

Abstract
Therapeutic choices are limited for undifferentiated metastatic thyroid carcinomas. Although implanted subcutaneous thyroid tumors are standard preclinical models to examine the efficacy of new therapeutic agents, these xenograft models frequently fail to predict the outcomes of clinical trials in patients with metastatic thyroid carcinomas. Genetically engineered mouse models with alterations similar to human cancers in their pathological progression and in an immunocompetent environment offer unparalleled opportunities for evaluating novel potential molecular targets. We review recent advances in the modeling of follicular thyroid carcinoma with distant metastasis and in the use of these mouse models in preclinical studies, emphasizing the significance of genetically engineered mouse models in clinical applications.
Keywords: Thyroid cancer, thyroid hormone receptors, preclinical studies, mouse models, thyroid hormone receptor mutations
Introduction
Thyroid cancer arisen from the follicular epithelium consists mainly of differentiated thyroid carcinoma (DTC) and anaplastic thyroid cancer (ATC). Papillary thyroid carcinoma is the most frequent subtype of DTC (~80%); follicular thyroid carcinoma (FTC) is less common (~15%). Most patients with DTC have an excellent prognosis after standard treatment including surgery, adjuvant radioiodine, and L-thyroxine suppression therapy. But recurrent disease occurs in 10-15% patients with treatment, half of these patients became nonresponsive to radioiodine therapy and tumor cells display a poorly differentiated phenotype. Currently, no effective therapy is available for this subgroup of patients, and so their survival rate is poor [1-3]. Although distant metastases are rare at the time of diagnosis of DTC, follicular thyroid carcinoma metastasizes via the vascular system to distant organs and often has a poor prognosis with a high recurrence rate [4, 5]. Though thyroid cancer patients have better 10-year survival rates than patients with other cancers owing to effective surgery and radiation therapy against localized differentiated carcinomas, the choices of successful treatments are limited for dedifferentiated, invasive, or metastatic thyroid cancers [6]. For these advanced carcinomas, new therapeutics and treatment modalities are needed.
Small molecule inhibitors against the critical mediators in the proliferation signaling pathways such as RAS and extracellular signal-regulated kinases (ERK) have been extensively explored. These inhibitors are usually evaluated in ectopic implanted thyroid tumors in mice (i.e., xenograft models) before being tested in clinical trials. Testing therapeutic targets using xenograft models is relatively easy and usually not overly time-consuming. Accumulating evidence, however, suggests that results with these xenograft models often are not concordant with the outcome of clinical trials in patients with advanced metastatic tumors.
Genetically engineered mouse models are created through genetic manipulation to reflect changes in human tumorigenic processes. Although creating a mouse model is labor-intensive, time-consuming, and costly, it offers the advantages of providing an immunocompetent host and a bona fide microenvironment with stroma and vasculatures in which tumors can develop. Consequently, genetically engineered models are being used more and more to test the efficacy of experimental drugs. An excellent correlation between the efficacy of drugs used in mouse models and in clinical trials was shown retrospectively for progression-free survival and overall survival of patients with non-small cell lung cancer and pancreatic ductal adenocarcinoma [7].
Genetically engineered mouse models, however, have not been commonly used as clinically predictive tools to evaluate the efficacy of therapeutics to treat metastatic diseases. One reason is that most mouse models generally show a low incidence of distant metastasis or they require a long latency for metastasis to occur, thus making it difficult to monitor the cancer progression in vivo. Despite such daunting issues, promising advances are being made with the use of mouse models of FTC.
Mouse models derived from alterations in thyroid hormone receptor genes
Thyroid hormone receptors (TRs) are members of the nuclear receptor superfamily that are encoded by the THRA and THRB genes. Alternative splicing of the primary transcripts generates several thyroid hormone (T3) binding TR isoforms, including TRα1 and TRβ1. Mutations of the THRB gene are associated with thyroid, pituitary, liver, and kidney cancers [8-12]. Reduced expression of THRB mRNA or promoter hypermethylation of the THRB gene has also been implicated in the carcinogenesis of human papillary thyroid carcinoma, kidney cancer, and breast cancer [12-17]. Moreover, retroviral v-erbA is a highly mutated chicken TRα1 with no T3 binding activity, and has lost the ability to activate gene transcription. It interferes with the transcriptional activity of liganded TRs [18, 19] and induces acute erythroleukemia and sarcomas in birds [20-22]. Both TRα1 and TRβ1 strongly repress Hrasval12-induced transformation of NIH3T3 fibroblasts and reduce tumor growth of hepatocarcinoma and breast cancer cells [23-25]. To understand the role of TRβ mutations in cancer, mouse models harboring mutated thyroid hormone β receptor have been used to understand the molecular basis of FTC.
The ThrbPV/PV mouse
The ThrbPV/PV knockin mouse, created by targeting the PV mutation to the Thrb gene locus, was initially created to study an inheritable disease with reduced tissue sensitivity to thyroid hormone known as resistance to thyroid hormone (RTH) [26]. The PV mutation was identified in an RTH patient with a frameshift mutation in the C-terminal 14 amino acids of TRβ, resulting in a complete loss of T3 binding and transcriptional capacity [27]. This ThrbPV/PV mouse faithfully recapitulates human RTH with the dysregulation of the hypothalamus-pituitary-thyroid axis, leading to elevated serum thyroid hormone accompanied by nonsuppressible high serum thyroid-stimulating hormone (TSH) [26]. As ThrbPV/PV mice age, they spontaneously develop FTC resembling the pathological progression of human thyroid cancer. Pathological changes progress from hyperplasia, capsular invasion, vascular invasion, and anaplasia to eventual distal metastasis (examples shown in Figure 1A, panels a, b, and c). Metastasis occurs mainly in the lung and occasionally in the endocardium, but not in the local lymph nodes [28]. The findings that ThrbPV/PV mice spontaneously develop FTC similar to human cancer indicate that these mice could be used as a model to elucidate the molecular genetic changes underlying FTC and to identify potential molecular targets for treatment and diagnosis. Consistent with human thyroid cancer, aberrant activation of cyclin D1, β-catenin, the phosphatidylinositol 3-kinase (PI3K), protein kinase B (AKT), pituitary tumor transforming gene, and Src-focal adhesion kinase were shown to promote thyroid carcinogenesis of ThrbPV/PV mice [29-33].
Figure 1.

