

Abstract
Persistent infection with a high risk (hr) human papillomavirus (HPV) has been established as the main cause of cervical cancer and high-grade cervical intraepithelial neoplasia (CIN3). Because most infections are transient, testing for hrHPV lacks specificity and has a low positive predictive value. It has been suggested that additional parameters like viral load and physical status of the viral genome could improve the effectiveness of HPV-based screening. We investigated the association between HPV16 viral load and physical state with viral persistence or risk of incident CIN3 or worse in a population-based prospective cohort study comprising 8656 women (20-29 years). All participants had two gynecological examinations two years apart and were followed through the nationwide Danish Pathology Data Bank (median follow-up: 12.9 yrs). Seventynine cervical swabs from women with a persistent HPV16 infection were available for analysis. For comparison we selected a random age-matched sample of transiently HPV16 infected women (N=91). Persistently infected women with incident CIN3 or cancer (CIN3+; N=31) were compared to women with normal cytology during follow up (non-progressors; N=39). Quantitative real-time PCR for HPV16E6, E2 and IFNb1 was done to determine the HPV16 viral load and the E2/E6 ratio was used as a surrogate marker for integration. Women with normal cytology who became persistently HPV16 infected had a significantly lower HPV16 load at baseline than women who cleared the infection (median 4.72 copies/cell versus median 20.0 copies/cell, respectively; p=0.0003). There was no difference in viral load at enrollment between women who progressed to CIN3+ and women who stayed cytologically normal (p=0.85). At the second examination viral load tended to be higher in women who progressed, but the difference was not statistically significant (p=0.39). The E2/E6 ratio was shown to be lower in the persistently infected group (p<0.0001) already at the first examination, but no difference between non-progressors and CIN3+ cases was observed at any of the two examinations (p=0.61 and 0.86). Lower viral load and integration of the viral genome are predictive for the persistence of HPV16 DNA, but not for the progression of a persistent HPV16 infection to CIN3+ in women with normal cytology.
Keywords: Cervical cancer, HPV, viral load, viral integration
Introduction
The observation that a persistent infection with high risk (hr) human papillomavirus (HPV) types is a necessary cause of cervical cancer [1] offers not only the prospect of an effective primary prevention, but also the possibility of testing for hrHPV types to improve the efficiency of cervical cancer screening programmes. The continued presence of hrHPV DNA for ≥ 24 months identifies women at particular risk for cervical intraepithelial neoplasia of grade 3 or higher (CIN3+) [2]. In contrast, a negative HPV result has been shown to be associated with a greater long-term protection from CIN3 compared with a normal cytology result [3-5]. HPV16 is the most common carcinogenic type in preinvasive and cervical cancer and is detectable in roughly 50% of women with CIN3+ [6, 7]. It was shown to cause persistent infections in 30% of the women [8, 9].
The inability of a single hrHPV DNA test to discriminate between persistent and transient infections urges the need for additional parameters that enable the identification of hrHPV-positive women who are indeed at risk of CIN3+. In this context, several studies have investigated the value of viral DNA load or the integration status of the viral DNA as risk determinants for high-grade CIN. While some case control studies found no association of viral load with disease status [10, 11] others reported higher load in CIN2+ cases [12-16]. In one study this was only due to the inclusion of invasive squamous cervical cancer in the case group [17]. Studies with a prospective design reported higher viral load to be associated with a modest increase in the incidence of cytological abnormalities [18] and to be predictive for incident CIN2/3+, but restricted these observations only to cases of infections with HPV16 [19, 20] or HPV16, 31 and 33 [21].
The physical state of the viral genome was almost exclusively investigated in case control studies and the majority reported an association between integration and increasing severity of the lesions [11, 22-25]. However, it has to be noted that integrated viral DNA was already found in up to 44% of control samples with normal cytology [17, 26] which does not support the use of this parameter as a biomarker. These early integration events were rather frequently observed for HPV16 (26%) and HPV18 (58%) already in young women soon after sexual debut [27].
Integration of the viral DNA usually disrupts the E2 gene and causes the lack of the E2 repressor protein or the more potent E8^2C repressor [28, 29], which leads to subsequent overexpression of the viral oncogenes E6 and E7. However, frequently mixed levels of episomal and integrated forms have been described especially in normal and mildly abnormal epithelium. Purely integrated forms were found enriched in CIN3+, where the extinction of episomal forms and thereby the exclusion of trans-repression of the integrated genomes by the E2/E8^E2C repressor proteins provides the infected cell with a clear growth advantage.
Therefore studies are required that combine a robust measurement of viral load of the most relevant hrHPV types in conjunction with the physical state of the viral genome in the frame of a prospective cohort study.
In this study, we measured E2, E6 and IFNb1 DNA levels using quantitative real-time PCR (qPCR) in HPV16 positive cervical samples from a large population-based prospective cohort [2]. The aim of our work was to evaluate viral load and physical state as potential predictive biomarkers to assess the risk for persistence and the risk for progression in persistently infected women.
Materials and methods
The Danish HPV cohort
The present paper is based on women (20-29 years of age at enrollment) participating in a Danish prospective cohort study. The data collection procedures have previously been described in detail [2]. In brief, the women were selected at random from the general female population of Copenhagen, Denmark where every citizen has a unique 10-digit personal identification number, which is universally used in all health registries. These identification numbers, which contain information on sex and date of birth, are registered in the computerized Danish Central Population Register, which includes information on vital status, emigration and current address. Between May 15, 1991 and January 31, 1993, 11088 women were enrolled in the study for the first gynaecological examination (Figure 1). Before entering the study, all participants were informed verbally and in writing about the study and all participants signed a written informed consent. The study was approved by the national Scientific Ethical Committee and the national Data Protection Board.
Figure 1.

Overview of the study design and the cohort. Out of 7 482 eligible women 79 women were persistently infected with HPV16. Among the women who had a negative HPV test at the second examination (transient infections), we randomly selected 91 age matched women as controls. In the persistently infected group 39 women showed no progression within the median follow-up time of 12.9 years. Among the women eligible for analysis 4 developed CINl, 5 developed CIN2 and 31 progressed to CIN3 or cancer.
