Proteasome inhibition in human breast cancer cells with high catechol-O-methyltransferase activity by green tea polyphenol EGCG analogs. Bioorg Med Chem. 2010;18:1252–1258. doi: 10.1016/j.bmc.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith DM, Daniel KG, Wang Z, Guida WC, Chan TH, Dou QP. Docking studies and model development of tea polyphenol proteasome inhibitors: Applications to rational drug design. Proteins. 2004;1:54–58. doi: 10.1002/prot.10504. [DOI] [PubMed] [Google Scholar]
- 23.Chen D, Daniel KG, Chen SM, Khun DJ, Landis-Piwowar KR, Dou QP. Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem Pharmacol. 2005;69:1421–1432. doi: 10.1016/j.bcp.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 24.Morris GM, Goodsell DS, Halliday RS, RH, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 25.Dym O, Xenarios I, Ke H, Colicelli J. Molecular docking of competitive phosphodiesterase inhibitors. Mol Pharmacol. 2002;61:20–25. doi: 10.1124/mol.61.1.20. [DOI] [PubMed] [Google Scholar]
- 26.Rao MS, Olson AJ. Modelling factor Xa-inhibitor complexes: a computational flexible docking approach. Proteins. 1999;34:173–183. doi: 10.1002/(sici)1097-0134(19990201)34:2<173::aid-prot3>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 27.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 Å resolution. Nature. 1997;386:463–471. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- 29.Kisselev AF, Goldberg AL. Proteasome inhibitors: from research tools to drug candidates. Chem Biol. 2001;8:739–758. doi: 10.1016/s1074-5521(01)00056-4. [DOI] [PubMed] [Google Scholar]
- 30.Delano WL. The PyMOL Molecular Graphics System. Delano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 31.Dou QP, Li B. Proteasome inhibitors as potential novel anticancer agents. Drug Resist Updat. 1999;4:215–223. doi: 10.1054/drup.1999.0095. [DOI] [PubMed] [Google Scholar]
- 32.Chen MS, Chen D, Dou QP. Inhibition of proteasome activity by various fruits and vegetables is associated with cancer cell death. In vivo. 2004;1:73–80. [PubMed] [Google Scholar]