(A). LY treatment delays pathological progression and blocks of metastasis in ThrbPV/PV mice. Representative H&E stained sections from thyroid of treated (b, d, f) or untreated (a, c, and e) mice for 150 days (7-month-old) are shown. Arrows show vascular invasion in an untreated mouse (c), and apoptosis and cell death in hyperplastic thyroid of a ThrbPV/PV mouse in response to LY treatment (d). Panel e shows metastatic thyroid carcinoma lesions in the lung (arrow). No metastases were detected in ThrbPV/PV mice treated with LY (panel f). (B). Reduced occurrence of vascular invasion and metastasis in mice treated with LY. Sections of thyroids and lungs from moribund ThrbPV/PV mice treated with vehicle (n=23) or LY (n=24) were stained with H&E and analyzed for pathological progression of vascular invasion and metastasis in lung. “*” denotes that the p values cannot be determined as no lung metastases were detected in LY-treated mice [30].
The findings that the PI3K-AKT signaling pathway is aberrantly activated in thyroid carcinogenesis of the ThrbPV/PV mouse suggests that this knockin mutant mouse can be used as a preclinical mouse model to test the efficacy of novel molecular targets uncovered in this pathway. To test this hypothesis, Furuya et al. treated ThrbPV/PV mice with LY294002 (LY), a potent inhibitor of PI3K, and monitored its effects on the spontaneous development of thyroid cancer in ThrbPV/PV mice [30]. LY treatment inhibits the phosphorylation cascade of PI3K-AKT-mammalian target of rapamycin (mTOR)-p70S6K signaling. Such treatment reduces tumor growth by inhibiting tumor cell proliferation (compare panels a and b, Figure 1A) and induces apoptosis (panel d, Figure 1A). Remarkably, in addition to a marked decrease in the occurrence of vascular invasion in the thyroid of LY-treated mice (Figure 1B), no metastasis is evident in the lung of LY-treated ThrbPV/PV mice (Figure 1A, panel f; Figure 1B) whereas the lung metastasis is apparent in vehicle-treated controls (Figure 1A, panel e; Figure 1B). Thus, by inhibiting tumor growth and blocking tumor invasion and metastasis, the survival of LY-treated mice is significantly prolonged. These findings indicate that PI3K is a potential molecular target in FTC. Though its toxicity profile has precluded the use of LY in clinical trails, the present preclinical data suggest that other minimally toxic inhibitors of PI3K could be considered as treatment strategies for thyroid cancer.
The ThrbPV/PV mouse is also valuable in preclinical testing of novel genes uncovered in thyroid carcinogenesis. Gene expression profiling of thyroid tumors in ThrbPV/PV mice has identified the repression of the peroxisome proliferator-activated receptor γ (PPARγ)-signaling pathway as one of the altered pathways that contribute to thyroid carcinogenesis [34]. PPARγ is also a member of the nuclear hormone receptor super-family and plays an important role in adipogenesis, cell cycle control, apoptosis, and carcinogenesis [35]. Its involvement in follicular thyroid carcinoma was demonstrated by the identification of a chromosomal rearrangement t(2;3) (q13;p25), yielding paired box gene 8 (PAX8)-PPARγ fusion gene in human follicular carcinomas [36-38]. When fused to PAX8, PPARγ not only loses its ability to stimulate thiazolidinedione-induced transcription, but also acts to inhibit PPARγ transcriptional activity [36, 37], raising the possibility that PPARγ could act as a tumor suppressor in thyroid carcinoma. Consistent with this notion, the expression of the Pparγ gene is inhibited and transcription activity of PPARγ is repressed in thyroid carcinogenesis of ThrbPV/PV mice [39]. These observations raise the possibility that PPARγ could be tested as a therapeutic target. Indeed, treatment of ThrbPV/PV mice with a PPARγ agonist, rosiglitazone, delayed the progression of thyroid carcinogenesis by decreasing tumor growth (Figure 2A) and activation of apoptosis (Figure 2B, panels b and d). Moreover, the lung metastasis is blocked (Figure 2C) [39]. That activation of PPARγ delays thyroid cancer progression is further supported by a recent study in another mouse model in which the Pax8-Pparγ (PPFP) gene was targeted to the thyroid deficient in PTEN (phosphatase and tensin homologue deleted from chromosome 10). Treatment of this mouse with another PPARγ ligand, pioglitazone, decreased thyroid growth and prevented metastatic disease [40].
Figure 2.
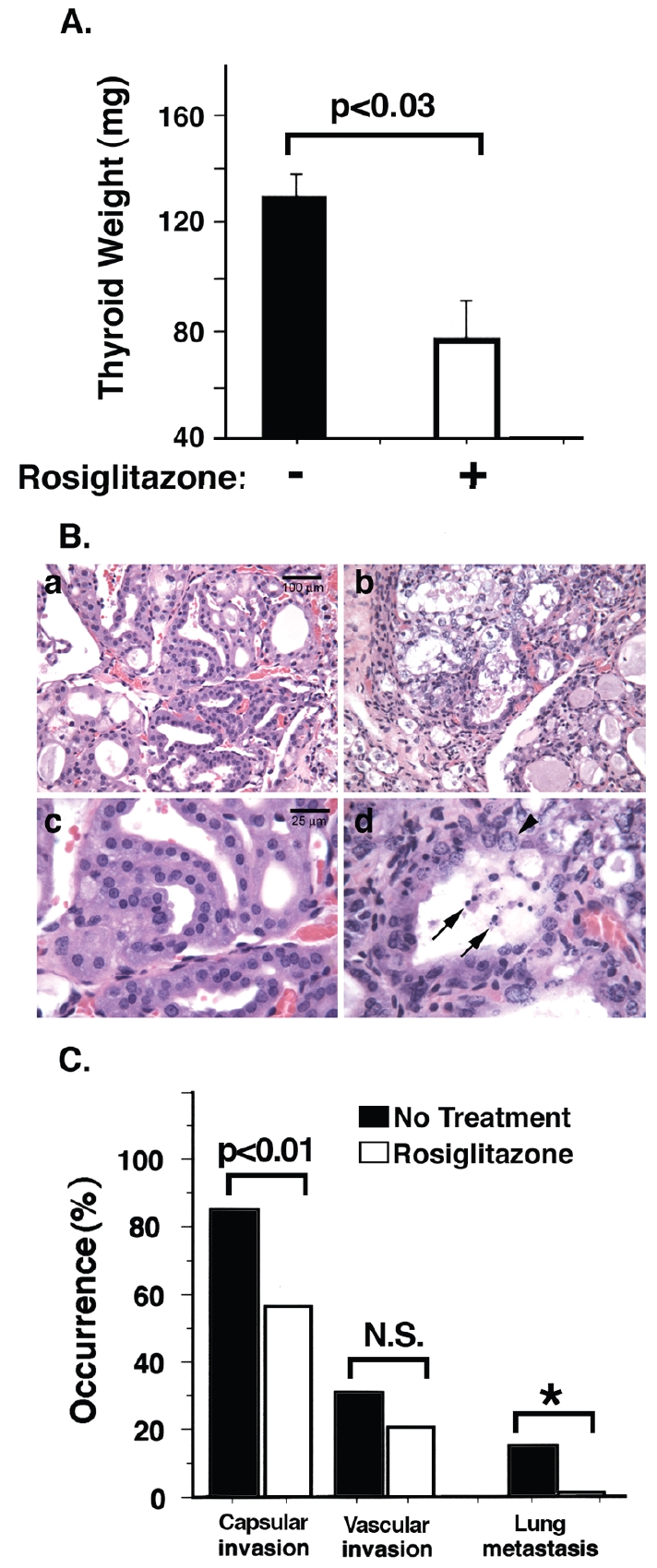
Rosiglitazone treatment delays thyroid cancer progression and blocks metastasis of ThrbPV/PV mice. (A). Rosiglitazone treatment decreases tumor growth (open bar) as compared with untreated mice (closed bar). (B). Rosiglitazone treatment induces apoptosis of tumor cells. Representative H&E stained sections of thyroid of treated (panels b and d) or untreated (panels a and c) ThrbPV/PV mice are shown. The hyperplastic thyroids in untreated mice show extensive hyperplasia of follicular epithelia, shown at low (panel a) and high (panel c) magnification. Similar fields from mice treated with rosiglitazone show epithelial cellular damage, including cell and nuclear swelling (arrowhead, panel d) and nuclear chromatin condensation typical of apoptotic cell death (arrows, panel d). (Mags: panels a, b = ×75; panels c, d= ×300). (C). Rosiglitazone treatment decreases occurrence of capsular invasion, vascular invasion, and metastasis in ThrbPV/PV mice. “*” denotes that p values could not be calculated since no metastasis was observed in drug-treated mice [39].