Approximately 2 years after enrollment (from October 1, 1993 to January 31, 1995), the study participants were re-invited, in the same order in which they were originally enrolled, for a second gynecological examination. A total of 8656 women (78%) participated in this second examination. At both gynaecological examinations, a Pap smear was obtained, and a cervical swab containing ecto- and endocervical cells was taken and placed in a tube containing phosphate-buffered saline and stored at -80°C for subsequent HPV DNA testing.
After the second examination the cohort was followed passively through the Pathology Data Bank which is a nationwide pathology register that contains information on all cervical cytology examinations and all cervical biopsy specimens, cones and hysterectomies performed in Denmark in the last 20 years. Clinical communication between pathology departments and the Pathology Data Bank is ensured through an online real-time data reporting system. Abnormal cervical diagnoses are usually reported as atypia/koilocytosis, mild dysplasia, moderate dysplasia, severe dysplasia, or carcinoma in situ. The histological diagnoses were translated into CIN nomenclature as follows: atypia/koilocytosis and mild dysplasia were grouped as CIN1, moderate dysplasia was categorized as CIN2 and severe dysplasia and carcinoma in situ were categorized as CIN3.
The existence of the nationwide Central Population Register and the unique personal identification numbers ensures correct linkages between registries and makes it possible to conduct follow-up studies with virtually no loss of follow-up.
Using the personal identification number for each woman, the study cohort was linked to the Pathology Data Bank and followed until March 6, 2007 to identify all cervical pathological lesions and diagnostic or treatment procedures. As HPV DNA testing took place several years after the study examinations were performed, the women were unaware of the results obtained in the study, and results of the HPV DNA testing were not used for referrals or clinical management of the women.
All women in the cohort were tested at both examinations for the presence of hrHPV DNA by the Hybrid Capture 2 test (HC2, Qiagen, Hilden, Germany) using the high risk probe cocktail and samples positive in the HC2 high risk probe were genotyped using the LiPav2 reverse line blot test system (Innogenetics) [30-32].
The study population (Figure 1) for the analyses presented here comprises women who participated in both examinations (N=8656). We excluded a total of 977 women who participated in the second examination only through a telephone interview, had abnormal smear at the first or the second examination or had no cervical swab (e.g. due to menstruation at the time of examination or a cervical swab that was inadequate for HPV testing). In addition, we excluded 197 women with no follow-up examination after the second examination, leaving 7482 women. A total of 296 women were HPV16 positive at the first examination and of these, 84 women were also HPV16 positive at the second examination (defined as persistent HPV16 infection). Five women were excluded due to insufficient material for further analysis leaving 79 women. Among the women who were HPV16 positive at the first examination and became HPV16 negative at the second examination (transient HPV16 infection), we randomly selected 91 women so that the age distribution was similar to that of the HPV16 persistors. Among the 79 women with persistent HPV16 infection, a total of 40 women developed abnormal cytology during follow-up. The corresponding histological diagnoses comprised CIN1 (N=4), CIN2 (N=5), CIN3 (N=29) and cervical cancer (N=2). This study focused on CIN3 or worse, so in the analysis of risk of progression among women with a persistent HPV16 infection we included 31 women with incident CIN3 or cancer (CIN3+), and 39 women with normal cytology during follow-up.
DNA extraction and qPCR
DNA was extracted from residual material from the HC2 assay using a Magna Pure device (Roche Molecular Systems, Pleasanton, CA). Quantification of the human IFNBl gene, the HPV16 E6 and E2 genes was performed with a Light Cycler 480 (Roche Applied Science, Mannheim, Germany). A predesigned primer pair (QT00203763, Qiagen, Hilden, Germany) was used for detection of IFNB1. The HPV16 E2 and E6 primers have been previously described [33]. PCR was performed in a final volume of 20 μl containing 1× Light Cycler 480 SYBR green I Master Mix (Roche Diagnostics, Mannheim, Germany), 0.3 μM primer and 5 μl of DNA. The amplification conditions were 10 min at 95°C, and then 10 sec at 95 °C, 15 sec at 55 °C, 15 sec at 72°C for 45 cycles. Amplification products were analysed by melting curves with a thermal profile of 10 sec at 95 °C, 30 sec at 60°C followed by heating to 90 °C.
Amplification of the single-copy IFNB1 gene was used to correct HPV copy numbers for the amount of cells in the cervical swab material. A standard curve for IFNB1 was generated using serial dilutions of total genomic DNA isolated from cultured normal human keratinocytes (NHK). Cell numbers were calculated assuming a DNA content of 6.6pg per human diploid cell. Standard curves used to quantify the copy numbers of the E2 and the E6 genes of HPV16 were generated using 107, 106, 105, 104, 103, 102 and 101 copies of a cloned HPV16 genome as previously described [33]. Copy numbers in samples were calculated using the Light Cycler 480 software version 1.5.0 second-derivative maximum algorithm. All qPCR reactions were run in duplicate showing extremely high concordance and values were then averaged for analysis. In each run non-template controls and positive controls were included.
Viral load and physical state determination
HPV16 copy numbers per cell were calculated by dividing E6 copies by the cell numbers calculated from the IFNB1 amplifications.
The physical state of HPV16 was determined using real-time PCR targeting the E6 and E2 open reading frame of HPV16, because the E2 gene is often lost during viral integration [34]. Thus, E2 and E6 gene segments are present in equivalent amounts in episomal HPV genomes, whereas integrated HPV genomes would have E6 present and the E2 gene absent. Assignment of integrated, mixed or episomal physical state was calculated as described elsewhere [35]. Integration was defined as an E2/E6 ratio between zero and 0.15 [36]. An E2/E6 ratio between 0.15 and 0.9 indicates the presence of both integrated and episomal forms and ratios of greater than 0.9 indicated the predominance of episomal forms [12]. However, it has to be taken into account that tandem integrated head -to-tail viral genomes would also result in values ≥ 0.9 falsely indicating episomal genomes. We therefore concentrated only on the reliable cut-off ≤ 0.15 as a surrogate marker for integration.
Statistical analysis
In the analysis of viral load and physical state as risk factors for persistence, we compared transiently HPV16 infected women with persistently HPV16 infected women using cervical samples taken at the first examination.
We also assessed the importance of viral load and physical state for progression within the group of persistently HPV16 infected women. For analysis of those with incident CIN3+ versus those who stayed cytologically normal during follow-up we investigated samples both from the first and the second examination, separately. The Mann-Whitney test was used to test for significant differences. P values were considered significant if less than 0.05. Receiver operating characteristics (ROC) curves were calculated using GraphPad Prism 5 ® (GraphPad Software, San Diego).
