

Abstract
Both genetic and environmental interactions affect systemic lupus erythematosus (SLE) development and pathogenesis. One known genetic factor associated with lupus is a haplotype of the interferon regulatory factor 5 (IRF5) gene. Analysis of global gene expression microarray data using gene set enrichment analysis identified multiple interferon- and inflammation-related gene sets significantly overrepresented in cells with the risk haplotype. Pathway analysis using expressed genes from the significant gene sets impacted by the IRF5 risk haplotype confirmed significant correlation with the interferon pathway, Toll-like receptor pathway, and the B-cell receptor pathway. SLE patients with the IRF5 risk haplotype have a heightened interferon signature, even in an unstimulated state (P = 0.011), while patients with the IRF5 protective haplotype have a B cell interferon signature similar to that of controls. These results identify multiple genes in functionally significant pathways which are affected by IRF5 genotype. They also establish the IRF5 risk haplotype as a key determinant of not only the interferon response, but also other B-cell pathways involved in SLE.
1. Introduction
Systemic lupus erythematosus is a complex disease with multifactorial etiology and pathogenesis. Studies in identical twins indicate that concordance for lupus is approximately 40%, indicating a strong but not exclusive genetic component [1, 2]. Recent genetic analyses have identified more than thirty candidate genes that are associated with lupus risk [3–18]. IRF5 was found to be associated with lupus by multiple independent groups in a variety of populations [10, 13–15, 19, 20]. IRF5 risk haplotypes may function at the crossroads of environmental risk, such as virus infection, and cellular immune responses. At least three polymorphisms of IRF5 have been identified that contribute independently to the risk for lupus, which together constitute the lupus risk haplotype [10, 21]. Although the majority of the polymorphisms that have been associated with lupus are in nontranslated regions, they may affect several facets of IRF5 activity, including splicing, RNA stability, transcription factor binding, and apoptosis [9, 10, 15, 21, 22].
IRF5 is important in the production of and response to interferon alpha (IFNα), which is heightened in lupus. IFNα is produced by dendritic cells, macrophages, B cells, and other cell types, primarily in response to virus infection [23, 24]. Dendritic cells have been shown to produce IFNα in response to incubation with immune complex-containing sera from SLE patients [25], especially patients that have the risk haplotype for IRF5 [19]. Additionally, serum interferon levels, as well as the interferon response signature, are increased in patients with the risk haplotype [19, 26]. IRF5 is an especially interesting candidate for a genetic risk factor in lupus because it acts in pathways that control many of the cellular and immune responses to environmental factors, such as infection, which may contribute to lupus.
One putative environmental agent that is strongly associated with risk for lupus is Epstein-Barr virus (EBV) infection. Lupus has been associated with prior EBV infection in both pediatric and adult populations [27–33]. EBV expresses antigens that are immunologically cross-reactive with significant lupus autoantigens such as Sm and nRNP [34–39]. However, since over 95% of adults are infected with EBV, determining why EBV could contribute to lupus in certain individuals but not others has proven challenging. The identification of IRF5 and other genetic risk factors for lupus open the possibility that the lupus-associated genetic polymorphisms in one or more of these genes works in concert with environmental factors culminating in the increased observed risk for developing lupus. Previous work has shown that pediatric lupus patients have broadened, more cross-reactive humoral immune responses to EBV than controls [40]. EBV is also not as well controlled in lupus patients as it is in controls, with increased viral load and altered T-cell responses [41, 42]. Differences in viral infection or the response to viral infection conferred by genetic factors such as IRF5 polymorphisms may in part explain these observations.
Since B cells are the primary host cell for EBV infection, we used B cells and EBV interactions as a model to study the impact of IRF5 genotype on downstream B-cell responses. For this study, we examined differences in B-cell gene expression between naïve B cells from individuals with the IRF5 risk haplotype and those with the protective or neutral haplotypes at both basal levels and after exposure to EBV. Naïve B cells were chosen because they are the cell type in which EBV establishes latent infection [43]. We found multiple networks of genes that were enriched for differential expression, as well as individual gene expression differences. Most importantly, we identified different expression patterns of interferon response genes in lupus patients based on the IRF5 risk haplotype. Understanding these differences will aid in determining mechanisms through which the genetic risk conferred by the IRF5 risk haplotype is manifested.