The pituitary tumor transforming gene 1 (Pttg1) was found to be highly elevated in the thyroid tumors of ThrbPV/PV mice. Further studies have indicated that the over-expressed PTTG1 not only inhibits mitotic progression and causes chromosomal aberrations, but also promotes angiogenesis in thyroid carcinogenesis [41]. In the thyroid tumors of ThrbPV/PV mice, sustained β-catenin signaling was also observed to promote thyroid cancer progression [29]. These findings suggest that PTTG1 and β-catenin could be tested as potential molecular targets in thyroid cancer.
The ThrbPV/- mouse
The ThrbPV/- mouse was generated by crossbreeding heterozygous ThrbPV/+ mice with Thrb knockout mice [42]. Remarkably, in contrast to ThrbPV/+ mice, ThrbPV/- mice spontaneously develop FTC [43]. The pathological progression in the thyroids of ThrbPV/- mice is indistinguishable from that in ThrbPV/PV mice. These findings indicate that one mutated Thrb allele in the absence of the other wild-type allele is sufficient to induce spontaneous thyroid carcinoma. Thyroid carcinoma occurs either when both Thrb alleles are mutated or when one allele is mutated and there is ablation of the other wild-type allele. Importantly, there are similarities in the altered expression patterns of key regulators in signaling pathways such as the repression of the tumor suppressor Pparγ and the activation of the cyclin D1 gene in the thyroids of ThrbPV/- and ThrbPV/PV mice [43]. Thus, this ThrbPV/- mouse model has provided direct in vivo evidence to indicate that the Thrb gene can function as a tumor suppressor and raises the possibility that the Thrb gene could serve as a novel therapeutic target in thyroid cancer.
The TR-double knockout (Thra1-/-Thrb-/-) mouse
Prompted by the observations that the expression of TRs is frequently silenced in tumors [12-17], Zhu et al. used Thra1-/-Thrb-/- mice [42, 44] to delineate whether total loss of all functional TRs could lead to thyroid cancer. These mice spontaneously develop FTC with pathological progression from hyperplasia to capsular invasion, vascular invasion, anaplasia, and metastasis, similar to human thyroid cancer [45]. Consistent with human FTC, aberrant activation of AKT and activation of vascular growth factor and its receptors were observed to drive tumor progression. The over-expression of known tumor promoters such as Pttg1 gene and the suppression of tumor suppressors such as the PPARγ and p53 also were detected in thyroid tumors of Thra1-/-Thrb-/- mice. These findings provided direct in vivo evidence to show that functional loss of both Thra1 and Thrb genes promotes thyroid tumor development and metastasis [45].
The observations that ThrbPV/PV and Thra1-/-Thrb-/- mice both spontaneously develop FTC indicate that loss of normal TR functions via a mutation of TRβ as in ThrbPV/PV mice and via deletion of the TR genes as in Thra1-/-Thrb-/- mice contribute to thyroid carcinogenesis. These findings further support the notion that TR functions as a tumor suppressor in thyroid cancer. However, the ThrbPV/PV and Thra1-/-Thrb-/- mice exhibit similarly elevated serum levels of TSH and thyroid hormones [46], but intriguingly the Thra1-/-Thrb-/- mouse develops FTC with a slower progression and a less aggressive malignant phenotype [28, 45]. These observations suggested that in addition to the loss of the normal tumor suppressor functions of wild-type TRβ, PV could acquire additional oncogenic activity via gain-of-function through mutation. Indeed, analysis of the cDNA microarray data derived from micro-dissected thyroid tumor cells of these two mice showed contrasting global gene expression profiles [47]. Among the 241 genes identified with altered gene expression, nearly half of those in the thyroid tumor cells of ThrbPV/PV mice were associated with tumorigenesis and metastasis. Some of these genes function as oncogenes in human thyroid cancers. The remaining genes were found to function in transcriptional regulation, RNA processing, cell proliferation, apoptosis, angiogenesis, and cytoskeleton modification [47]. These results indicate that the more aggressive thyroid tumor progression in ThrbPV/PV mice is not due simply to the loss of tumor suppressor functions of TR via mutation but also, importantly, to gain-of-function in the oncogenic activities of PV to drive thyroid carcinogenesis. Testing molecular targets in both mice will reveal new mechanist insights into how the molecular targets act via loss of TR functions or via gain-of-function of a mutated TRβ.
Mouse models derived from multiple genetic alterations
Metastasis is a complex process that requires the collaboration of many key players to coordinate the invasion and migration of tumor cells to the distant sites [48]. Deregulation of many molecular pathways of tumor cells initiated by multiple genetic alterations is responsible for the transformation of a single normal cell into life-threatening malignant tumor cells [49, 50].