Results
Viral loads in cervical swabs taken at the first examination from women infected with HPV16 ranged from <1 to 2010 viral DNA copies per cell (c/c) (median: 20.0) in women who cleared the infection till the second examination (transient infections) and from <1 to 1958 viral DNA c/c (median: 4.72) in women who were HPV16 positive at both examinations (persistent infections; Table 1). The median viral load at the first examination was significantly higher in women with a transient HPV16-infection versus women that developed a persistent infection (p=0.0003; Figure 2A), although a wide range was observed within both groups.
Table 1.
Comparison of viral load in women who cleared an HPV16 infection (transient infection) and women with persistent HPV16 infection
| Viral load in Samples taken at | Transience (N=91) | Persistence (N = 79) |
|---|---|---|
| First examination: | ||
| Median (copies/cell) | 20 | 4.72 |
| 25-75% quartile | 4.5-65.8 | 1.8-21.36 |
Figure 2.
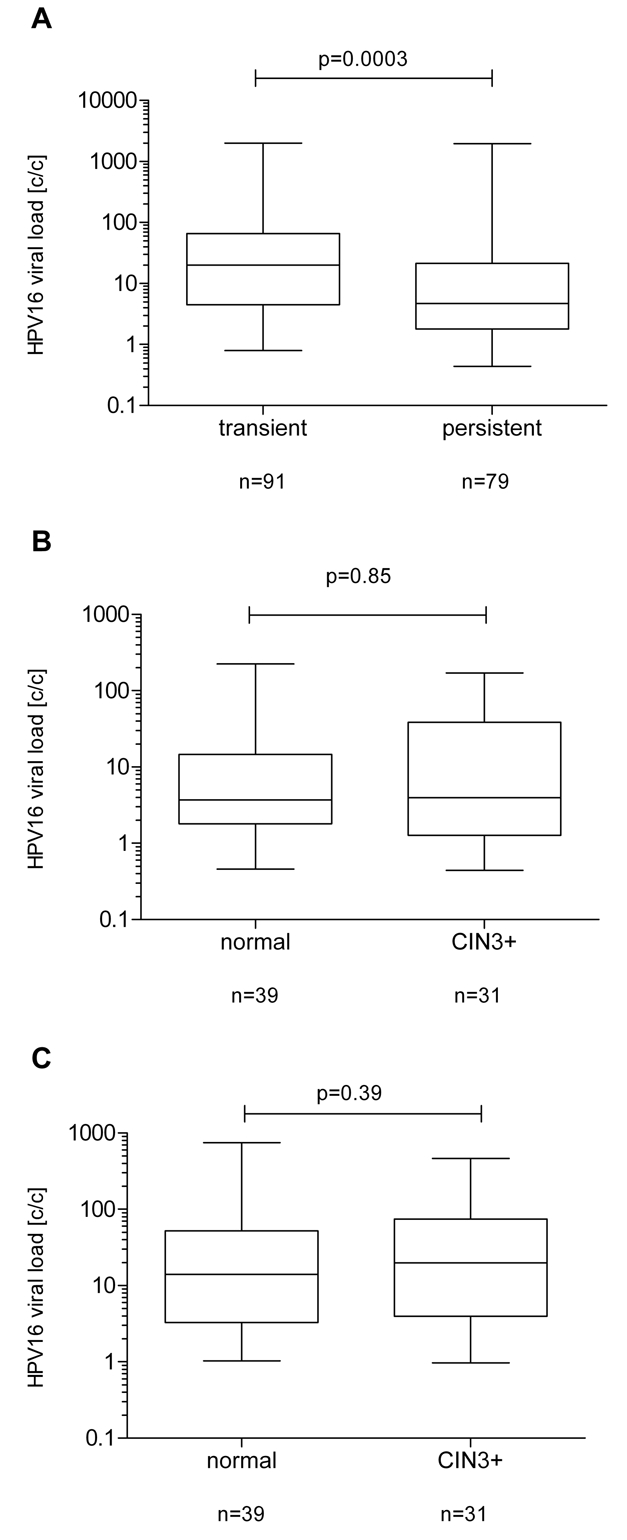
HPV16 viral load in persistent versus transient infections. Viral load values are shown on the Y-axis in HPV copies per cell (c/c) and are log transformed. The upper and lower boundaries of the boxes represent the 75th and 25th percentiles, respectively. The black line within the box represents the median; the whiskers represent the minimum and maximum values. A. LoglO HPV16 DNA loads were compared between women with transient infections (n=91) and persistent infections (N=79). A significant difference between transiently (median 20c/c; 25-75% quar-tiles of 4.5-65.8) and persistently (median: 4.72/c; 25-75% quartiles of 1.8-21.36) infected women (p=0.0003) was observed. B. HPV16 DNA loads were compared between samples taken at the first examination from persistently infected women who did (N=31) or did not (N=39) progress to CIN3+. No difference (p=0.85) between non-progressors (median 3.7c/c; 25-75% quartiles of 1.8-14.61) and progressors (median: 3.96c/c; 25-75% quartiles of 1.27-38.5) was observed. C. LoglO HPV16 DNA loads were compared between samples taken at the second examination from persistently infected women who did (N=31) or did not (N=39) progress to CIN3+. A non significant difference (p=0.39) between non-progressors (median 13.93c/c; 25-75% quartiles of 3.28-52.11) and progressors (median: 19.81c/c; 25-75% quartiles of 3.96-74.61) was observed.
To investigate whether viral load could be predictive for the development of incident CIN3+, we compared persistently infected women who stayed cytologically normal (non-progressors; N=39) to those women who developed CIN3+ after the second examination during the follow up time (N=31). The measurements of viral load in samples taken from non-progressor women at the first examination ranged from <1 to 225 viral DNA c/c (median: 3.7) and in those who developed CIN3+ from <1 to 171 c/c (median: 3.96; Table 2). No significant difference in the median viral load between non-progressors and women that progressed to CIN3+ was observed (p=0.85; Figure 2B).
Table 2.