Aberrant activation of the PI3K-AKT pathway is frequent in follicular and anaplastic thyroid cancers and promotes the progression from benign adenomas to cancer cells [51, 52]. PTEN functions as a tumor suppressor by opposing the PI3K-AKT signaling pathway [53]. Many studies using either primary tumor tissues or established tumor cell lines have revealed high frequencies of PTEN somatic mutations or deletion in various human tumors, including thyroid tumors [54], making PTEN the second most frequently mutated human tumor suppressor gene, after the TP53 gene. Accordingly, the impact of the lack of PTEN in thyroid carcinogenesis has been explored in mice harboring oncogenes shown to be involved in cancer development.
The ThrbPV/PVPten+/- mouse
ThrbPV/PV mice were crossed with Pten haplodeficient mice to elucidate the role of overactivated PI3K signaling in thyroid carcinogenesis. PTEN deficiency accelerated the progression of the thyroid tumor and increased the occurrence of metastatic spread to the lung, thereby significantly reducing their survival as compared with ThrbPV/PV mice [55]. Molecular studies have indicated that the loss of the negative regulation of PI3K leads to additional activation of AKT and its downstream mTOR-p70S6K signalling and decreases activity of the forkhead family member FOXO3a. Consistently, cyclin D1 expression is also increased. Apoptosis is decreased as indicated by the increased expression of NF-κB and decreased caspase-3 activity in the thyroids of ThrbPV/PVPten+/- mice. These findings indicate that the Pten gene acts as a tumor suppressor in the thyroid carcinogenesis of ThrbPV/PV mice.
The aggressive cancer progression has made this ThrbPV/PVPten+/- mouse an attractive preclinical model for more efficient testing molecular targets in the PI3K-AKT signalling pathways. A specific mTORC1 inhibitor-RAD001 was used to examine the efficacy of mTOR inhibition against thyroid carcinoma in ThrbPV/PVPten+/-mice. RAD001 effectively decreased cell proliferation as shown by Ki-67 staining (Figure 3A) in which the staining intensities were ~50% lower in treated thyroid than in control. The decreased proliferation markedly slows thyroid tumor growth, thereby prolonging survival (Figure 3B) [55]. However, inhibition of mTOR activity did not prevent capsular and vascular invasion of the thyroid or reduce the occurrence of lung metastasis. These data demonstrate that treatment against mTOR signaling can effectively arrest cancer growth [56]. This mouse model can be further used to test other effectors in the PI3K-AKT-mTOR signaling pathway as molecular targets in treatment.
Figure 3.
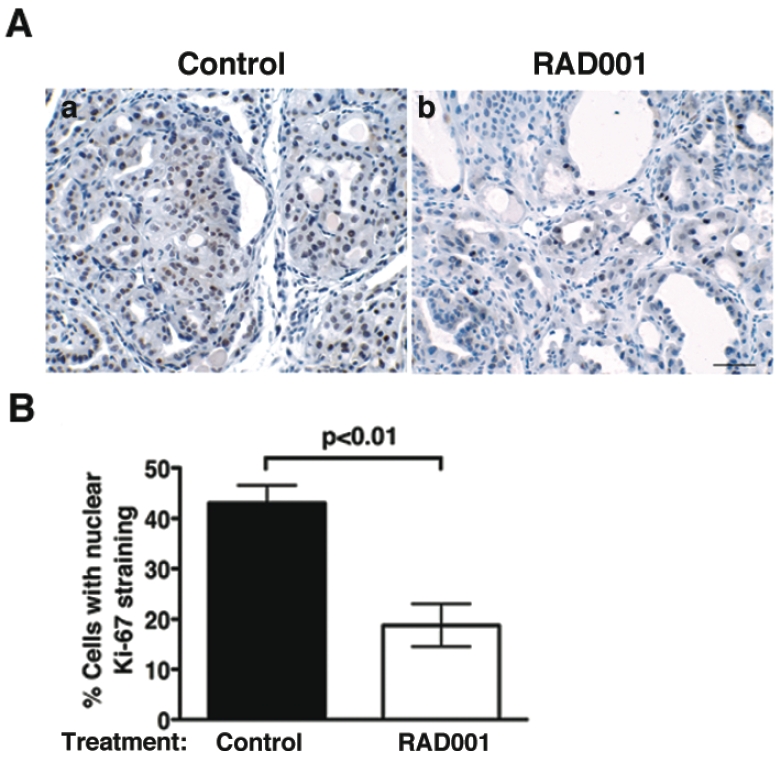
RAD001 treatment inhibits thyroid tumor cell proliferation of ThrbPV/PVPten+/- mice. (A). Representative microphotographs of Ki-67 immunohisto-chemistry on thyroid sections of placebo (control; panel a) and RAD001-treated ThrbPV/PVPten+/- mice (panel b). (B). Thyroid cell proliferative index, determined by Ki-67 immunohistochemistry in the control and treated groups, shows a significant reduction in the percentage of proliferating cells in the thyroids of RAD001-treated ThrbPV/PVPten+/- mice (open bar) as compared with placebo-treated ThrbPV/PVPten+/- mice (closed bar) [56].
The mouse with targeted KrasG12DPten-/- in the thyroid
This mouse was generated to understand whether PI3K cooperates with Kras mutations to promote thyroid cancer progression [57]. The KrasG12D mutant gene was targeted to the thyroid via a thyroid peroxidase (TPO)-Cre recombinase (Cre) -mediated mechanism, but constitutive expression of the KrasG12D mutant in the thyroid did not lead to morphological and functional alterations of the thyroid gland. Mice with deletion of both alleles of the Pten gene together with the KrasG12D mutant in the thyroid (i.e., double mutant mice) were then generated to explore the collaborative role of PI3K-AKT signaling with constitutive activation of the KrasG12D mutant. Although mice with only targeted deletion of both alleles of the Pten gene in the thyroid exhibited no thyroid carcinoma, all double mutant mice rapidly developed FTC with only 50% of the mice still alive at 7 weeks of age. No survival of double mutant mice was observed beyond 5 months of age [57]. These observations suggest that a collaboration of the activated Kras- and PI3K-AKT signaling pathways is required to drive thyroid carcinogenesis.
To dissect how each pathway contributes to thyroid carcinogenesis, cultured cells were generated from the thyroids of double mutant mice, and the effects on tumor cell growth by LY, the inhibitor of PI3K, and by PD98059, the inhibitor of MAPK/extracellular signal-regulated kinase kinase 1 (MEK), were evaluated. LY was much more effective than PD98059 in inhibiting tumor cell growth. Prolonged treatment of cells with LY led to the inhibition of ERK phosphorylation to the same extent as did direct MEK inhibition and induction of cell senescence. Combined inhibition of PI3K and MAPK completely stopped the growth of cultured cancer cells. The effect of LY was further tested in vivo with the double mutant mice. LY treatment prolonged the survival of those mice, suggesting that continuous PI3K signaling is necessary to facilitate the transforming activity of oncogenic Kras mutations.