Comparison of viral load in women stayingcytologically normal (non-progressors) and women developing CIN3 or worse (progressors)
| Viral load in samples taken at | No progression (N=39) | CIN3+ (N=31) |
|---|---|---|
| First examination: Median (copies/cell) | 3.7 | 3.96 |
| 25-75% quartile | 1.8-14.61 | 1.27-38.5 |
| Second examination: Median (copies/cell) | 13.93 | 19.81 |
| 25-75% quartile | 3.28-52.11 | 3.96-74.61 |
When we repeated the experiment with samples from the second examination, we observed a viral load ranging in non-progressors from 1 to 745 viral DNA c/c (median: 13.93) and in women who developed CIN3+ from <1 to 464 c/c (median: 19.81; Table 2). This difference was, however, not statistically significant (p=0.39; Figure 2C).
The analysis of the E2/E6 ratio of HPV16, as a proxy for physical status, showed values ranging from zero to 1.24 (median: 0.68) in samples taken at the first examination from women with transient infections, while the values of the E2/E6 ratio were significantly lower (p<0.0001; Figure 3A) in persistently infected women (median: 0.53, range: 0-0.83; Table 3).
Figure 3.
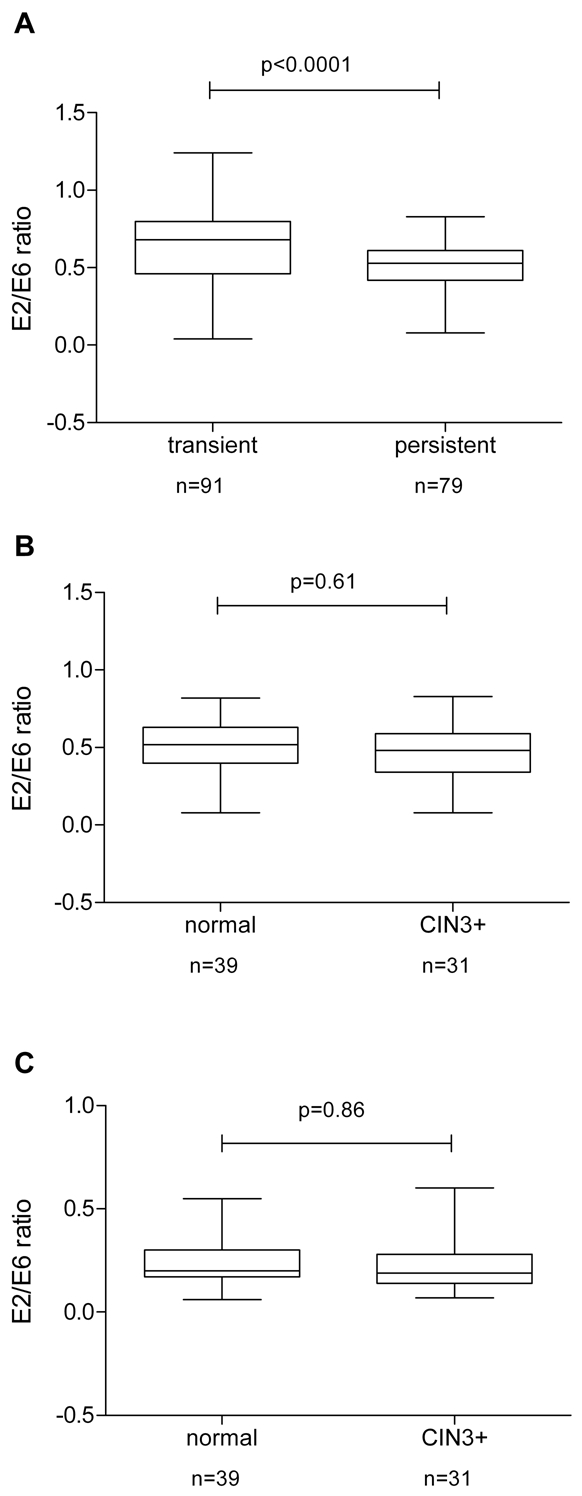
HPV16 physical status in persistent versus transient infections. E2/E6 ratios are shown on the Y-axis. The upper and lower boundaries of the boxes represent the 75th and 25th percentiles, respectively. The black line within the box represents the median; the whiskers represent the minimum and maximum values. A. E2/E6 ratios were compared between women with transient infections (N=91) and persistent infections (N=79). A significant difference (p<0.0001) between transiently (median 0.68; 25-75% quartiles of 0.46-0.80) and persistently (median: 0.53; 25-75% quartiles of 0.42-0.61) infected women was observed. Within the group of women with a transient infection (N=91) two samples showed a E2/E6 ratio below 0.15, 80 women showed mixed episomal and integrated forms (E2/E6 ratio between 0.15-0.90) and nine women had an E2/E6 ratio above 0.9. The group of persistently infected women (N = 79) consisted of six women with integrated forms (E2/E6>0.15), 73 women showed mixed episomal and integrated forms (E2/E6 ratio between 0.15-0.90) and no women had an E2/E6 ratio above 0.9. B. E2/E6 ratios were compared between persistently infected women who did (N=31) or did not (N=39) progress to CIN3+ from samples taken at the first examination. No significant difference (p=0.61) between non-progressors (median 0.52; 25-75% quartiles of 0.4-0.63) and progressors (median: 0.48; 25-75% quartiles of 0.34-0.59) was observed. Within the group of women who showed no progression (N=39) three samples showed an E2/E6 ratio below 0.15, 36 women showed mixed episomal and integrated forms (E2/E6 ratio between 0.15-0.90) and no woman had an E2/E6 ratio above 0.9. The group of women with CIN3+ (N=31) consisted of three women with integrated forms (E2/E6>0.15), 28 women showed mixed episomal and integrated forms (E2/E6 ratio between 0.15-0.90) and no woman had an E2/E6 ratio above 0.9. C. E2/E6 ratios were compared between persistently infected women who did (N=31) or did not (N=39) progress to CIN3+ from samples taken at the second examination. No significant difference (p=0.86) between non-progressors (median 0.2; 25-75% quartiles of 0.17-0.30) and progressors (median: 0.19; 25-75% quartiles of 0.14-0.28) was observed. Within the group of non-progressors (N=39) eight samples had integrated genomes (E2/E6 ratio below 0.15) and 31 women showed mixed episomal and integrated forms (E2/E6 ratio between 0.15-0.90). The group of women with CIN3+ (N=31) consisted of eight women with integrated forms (E2/E6>0.15), 23 women showed mixed episomal and integrated forms (E2/E6 ratio between 0.15-0.90) and no woman had an E2/E6 ratio above 0.9.
Table 3.
Comparison of E2/E6 ratio in women who cleared an HPV16 infection (transient infection) and women with persistent HPV16 infection
| E2/E6 ratio in Samples taken at | Transience (N=91) | Persistence (N = 79) |
|---|---|---|
| First examination: Median (copies/cell) | 0.68 | 0.53 |
| 25-75% quartile | 0.46-0.8 | 0.42-0.61 |
| Integrated <0.15 | 2 | 6 |
| Mixed 0.15-0.90 | 80 | 73 |
| Episomal >0.9 | 9 | 0 |
To investigate whether the E2/E6 ratio could be a predictive marker for the development of CIN3+ we compared women with persistent HPV16 infection who stayed cytologically normal with those who developed CIN3+ during follow up. In samples taken at the first examination the E2/E6 ratio in non-progressor women ranged from 0.1 to 0.82 (median: 0.52) and in women with persistent infections who developed incident CIN3+ from 0.1 to 0.83 (median: 0.48; Table 4). No significant correlation between the E2/E6 ratio as a proxy for physical state of the viral genome and progression to CIN3+ was found (p=0.61; Figure 3B).
Table 4.
Comparison of E2/E6 ratio in women stayingcytologically normal (non-progressors) and women developing CIN3 or worse (progressors)
| E2/E6 ratio in samples taken at | No progression (N=39) | CIN3+ (N=31) |
|---|---|---|
| First examination: Median (copies/cell) | 0.52 | 0.48 |
| 25-75% quartile | 0.4-0.63 | 0.34-0.59 |
| Integrated <0.15 | 3 | 3 |
| Mixed 0.15-0.90 | 36 | 28 |
| Episomal >0.9 | 0 | 0 |
| Second examination: Median (copies/cell) | 0.2 | 0.19 |
| 25-75% quartile | 0.17-0.3 | 0.14-0.28 |
| Integrated <0.15 | 8 | 8 |
| Mixed 0.15-0.90 | 31 | 23 |
| Episomal >0.9 | 0 | 0 |
Repeating the analysis using samples from the second examination showed E2/E6 ratios in non-progressors from 0.06 to 0.55 (median: 0.20) and in women with persistent infections who developed CIN3+ from 0.07 to 0.6 (median: 0.19; Table 4). Again no significant correlation between E2/E6 ratio and progression to CIN3+ was found (p=0.86; Figure 3C).
Interestingly an analysis of the E2/E6 ratio combined with the viral load quantification in samples from persistently infected women comparing the first and the second examination time point confirmed an increase of integrated genomes (E2/E6 ratio <0.15) over time. In addition a trend to lower E2/E6 ratios was visible when comparing samples from the first examination to samples from the second. While the E2/E6 ratio of HPV16 at the first examination ranged from 0-0.83 (median 0.53) with only 7.6% integrated genomes and 92.4% samples had a mix of integrated and probably episomal genomes (Figure 4) we observed at the second examination an E2/E6 ratio from 0.06 to 0.6 (median: 0.21) with 21.5% integrated genomes and only 78.4% mixed forms.
Figure 4.

Correlation of HPV16 viral load and physical status from persistently HPV16 infected women (N=79). HPV16 viral loads are displayed on the y-axis in copies/cells (c/c). The corresponding E2/E6 ratios are plotted on the x-axis. In contrast to samples taken at the first examination with only six samples with integrated genomes, samples taken at the next examination two years later revealed a higher number of samples with integrated HPV16 genomes (N=17).
When we performed receiver operating characteristic (ROC) curve analysis in order to analyze the utility of viral load or physical state measurements for diagnostic purposes predicting clearance or persistence of HPV16 infections we obtained comparable values for viral load (p=0.00028, AUC=0.662) and physical state (p<0.0001, AUC=0.6968; data not shown).
Similarly, a ROC curve analysis for the viral load and E2/E6 ratio predicting progression to CIN3+ showed that neither viral load (first examination: p=0.84, AUC=0.514; second examination: p=0.388, AUC=0.56) nor physical state (first examination: p=0.607, AUC=0.536; second examination: p=0.855, AUC=0.513) enabled discrimination between development of CIN3+ or not (data not shown).
Discussion
To measure viral load and the physical status of the genome we used quantitative real time PCR for the E6 and E2 genes of HPV16 and normalized the values to the cell number in each sample through additional measurement of the single copy gene IFNB1. This method is superior to semi-quantitative measurement of viral load by using e.g. the RLU/CO values of HC2, where the cell number of the analyzed sample is unknown and multiple infections can cause a viral load that is not attributable to a distinct HPV type or to dot blot analysis using estimated signal strength [37].
We find that HPV16 positive women who develop a persistent infection have a significantly lower median viral load (p=0.0003) at the first examination in comparison to women that are able to clear the infection. Interestingly a ROC curve analysis provided some evidence that indeed using low viral load could be possibly used as a diagnostic parameter. Moreover if HPV testing and genotyping would be combined with viral load measurement this may help to identify women with greatest risk for cervical cancer as persistence of infection is a major risk factor besides the prevailing HPV type [2]. The group of women with lower viral load already may have integrated viral genomes in their infected cells that do not support virus replication any longer. We observed integrated, a mixture of integrated and episomal forms and potentially episomal DNA in respectively 3.3%, 86.81% and 9.89% of the samples from the 91 women who cleared the infection. Samples from women who developed a persistent infection showed in contrast 7.59% integrated and 92.41% mixed HPV16 genomes and no episomal DNA. Our data confirm that integration can be an early event, because a high number of samples with normal cytology harboured already integrated HPV16 genomes. Those women with integrated genomes might have a higher risk of becoming persistently infected as fewer viral epitopes derived from E1 and especially E2 [38] are presented to the immune system compared to cells with episomal and potentially productive infections and therefore may be less likely eliminated by cell mediated immunity. In most cells with integrated HPV only the transforming genes E6/E7 are expressed and required for the transformed state. The expression of the viral oncogenes indeed seems to be tolerated by the immune system of some women as it has been shown earlier that the lack of response to E6 by cell mediated immunity is important in the persistence of an HPV16 infection [39].