These preclinical findings provided additional support that PI3K inhibitors are effective agents to treat FTC. Moreover, these results suggest dual targeting of the PI3K and Kras pathways could be beneficial and that this mouse model would be well suited for testing the efficacy of dual targeting for thyroid cancer while newer generations of PI3K and Ras are continuously being developed [58-60].
The mouse with targeted PPFP and Pten-/- in the thyroid
A chromosomal translocation leading to the fusion of PAX8 with PPARγ gene (PAX8-PPARγ gene; PPFP) [37] was detected in 35% of FTCs and occasionally in thyroid adenomas [61-63]. To elucidate the role of PPFP in the oncogenesis of FTC, PPFP was targeted to the thyroid via a Cre-dependent expression system. The expression of PPFP alone did not result in thyroid carcinoma, but when combined with homozygous deletion of the Pten gene, the double mutant mice (PPFP;Pten-/-;Cre mice) developed metastatic thyroid cancer [40]. With the use of ThrbPV/PV mice, it has been shown that PPARγ acts as a tumor suppressor [39]. The PPFP;Pten-/-;Cre mice provided another model to reinforce this notion. Indeed, treatment of PPFP;Pten-/-;Cre mice with a PPARγ ligand, pioglitazone, led to a 7-fold reduction of thyroid growth and prevention of metastatic lung tumor [39]. Interestingly, pioglitazone treatment induced proadipogenic responses by up-regulation of adipocyte PPARγ target genes and lipid accumulation in thyroids. These data indicate that PPARγ agonists will likely be effective against carcinomas with the expression of the translocated PPARγ gene. Several PPARγ ligands are being used to treat patients with type II diabetes mellitus. Thus, the availability of the PPFP;Pten-/-;Cre mouse will facilitate the testing of PPARγ ligands as therapeutics in thyroid cancers, particularly in those harboring PPFP.
Perspectives in modeling follicular thyroid cancer
The mouse models of FTC have significantly advanced our understanding of the molecular genetics of thyroid cancer. With these models, the altered signaling pathways reported for human thyroid cancer not only have been validated, but also have led to a better understanding of the mechanisms by which the key effectors in the altered signaling act to affect thyroid cancer development and progression. These models also serve to address long-standing challenging issues. One of which is the role of TSH in thyroid cancer. Although several epidemiological studies have reported that a high level of TSH is a risk factor for thyroid cancer [64, 65], other studies have argued against its role as an initiator of thyroid cancer [66, 67]. The elevated TSH in ThrbPV/PV mice has provided an opportunity to address the role of TSH in thyroid cancer. Indeed, by blocking the action of TSH in the progeny of crosses of ThrbPV/PV mice with TSH receptor (Tshr) knockout (ThrbPV/PVTshr-/- mice), it was shown that TSH is required, but alone is not sufficient, to induce thyroid cancer. Additional oncogenic changes are needed to propel the TSH-stimulated hyperplastic cells to undergo transformation to cancer cells [68]. Still, the question of what threshold concentration of TSH in the presence of oncogenic mutations poses a risk for thyroid cancer remains unanswered. Heterozygous ThrbPV/+ mice [26] as well as homozygous Thrb-/- knockout mice [42] could be useful in clarifying this issue. These mutant mice displayed 2- to 3-fold higher TSH levels due to the resistance to thyroid hormone [26, 42]. Known oncogenic mutations in thyroid cancer, such as PIK3CA (phosphoinositide-3-kinase, catalytic, alpha polypeptide), CTNNB1, RAS, and TP53, could be targeted to the thyroids of these mice to ascertain the effects on thyroid carcinogenesis of relatively low elevations in TSH levels (2- to 3-fold elevation). Such studies could provide new insights into how chronic long-term stimulation by marginally elevated TSH, such as in patients in regions with dietary iodide deficiency, affects thyroid carcinogenesis.
Human thyroid cancer frequently results from somatic mutations such as RAS, PIK3CA, and PTEN [54, 69, 70]. To model thyroid cancer due to somatic mutations, an inducible system would be a better approach in that mouse models with germ line mutations of the genes of interest could affect the development of thyroid gland and thereby not faithfully reflecting the somatic mutation-induced carcinogenesis of the adult thyroid. This issue is exemplified by a mouse model of papillary thyroid carcinoma in which the BRafV600E mutation was targeted to thyroid of mice with thyrocyte-specific expression of a conditional Cre (CreERT2) under the control of the thyroglobulin promoter (Thyro::CreERT2) [71]. Adult-onset and thyroid-specific expression of BRafV600E gene led to increased thyroid size and alterations in thyroid architecture. After 1 year, all mice developed papillary thyroid carcinoma with nuclear atypia and expression of markers characteristic of the human disease [71]. A similar approach could be used to model inducible adult-onset somatic mutations of the relevant genes of interest and to understand the kinetics of induction and latency for full phenotypic manifestation of FTC. During carcinogenesis, the sequential changes in genetic profiles can be continuously monitored and identified such that cause and effect relationships can be easily established. In addition, such inducible system also provides the opportunity to evaluate the most optimal timing for treatment and prevention.