The substantial overlap of the viral load measurements between women that cleared versus those that with persistent infections, however, makes it difficult to determine cut-off values for risk prediction.
In the present study we did not observe a significant difference in the median levels of viral load between women persistently infected with HPV16 who developed a CIN3+ or not during follow up. This is in line with some studies that found no association of higher viral load with cases, although others reported such an association [13, 40-42]. It has to be noted that we only genotyped women that were positive in the HC2 test using the high risk probe since women that test negative by the HC2 test high risk probe have virtually no risk of developing CIN3+ within 6 years of follow up [5]. Other studies that used more sensitive detection methods with no clinically validated cut-off might find stronger associations between higher viral load and high grade disease [12, 14-16]. In addition it is possible that at the second examination women already had cervical lesions that were not detected by cytology, as the average sensitivity of a single cytology screen is not exceeding 50% for detection of a HSIL lesion [43].
When we measured the E2/E6 ratio as a surrogate marker for the physical status of the viral genome we considered only ratios ≤ 0.15 as a reliable indicator for the presence of integrated genome copies as ratios above 0.9 might be indicative for an episomal state, but are not excluding the presence of integrated head to tail tandem genome copies. We observed that women that became persistently infected had a significantly lower median E2/E6 ratio (higher integration rate) in comparison to women that were able to clear the infection. A ROC curve analysis supported this finding showing that using an E2/E6 ratio of ≤ 0.15 could possibly be used as diagnostic parameter with a sensitivity of approximately 91%, but at the cost of a specificity as low as 48% (data not shown). One possible explanation for the low specificity of the E2/E6 ratio are findings by others and by us that already in 29%-89% of cytologically normal samples integrated viral genome copies could be found [11, 17, 35]. The suboptimal performance of the parameter “E2/E6 ratio” could also be due to the fact, that we did not exclude women that were infected by other HPV types with undefined physical status of the genome in addition to HPV16, which finally may have caused the CIN3+ lesion. However, when we repeated the analysis with just single HPV16 infections we observed no difference to our initial observation (data not shown). Again it can be hypothesized that cells with predominantly integrated viral genome copies are at lower risk to be cleared by cell mediated immunity and that the loss of the negative regulator E2 and especially E8^E2C [29] increases viral oncogene expression, which fosters persistence. In conclusion a combination of low viral load and an E2/E6 ratio of ≤ 0.15 may be a useful marker to attach to a single HPV test for the risk stratification of women in cervical cancer screening.
In contrast to other studies with a case control design we could not find a significant difference in the E2/E6 ratio between cases (women that developed CIN3+ during prospective follow up) and non-progressors. That was true for samples taken at the first and at the second phase. When we, however, plotted the viral load against E2/E6 ratio for the persistent cases and compared the results for the first versus the second examination it became obvious that a shift towards lower E2/E6 ratios was visible over time. This supports earlier findings from us [2] that HPV16 has the highest tendency for persistence among all hr HPV types, which might be due to a high integration rate.
In conclusion our data show that lower viral load at baseline and integration of the viral genome are predictive for the persistence of HPV16 DNA, but not for the progression of a persistent infection with HPV16 to development of CIN3+ during follow up in women with normal cytology at baseline.
Acknowledgments
This work was supported by the Mermaid project (MERMAID-2), SavvÆrksejer Jeppe Juhl og Hustru Ovita Juhls Mindelegat, and the Danish Cancer Society.
References
- 1.Clifford G, Franceschi S, Diaz M, Munoz N, Villa LL. Chapter 3: HPV type-distribution in women with and without cervical neoplastic diseases. Vaccine. 2006;3(24 Suppl):S3/26–34. doi: 10.1016/j.vaccine.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 2.Kjaer SK, Frederiksen K, Munk C, Iftner T. Long-term absolute risk of cervical intraepithelial neoplasia grade 3 or worse following human papillomavirus infection: role of persistence. J Natl Cancer Inst. 2010;102:1478–1488. doi: 10.1093/jnci/djq356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bulkmans NW, Berkhof J, Bulk S, Bleeker MC, van Kemenade FJ, Rozendaal L, Snijders PJ, Meijer CJ. High-risk HPV type-specific clearance rates in cervical screening. Br J Cancer. 2007;96:1419–1424. doi: 10.1038/sj.bjc.6603653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cuzick J, Szarewski A, Mesher D, Cadman L, Austin J, Perryman K, Ho L, Terry G, Sasieni P, Dina R, Soutter WP. Long-term follow-up of cervical abnormalities among women screened by HPV testing and cytology-Results from the Hammersmith study. Int J Cancer. 2008;122:2294–2300. doi: 10.1002/ijc.23339. [DOI] [PubMed] [Google Scholar]
- 5.Dillner J, Rebolj M, Birembaut P, Petry KU, Szarewski A, Munk C, de Sanjose S, Naucler P, Lloveras B, Kjaer S, Cuzick J, van Ballegooijen M, Clavel C, Iftner T. Long term predictive values of cytology and human papillomavirus testing in cervical cancer screening: joint European cohort study. BMJ. 2008;337:a1754. doi: 10.1136/bmj.a1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosch FX, Manos MM, Munoz N, Sherman M, Jansen AM, Peto J, Schiffman MH, Moreno V, Kurman R, Shah KV. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J Natl Cancer Inst. 1995;87:796–802. doi: 10.1093/jnci/87.11.796. [DOI] [PubMed] [Google Scholar]