Acknowledgments
We regret any reference omissions due to length limitation. We wish to thank all colleagues and collaborators who have contributed to the work described in this review. The research described in this review by the authors and their colleagues at National Cancer Institute was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
References
- 1.Cooper DS, Doherty GM, Haugen BR, Kloos RT, Lee SL, Mandel SJ, Mazzaferri EL, McIver B, Pacini F, Schlumberger M, Sherman SI, Steward DL, Tuttle RM. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19:1167–1214. doi: 10.1089/thy.2009.0110. [DOI] [PubMed] [Google Scholar]
- 2.Pacini F, Schlumberger M, Dralle H, Elisei R, Smit JW, Wiersinga W. European consensus for the management of patients with differentiated thyroid carcinoma of the follicular epithelium. Eur J Endocrinol. 2006;154:787–803. doi: 10.1530/eje.1.02158. [DOI] [PubMed] [Google Scholar]
- 3.Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, Travagli JP, Caillou B, Ricard M, Lumbroso JD, De Vathaire F, Schlumberger M. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91:2892–2899. doi: 10.1210/jc.2005-2838. [DOI] [PubMed] [Google Scholar]
- 4.Gulcelik MA, Gulcelik NE, Kuru B, Camlibel M, Alagol H. Prognostic factors determining survival in differentiated thyroid cancer. J Surg Oncol. 2007;96:598–604. doi: 10.1002/jso.20845. [DOI] [PubMed] [Google Scholar]
- 5.Passler C, Scheuba C, Prager G, Kaczirek K, Kaserer K, Zettinig G, Niederle B. Prognostic factors of papillary and follicular thyroid cancer: differences in an iodine-replete endemic goiter region. Endocr Relat Cancer. 2004;11:131–139. doi: 10.1677/erc.0.0110131. [DOI] [PubMed] [Google Scholar]
- 6.Cornett WR, Sharma AK, Day TA, Richardson MS, Hoda RS, van Heerden JA, Fernandes JK. Anaplastic thyroid carcinoma: an overview. Curr Oncol Rep. 2007;9:152–158. doi: 10.1007/s11912-007-0014-3. [DOI] [PubMed] [Google Scholar]
- 7.Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V, Thompson JD, Cheng JH, Bou Reslan H, Ho CC, Cao TC, Lee CV, Nannini MA, Fuh G, Carano RA, Koeppen H, Yu RX, Forrest WF, Plowman GD, Johnson L. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol. 2010;28:585–593. doi: 10.1038/nbt.1640. [DOI] [PubMed] [Google Scholar]
- 8.Ando S, Sarlis NJ, Krishnan J, Feng X, Refetoff S, Zhang MQ, Oldfield EH, Yen PM. Aberrant alternative splicing of thyroid hormone receptor in a TSH-secreting pituitary tumor is a mechanism for hormone resistance. Mol Endocrinol. 2001;15:1529–1538. doi: 10.1210/mend.15.9.0687. [DOI] [PubMed] [Google Scholar]
- 9.Ando S, Sarlis NJ, Oldfield EH, Yen PM. Somatic mutation of TRbeta can cause a defect in negative regulation of TSH in a TSH-secreting pituitary tumor. J Clin Endocrinol Metab. 2001;86:5572–5576. doi: 10.1210/jcem.86.11.7984. [DOI] [PubMed] [Google Scholar]
- 10.Safer JD, Colan SD, Fraser LM, Wondisford FE. A pituitary tumor in a patient with thyroid hormone resistance: a diagnostic dilemma. Thyroid. 2001;11:281–291. doi: 10.1089/105072501750159750. [DOI] [PubMed] [Google Scholar]
- 11.Lin KH, Shieh HY, Chen SL, Hsu HC. Expression of mutant thyroid hormone nuclear receptors in human hepatocellular carcinoma cells. Mol Carcinog. 1999;26:53–61. doi: 10.1002/(sici)1098-2744(199909)26:1<53::aid-mc7>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 12.Puzianowska-Kuznicka M, Krystyniak A, Madej A, Cheng SY, Nauman J. Functionally impaired TR mutants are present in thyroid papillary cancer. J Clin Endocrinol Metab. 2002;87:1120–1128. doi: 10.1210/jcem.87.3.8296. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Meng ZH, Chandrasekaran R, Kuo WL, Collins CC, Gray JW, Dairkee SH. Biallelic inactivation of the thyroid hormone receptor beta1 gene in early stage breast cancer. Cancer Res. 2002;62:1939–1943. [PubMed] [Google Scholar]
- 14.Puzianowska-Kuznicka M, Nauman A, Madej A, Tanski Z, Cheng S, Nauman J. Expression of thyroid hormone receptors is disturbed in human renal clear cell carcinoma. Cancer Lett. 2000;155:145–152. doi: 10.1016/s0304-3835(00)00416-x. [DOI] [PubMed] [Google Scholar]
- 15.Takano T, Miyauchi A, Yoshida H, Nakata Y, Kuma K, Amino N. Expression of TRbeta1 mRNAs with functionally impaired mutations is rare in thyroid papillary carcinoma. J Clin Endocrinol Metab. 2003;88:3447–3449. doi: 10.1210/jc.2003-030012. [DOI] [PubMed] [Google Scholar]
- 16.Jazdzewski K, Boguslawska J, Jendrzejewski J, Liyanarachchi S, Pachucki J, Wardyn KA, Nauman A, de la Chapelle A. Thyroid hormone receptor beta (THRB) is a major target gene for microRNAs deregulated in papillary thyroid carcinoma (PTC) J Clin Endocrinol Metab. 2010;96:E546–553. doi: 10.1210/jc.2010-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ling Y, Xu X, Hao J, Ling X, Du X, Liu X, Zhao X. Aberrant methylation of the THRB gene in tissue and plasma of breast cancer patients. Cancer Genet Cytogenet. 2010;196:140–145. doi: 10.1016/j.cancergencyto.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 18.Chen HW, Privalsky ML. The erbA oncogene represses the actions of both retinoid X and retinoid A receptors but does so by distinct mechanisms. Mol Cell Biol. 1993;13:5970–5980. doi: 10.1128/mcb.13.10.5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yen PM, Ikeda M, Brubaker JH, Forgione M, Sugawara A, Chin WW. Roles of v-erbA homodimers and heterodimers in mediating dominant negative activity by v-erbA. J Biol Chem. 1994;269:903–909. [PubMed] [Google Scholar]
- 20.Sap J, Munoz A, Damm K, Goldberg Y, Ghysdael J, Leutz A, Beug H, Vennstrom B. The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature. 1986;324:635–640. doi: 10.1038/324635a0. [DOI] [PubMed] [Google Scholar]
- 21.Thormeyer D, Baniahmad A. The v-erbA oncogene (review) Int J Mol Med. 1999;4:351–358. [PubMed] [Google Scholar]
- 22.Weinberger C, Thompson CC, Ong ES, Lebo R, Gruol DJ, Evans RM. The c-erb-A gene encodes a thyroid hormone receptor. Nature. 1986;324:641–646. doi: 10.1038/324641a0. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Silva S, Aranda A. The thyroid hormone receptor is a suppressor of ras-mediated transcription, proliferation, and transformation. Mol Cell Biol. 2004;24:7514–7523. doi: 10.1128/MCB.24.17.7514-7523.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Iglesias O, Garcia-Silva S, Tenbaum SP, Regadera J, Larcher F, Paramio JM, Vennstrom B, Aranda A. Thyroid hormone receptor beta1 acts as a potent suppressor of tumor invasiveness and metastasis. Cancer Res. 2009;69:501–509. doi: 10.1158/0008-5472.CAN-08-2198. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Silva S, Martinez-Iglesias O, Ruiz-Llorente L, Aranda A. Thyroid hormone receptor beta1 domains responsible for the antagonism with the ras oncogene: role of corepressors. Oncogene. 2011;30:854–864. doi: 10.1038/onc.2010.464. [DOI] [PubMed] [Google Scholar]
- 26.Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, Kazlauskaite R, Pankratz DG, Wynshaw-Boris A, Refetoff S, Weintraub B, Willingham MC, Barlow C, Cheng S. Mice with a targeted mutation in the thyroid hormone beta receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci USA. 2000;97:13209–13214. doi: 10.1073/pnas.230285997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parrilla R, Mixson AJ, McPherson JA, McClaskey JH, Weintraub BD. Characterization of seven novel mutations of the c-erbA beta gene in unrelated kindreds with generalized thyroid hormone resistance. Evidence for two “hot spot” regions of the ligand binding domain. J Clin Invest. 1991;88:2123–2130. doi: 10.1172/JCI115542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki H, Willingham MC, Cheng SY. Mice with a mutation in the thyroid hormone receptor beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid. 2002;12:963–969. doi: 10.1089/105072502320908295. [DOI] [PubMed] [Google Scholar]
- 29.Guigon CJ, Zhao L, Lu C, Willingham MC, Cheng SY. Regulation of beta-catenin by a novel nongenomic action of thyroid hormone beta receptor. Mol Cell Biol. 2008;28:4598–4608. doi: 10.1128/MCB.02192-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furuya F, Lu C, Willingham MC, Cheng SY. Inhibition of phosphatidylinositol 3-kinase delays tumor progression and blocks metastatic spread in a mouse model of thyroid cancer. Carcinogenesis. 2007;28:2451–2458. doi: 10.1093/carcin/bgm174. [DOI] [PubMed] [Google Scholar]
- 31.Furuya F, Hanover JA, Cheng SY. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proc Natl Acad Sci USA. 2006;103:1780–1785. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim CS, Ying H, Willingham MC, Cheng SY. The pituitary tumor-transforming gene promotes angiogenesis in a mouse model of follicular thyroid cancer. Carcinogenesis. 2007;28:932–939. doi: 10.1093/carcin/bgl231. [DOI] [PubMed] [Google Scholar]
- 33.Kim CS, Vasko VV, Kato Y, Kruhlak M, Saji M, Cheng SY, Ringel MD. AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology. 2005;146:4456–4463. doi: 10.1210/en.2005-0172. [DOI] [PubMed] [Google Scholar]
- 34.Ying H, Suzuki H, Zhao L, Willingham MC, Meltzer P, Cheng SY. Mutant thyroid hormone receptor beta represses the expression and transcriptional activity of peroxisome proliferator-activated receptor gamma during thyroid carcinogenesis. Cancer Res. 2003;63:5274–5280. [PubMed] [Google Scholar]
- 35.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4:61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- 36.Gregory Powell J, Wang X, Allard BL, Sahin M, Wang XL, Hay ID, Hiddinga HJ, Deshpande SS, Kroll TG, Grebe SK, Eberhardt NL, McIver B. The PAX8/PPARgamma fusion oncoprotein transforms immortalized human thyrocytes through a mechanism probably involving wild-type PPARgamma inhibition. Oncogene. 2004;23:3634–3641. doi: 10.1038/sj.onc.1207399. [DOI] [PubMed] [Google Scholar]
- 37.Kroll TG, Sarraf P, Pecciarini L, Chen CJ, Mueller E, Spiegelman BM, Fletcher JA. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected] Science. 2000;289:1357–1360. doi: 10.1126/science.289.5483.1357. [DOI] [PubMed] [Google Scholar]
- 38.Au AY, McBride C, Wilhelm KG Jr, Koenig RJ, Speller B, Cheung L, Messina M, Wentworth J, Tasevski V, Learoyd D, Robinson BG, Clifton -Bligh RJ. PAX8-peroxisome proliferator-activated receptor gamma (PPARgamma) disrupts normal PAX8 or PPARgamma transcriptional function and stimulates follicular thyroid cell growth. Endocrinology. 2006;147:367–376. doi: 10.1210/en.2005-0147. [DOI] [PubMed] [Google Scholar]
- 39.Kato Y, Ying H, Zhao L, Furuya F, Araki O, Willingham MC, Cheng SY. PPARgamma insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-kappaB signaling pathway. Oncogene. 2006;25:2736–2747. doi: 10.1038/sj.onc.1209299. [DOI] [PubMed] [Google Scholar]
- 40.Dobson ME, Diallo-Krou E, Grachtchouk V, Yu J, Colby LA, Wilkinson JE, Giordano TJ, Koenig RJ. Pioglitazone Induces a Proadipogenic Anti-tumor Response in Mice with PAX8-PPAR ﹛gamma﹜ Fusion Protein Thyroid Carcinoma. Endocrinology. 2011;152:4455–4465. doi: 10.1210/en.2011-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ying H, Furuya F, Zhao L, Araki O, West BL, Hanover JA, Willingham MC, Cheng SY. Aberrant accumulation of PTTG1 induced by a mutated thyroid hormone beta receptor inhibits mitotic progression. J Clin Invest. 2006;116:2972–2984. doi: 10.1172/JCI28598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forrest D, Erway LC, Ng L, Altschuler R, Curran T. Thyroid hormone receptor beta is essential for development of auditory function. Nat Genet. 1996;13:354–357. doi: 10.1038/ng0796-354. [DOI] [PubMed] [Google Scholar]
- 43.Kato Y, Ying H, Willingham MC, Cheng SY. A tumor suppressor role for thyroid hormone beta receptor in a mouse model of thyroid carcinogenesis. Endocrinology. 2004;145:4430–4438. doi: 10.1210/en.2004-0612. [DOI] [PubMed] [Google Scholar]