- 7.Herrero R, Hildesheim A, Bratti C, Sherman ME, Hutchinson M, Morales J, Balmaceda I, Greenberg MD, Alfaro M, Burk RD, Wacholder S, Plummer M, Schiffman M. Population-based study of human papillomavirus infection and cervical neoplasia in rural Costa Rica. J Natl Cancer Inst. 2000;92:464–474. doi: 10.1093/jnci/92.6.464. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen A, Kjaer SK, Munk C, Osler M, Iftner T. Persistence of high-risk human papillomavirus infection in a population-based cohort of Danish women. J Med Virol. 2010;82:616–623. doi: 10.1002/jmv.21750. [DOI] [PubMed] [Google Scholar]
- 9.Schiffman M, Herrero R, Desalle R, Hildesheim A, Wacholder S, Rodriguez AC, Bratti MC, Sherman ME, Morales J, Guillen D, Alfaro M, Hutchinson M, Wright TC, Solomon D, Chen Z, Schussler J, Castle PE, Burk RD. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology. 2005;337:76–84. doi: 10.1016/j.virol.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Guo M, Sneige N, Silva EG, Jan YJ, Cogdell DE, Lin E, Luthra R, Zhang W. Distribution and viral load of eight oncogenic types of human papillomavirus (HPV) and HPV 16 integration status in cervical intraepithelial neoplasia and carcinoma. Mod Pathol. 2007;20:256–266. doi: 10.1038/modpathol.3800737. [DOI] [PubMed] [Google Scholar]
- 11.Briolat J, Dalstein V, Saunier M, Joseph K, Caudroy S, Pretet JL, Birembaut P, Clavel C. HPV prevalence, viral load and physical state of HPV-16 in cervical smears of patients with different grades of CIN. Int J Cancer. 2007;121:2198–2204. doi: 10.1002/ijc.22959. [DOI] [PubMed] [Google Scholar]
- 12.Cricca M, Morselli-Labate AM, Venturoli S, Ambretti S, Gentilomi GA, Gallinella G, Costa S, Musiani M, Zerbini M. Viral DNA load, physical status and E2/E6 ratio as markers to grade HPV16 positive women for high-grade cervical lesions. Gynecol Oncol. 2007;106:549–557. doi: 10.1016/j.ygyno.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Swan DC, Tucker RA, Tortolero-Luna G, Mitchell MF, Wideroff L, Unger ER, Nisenbaum RA, Reeves WC, Icenogle JP. Human papillomavirus (HPV) DNA copy number is dependent on grade of cervical disease and HPV type. J Clin Microbiol. 1999;37:1030–1034. doi: 10.1128/jcm.37.4.1030-1034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Josefsson AM, Magnusson PK, Ylitalo N, Sorensen P, Qwarforth-Tubbin P, Andersen PK, Melbye M, Adami HO, Gyllensten UB. Viral load of human papilloma virus 16 as a determinant for development of cervical carcinoma in situ: a nested case-control study. Lancet. 2000;355:2189–2193. doi: 10.1016/S0140-6736(00)02401-6. [DOI] [PubMed] [Google Scholar]
- 15.Ylitalo N, Sorensen P, Josefsson AM, Magnusson PK, Andersen PK, Ponten J, Adami HO, Gyllensten UB, Melbye M. Consistent high viral load of human papillomavirus 16 and risk of cervical carcinoma in situ: a nested case-control study. Lancet. 2000;355:2194–2198. doi: 10.1016/S0140-6736(00)02402-8. [DOI] [PubMed] [Google Scholar]
- 16.van Duin M, Snijders PJ, Schrijnemakers HF, Voorhorst FJ, Rozendaal L, Nobbenhuis MA, van den Brule AJ, Verheijen RH, Helmerhorst TJ, Meijer CJ. Human papillomavirus 16 load in normal and abnormal cervical scrapes: an indicator of CIN ll/lll and viral clearance. Int J Cancer. 2002;98:590–595. doi: 10.1002/ijc.10232. [DOI] [PubMed] [Google Scholar]
- 17.Boulet GA, Benoy IH, Depuydt CE, Horvath CA, Aerts M, Hens N, Vereecken AJ, Bogers JJ. Human papillomavirus 16 load and E2/E6 ratio in HPV16-positive women: biomarkers for cervical intraepithelial neoplasia >or=2 in a liquid-based cytology setting? Cancer Epidemiol Biomarkers Prev. 2009;18:2992–2999. doi: 10.1158/1055-9965.EPI-09-0025. [DOI] [PubMed] [Google Scholar]
- 18.Constandinou-Williams C, Collins SI, Roberts S, Young LS, Woodman CB, Murray PG. Is human papillomavirus viral load a clinically useful predictive marker? A longitudinal study. Cancer Epidemiol Biomarkers Prev. 2010;19:832–837. doi: 10.1158/1055-9965.EPI-09-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gravitt PE, Kovacic MB, Herrero R, Schiffman M, Bratti C, Hildesheim A, Morales J, Alfaro M, Sherman ME, Wacholder S, Rodriguez AC, Burk RD. High load for most high risk human papillomavirus genotypes is associated with prevalent cervical cancer precursors but only HPV16 load predicts the development of incident disease. Int J Cancer. 2007;121:2787–2793. doi: 10.1002/ijc.23012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xi LF, Hughes JP, Castle PE, Edelstein ZR, Wang C, Galloway DA, Koutsky LA, Kiviat NB, Schiffman M. Viral Load in the Natural History of Human Papillomavirus Type 16 Infection: A Nested Case-control Study. J Infect Dis. 2011;203:1425–1433. doi: 10.1093/infdis/jir049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hesselink AT, Berkhof J, Heideman DA, Bulkmans NW, van Tellingen JE, Meijer CJ, Snijders PJ. High-risk human papillomavirus DNA load in a population-based cervical screening cohort in relation to the detection of high-grade cervical intraepithelial neoplasia and cervical cancer. Int J Cancer. 2009;124:381–386. doi: 10.1002/ijc.23940. [DOI] [PubMed] [Google Scholar]
- 22.Hudelist G, Manavi M, Pischinger Kl, Watkins-Riedel T, Singer CF, Kubista E, Czerwenka KF. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: different levels of viral integration are correlated with lesion grade. Gynecol Oncol. 2004;92:873–880. doi: 10.1016/j.ygyno.2003.11.035. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Wang W, Si M, Han L, Gao Q, Luo A, Li Y, Lu Y, Wang S, Ma D. The physical state of HPV16 infection and its clinical significance in cancer precursor lesion and cervical carcinoma. J Cancer Res Clin Oncol. 2008;134:1355–1361. doi: 10.1007/s00432-008-0413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tonon SA, Picconi MA, Bos PD, Zinovich JB, Galuppo J, Alonio LV, Teyssie AR. Physical status of the E2 human papilloma virus 16 viral gene in cervical preneoplastic and neoplastic lesions. J Clin Virol. 2001;21:129–134. doi: 10.1016/s1386-6532(01)00155-x. [DOI] [PubMed] [Google Scholar]
- 25.Cricca M, Venturoli S, Leo E, Costa S, Musiani M, Zerbini M. Molecular analysis of HPV 16 E6I/E6II spliced mRNAs and correlation with the viral physical state and the grade of the cervical lesion. J Med Virol. 2009;81:1276–1282. doi: 10.1002/jmv.21496. [DOI] [PubMed] [Google Scholar]
- 26.Kulmala SM, Syrjanen SM, Gyllensten UB, Shabalova IP, Petrovichev N, Tosi P, Syrjanen KJ, Johansson BC. Early integration of high copy HPV16 detectable in women with normal and low grade cervical cytology and histology. J Clin Pathol. 2006;59:513–517. doi: 10.1136/jcp.2004.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collins SI, Constandinou-Williams C, Wen K, Young LS, Roberts S, Murray PG, Woodman CB. Disruption of the E2 gene is a common and early event in the natural history of cervical human papillomavirus infection: a longitudinal cohort study. Cancer Res. 2009;69:3828–3832. doi: 10.1158/0008-5472.CAN-08-3099. [DOI] [PubMed] [Google Scholar]
- 28.Desaintes C, Demeret C, Goyat S, Yaniv M, Thierry F. Expression of the papillomavirus E2 protein in HeLa cells leads to apoptosis. EMBO J. 1997;16:504–514. doi: 10.1093/emboj/16.3.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stubenrauch F, Straub E, Fertey J, Iftner T. The E8 repression domain can replace the E2 transactivation domain for growth inhibition of HeLa cells by papillomavirus E2 proteins. Int J Cancer. 2007;121:2284–2292. doi: 10.1002/ijc.22907. [DOI] [PubMed] [Google Scholar]
- 30.Gravitt PE, Peyton CL, Apple RJ, Wheeler CM. Genotyping of 27 human papillomavirus types by using LI consensus PCR products by a single-hybridization, reverse line blot detection method. J Clin Microbiol. 1998;36:3020–3027. doi: 10.1128/jcm.36.10.3020-3027.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kjaer S, Hogdall E, Frederiksen K, Munk C, van den Brule A, Svare E, Meijer C, Lorincz A, Iftner T. The absolute risk of cervical abnormalities in high-risk human papillomavirus-positive, cytologically normal women over a 10-year period. Cancer Res. 2006;66:10630–10636. doi: 10.1158/0008-5472.CAN-06-1057. [DOI] [PubMed] [Google Scholar]
- 32.Kjaer SK, Breugelmans G, Munk C, Junge J, Watson M, Iftner T. Population-based prevalence, type- and age-specific distribution of HPV in women before introduction of an HPV-vaccination program in Denmark. Int J Cancer. 2008;123:1864–1870. doi: 10.1002/ijc.23712. [DOI] [PubMed] [Google Scholar]
- 33.Peitsaro P, Johansson B, Syrjanen S. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J Clin Microbiol. 2002;40:886–891. doi: 10.1128/JCM.40.3.886-891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalantari M, Karlsen F, Kristensen G, Holm R, Hagmar B, Johansson B. Disruption of the E1 and E2 reading frames of HPV 16 in cervical carcinoma is associated with poor prognosis. Int J Gynecol Pathol. 1998;17:146–153. doi: 10.1097/00004347-199804000-00009. [DOI] [PubMed] [Google Scholar]
- 35.Saunier M, Monnier-Benoit S, Mauny F, Dalstein V, Briolat J, Riethmuller D, Kantelip B, Schwarz E, Mougin C, Pretet JL. Analysis of human papillomavirus type 16 (HPV16) DNA load and physical state for identification of HPV16-infected women with high-grade lesions or cervical carcinoma. J Clin Microbiol. 2008;46:3678–3685. doi: 10.1128/JCM.01212-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arias-Pulido H, Peyton CL, Joste NE, Vargas H, Wheeler CM. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J Clin Microbiol. 2006;44:1755–1762. doi: 10.1128/JCM.44.5.1755-1762.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gravitt PE, Burk RD, Lorincz A, Herrero R, Hildesheim A, Sherman ME, Bratti MC, Rodriguez AC, Helzlsouer KJ, Schiffman M. A comparison between real-time polymerase chain reaction and hybrid capture 2 for human papillomavirus DNA quantitation. Cancer Epidemiol Biomarkers Prev. 2003;12:477–484. [PubMed] [Google Scholar]
- 38.Brandsma JL, Shlyankevich M, Zhang L, Slade MD, Goodwin EC, Peh W, Deisseroth AB. Vaccination of rabbits with an adenovirus vector expressing the papillomavirus E2 protein leads to clearance of papillomas and infection. J Virol. 2004;78:116–123. doi: 10.1128/JVI.78.1.116-123.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakagawa M, Stites DP, Patel S, Farhat S, Scott M, Hills NK, Palefsky JM, Moscicki AB. Persistence of human papillomavirus type 16 infection is associated with lack of cytotoxic T lymphocyte response to the E6 antigens. J Infect Dis. 2000;182:595–598. doi: 10.1086/315706. [DOI] [PubMed] [Google Scholar]
- 40.Lillo FB, Lodini S, Ferrari D, Stayton C, Taccagni G, Galli L, Lazzarin A, Uberti-Foppa C. Determination of human papillomavirus (HPV) load and type in high-grade cervical lesions surgically resected from HIV-infected women during follow-up of HPV infection. Clin Infect Dis. 2005;40:451–457. doi: 10.1086/427032. [DOI] [PubMed] [Google Scholar]
- 41.Heard I, Tassie JM, Schmitz V, Mandelbrot L, Kazatchkine MD, Orth G. Increased risk of cervical disease among human immunodeficiency virus-infected women with severe immunosuppression and high human papillomavirus load(1) Obstet Gynecol. 2000;96:403–409. doi: 10.1016/s0029-7844(00)00948-0. [DOI] [PubMed] [Google Scholar]
- 42.Nindl I, Greinke C, Zahm DM, Stockfleth E, Hoyer H, Schneider A. Human papillomavirus distribution in cervical tissues of different morphology as determined by hybrid capture assay and PCR. Int J Gynecol Pathol. 1997;16:197–204. doi: 10.1097/00004347-199707000-00002. [DOI] [PubMed] [Google Scholar]
- 43.McCrory DC, Matchar DB, Bastian L, Datta S, Hasselblad V, Hickey J, Myers E, Nanda K. Evaluation of cervical cytology. Evid Rep Technol Assess (Summ) 1999:1–6. [PMC free article] [PubMed] [Google Scholar]