- 44.Wikstrom L, Johansson C, Salto C, Barlow C, Campos Barros A, Baas F, Forrest D, Thoren P, Vennstrom B. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor alpha 1. EMBO J. 1998;17:455–461. doi: 10.1093/emboj/17.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu XG, Zhao L, Willingham MC, Cheng SY. Thyroid hormone receptors are tumor suppressors in a mouse model of metastatic follicular thyroid carcinoma. Oncogene. 2010;29:1909–1919. doi: 10.1038/onc.2009.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Furumoto H, Ying H, Chandramouli GV, Zhao L, Walker RL, Meltzer PS, Willingham MC, Cheng SY. An unliganded thyroid hormone beta receptor activates the cyclin D1/cyclin-dependent kinase/retinoblastoma/E2F pathway and induces pituitary tumorigenesis. Mol Cell Biol. 2005;25:124–135. doi: 10.1128/MCB.25.1.124-135.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu C, Mishra A, Zhu YJ, Meltzer P, Cheng SY. Global expression profiling reveals gain-of-function oncogenic activity of a mutated thyroid hormone receptor in thyroid carcinogenesis. Am J Cancer Res. 2011;1:168–191. [PMC free article] [PubMed] [Google Scholar]
- 48.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 49.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 51.Hou P, Liu D, Shan Y, Hu S, Studeman K, Condouris S, Wang Y, Trink A, El-Naggar AK, Tallini G, Vasko V, Xing M. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin Cancer Res. 2007;13:1161–1170. doi: 10.1158/1078-0432.CCR-06-1125. [DOI] [PubMed] [Google Scholar]
- 52.Wang Y, Hou P, Yu H, Wang W, Ji M, Zhao S, Yan S, Sun X, Liu D, Shi B, Zhu G, Condouris S, Xing M. High prevalence and mutual exclusivity of genetic alterations in the phosphatidylinositol-3-kinase/akt pathway in thyroid tumors. J Clin Endocrinol Metab. 2007;92:2387–2390. doi: 10.1210/jc.2006-2019. [DOI] [PubMed] [Google Scholar]
- 53.Wymann MP, Marone R. Phosphoinositide 3-kinase in disease: timing, location, and scaf-folding. Curr Opin Cell Biol. 2005;17:141–149. doi: 10.1016/j.ceb.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 54.Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 55.Guigon CJ, Zhao L, Willingham MC, Cheng SY. PTEN deficiency accelerates tumour progression in a mouse model of thyroid cancer. Oncogene. 2009;28:509–517. doi: 10.1038/onc.2008.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guigon CJ, Fozzatti L, Lu C, Willingham MC, Cheng SY. Inhibition of mTORC1 signaling reduces tumor growth but does not prevent cancer progression in a mouse model of thyroid cancer. Carcinogenesis. 2010;31:1284–1291. doi: 10.1093/carcin/bgq059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller KA, Yeager N, Baker K, Liao XH, Refetoff S, Di Cristofano A. Oncogenic Kras requires simultaneous PI3K signaling to induce ERK activation and transform thyroid epithelial cells in vivo. Cancer Res. 2009;69:3689–3694. doi: 10.1158/0008-5472.CAN-09-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trujillo JI. MEK inhibitors: a patent review 2008 - 2010. Expert Opin Ther Pat. 2011;21:1045–1069. doi: 10.1517/13543776.2011.577068. [DOI] [PubMed] [Google Scholar]
- 59.Hernandez-Aya LF, Gonzalez-Angulo AM. Targeting the phosphatidylinositol 3-kinase signaling pathway in breast cancer. Oncologist. 2011;16:404–414. doi: 10.1634/theoncologist.2010-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nikiforova MN, Biddinger PW, Caudill CM, Kroll TG, Nikiforov YE. PAX8-PPARgamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am J Surg Pathol. 2002;26:1016–1023. doi: 10.1097/00000478-200208000-00006. [DOI] [PubMed] [Google Scholar]
- 62.Cheung L, Messina M, Gill A, Clarkson A, Learoyd D, Delbridge L, Wentworth J, Philips J, Clifton-Bligh R, Robinson BG. Detection of the PAX8-PPAR gamma fusion oncogene in both follicular thyroid carcinomas and adenomas. J Clin Endocrinol Metab. 2003;88:354–357. doi: 10.1210/jc.2002-021020. [DOI] [PubMed] [Google Scholar]
- 63.Gustafson KS, LiVolsi VA, Furth EE, Pasha TL, Putt ME, Baloch ZW. Peroxisome proliferator-activated receptor gamma expression in follicular-patterned thyroid lesions. Caveats for the use of immunohistochemical studies. Am J Clin Pathol. 2003;120:175–181. doi: 10.1309/2E6Q-GJRG-GETV-T8K9. [DOI] [PubMed] [Google Scholar]
- 64.Fiore E, Rago T, Provenzale MA, Scutari M, Ugolini C, Basolo F, Di Coscio G, Berti P, Grasso L, Elisei R, Pinchera A, Vitti P. Lower levels of TSH are associated with a lower risk of papillary thyroid cancer in patients with thyroid nodular disease: thyroid autonomy may play a protective role. Endocr Relat Cancer. 2009;16:1251–1260. doi: 10.1677/ERC-09-0036. [DOI] [PubMed] [Google Scholar]
- 65.Haymart MR, Repplinger DJ, Leverson GE, Elson DF, Sippel RS, Jaume JC, Chen H. Higher serum thyroid stimulating hormone level in thyroid nodule patients is associated with greater risks of differentiated thyroid cancer and advanced tumor stage. J Clin Endocrinol Metab. 2008;93:809–814. doi: 10.1210/jc.2007-2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Santelli G, de Franciscis V, Portella G, Chiappetta G, D'Alessio A, Califano D, Rosati R, Mineo A, Monaco C, Manzo G, Pozzi L, Vecchio G. Production of transgenic mice expressing the Ki-ras oncogene under the control of a thyroglobulin promoter. Cancer Res. 1993;53:5523–5527. [PubMed] [Google Scholar]
- 67.Ribeiro-Neto F, Leon A, Urbani-Brocard J, Lou L, Nyska A, Altschuler DL. cAMP-dependent oncogenic action of Rap1b in the thyroid gland. J Biol Chem. 2004;279:46868–46875. doi: 10.1074/jbc.M406858200. [DOI] [PubMed] [Google Scholar]
- 68.Lu C, Zhao L, Ying H, Willingham MC, Cheng SY. Growth activation alone is not sufficient to cause metastatic thyroid cancer in a mouse model of follicular thyroid carcinoma. Endocrinology. 2010;151:1929–1939. doi: 10.1210/en.2009-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Garcia-Rostan G, Costa AM, Pereira-Castro I, Salvatore G, Hernandez R, Hermsem MJ, Herrero A, Fusco A, Cameselle-Teijeiro J, Santoro M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005;65:10199–10207. doi: 10.1158/0008-5472.CAN-04-4259. [DOI] [PubMed] [Google Scholar]
- 70.Xing M. Genetic alterations in the phosphatidylinositol-3 kinase/Akt pathway in thyroid cancer. Thyroid. 2010;20:697–706. doi: 10.1089/thy.2010.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Charles RP, Iezza G, Amendola E, Dankort D, McMahon M. Mutationally activated BRAF (V600E) elicits papillary thyroid cancer in the adult mouse. Cancer Res. 2011;71:3863–3871. doi: 10.1158/0008-5472.CAN-10-4463. [DOI] [PMC free article] [PubMed] [Google Scholar]