2. Materials and Methods
2.1. Study Participants
Genotypes were previously collected on samples obtained from the Oklahoma Rheumatic Disease Resource Cores Center (ORDRCC) at the Oklahoma Medical Research Foundation. Previously enrolled subjects were contacted for study participation based upon their IRF5 risk and protective haplotypes using genotypes at single nucleotide polymorphisms rs2004640 and rs10954213. Five IRF5 high-risk (3 controls, 2 patients) and five IRF5 nonrisk (2 controls, 3 patients) sex- and race- matched individuals were recruited. The study was approved by the institutional review board at OMRF and OUHSC, and informed consent was obtained from all subjects in the study.
2.2. B-Cell Stimulation
Peripheral blood mononuclear cells were separated by density gradient centrifugation from the peripheral blood of volunteers. Naïve B cells were isolated using the MACS Naïve B Cell Isolation Kit II (Miltenyi Biotec Inc). Untouched naïve B cells were incubated at a 1 : 1 (v/v) ratio with either virus-free media or infectious EBV for 16 hours. Virus preparations were in the form of B95-8 cell culture supernatant. The same preparation of supernatant was used for all assays.
2.3. Gene Expression Profiling
Total cellular mRNA was purified from lysates of infected and mock-infected cells using the Ambion RNaqueous-Micro Kit (Applied Biosystems, Austin, TX, USA) according to the manufacturer's protocol and quantified using a NanoDrop spectrophotometer (NanoDrop Technologies, Inc.). cRNA amplification and labeling with biotin were performed using the Illumina TotalPrep RNA amplification kit protocol (Ambion, Austin, TX, USA) on an aliquot of 200 ng of total RNA. Whole genome expression analysis was performed using the Illumina HumanRef-8 v.3 gene expression chip (24,526 transcripts) following the Illumina Whole-Genome Expression Protocol.
2.4. Statistical and Pathway Analysis
The microarray data were analyzed using gene set enrichment analysis, and pathway analysis to investigate changes in gene networks. These analyses were followed by comparison of individual gene expression differences inside these networks. Raw expression data was first normalized using the MDAT toolbox [44]. Gene Set Enrichment Analysis software (Molecular Signatures Database) was used to determine whether an a priori functionally defined set of genes showed statistically significant, concordant differences between two phenotypes (IRF5 risk and nonrisk haplotypes) [45, 46]. Significant gene sets were identified by an enrichment score, which reflects the degree to which a gene set is overrepresented at the top or bottom of a ranked list of genes, and a false discovery rate (FDR) of <25%. We focused our subsequent pathway analysis on the subset of enriched genes (n = 368) from the statistically significant gene sets.
Pathway analyses were generated through the use of Ingenuity Pathways Analysis (Ingenuity Systems, http://www.ingenuity.com/). A data set containing gene identifiers and corresponding expression values was uploaded into in the application. Each gene identifier was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base. The expression values entered were the normalized log (intensity) values of IRF5 nonrisk and risk haplotype individuals, respectively.
Canonical pathways analysis identified the pathways from the Ingenuity Pathways Analysis library of canonical pathways that were most significant to the data set. The significance of the association between the data set and the canonical pathway was measured in two ways: (1) a ratio of the number of genes from the data set that map to the pathway divided by the total number of genes that map to the canonical pathway; (2) Fisher's exact test was used to calculate a P value determining the probability that the association between the genes in the dataset and the canonical pathway is explained by chance alone. All associations are supported by at least one reference from the literature, from a textbook, or from canonical information stored in the Ingenuity Pathways Knowledge Base. Human and mouse orthologs of a gene are stored as separate objects in the Ingenuity Pathways Knowledge Base, but are represented as a single node in the network. Heat maps were created with previously mentioned expression values in Spotfire software.
3. Results
3.1. Enrichment of Lupus-Related Gene Sets by IRF5 Risk Haplotype
Using existing genotyping data for single-nucleotide polymorphism rs2004640 (T: risk, G: protective) and rs10954213 (A: risk, G: protective) [10, 11, 20, 21, 47] the frequency of the risk, protective, and neutral IRF5 haplotypes were examined in 1,390 SLE patients and 2,039 controls enrolled in a large cohort of European American female (EA) SLE patients from the Lupus Family Registry and Repository (LFRR: http://lupus.omrf.org/). A significant enrichment for the risk and risk-neutral haplotypes was found in lupus patients (P < 0.0001), as expected (Table 1). Using these results, five individuals with the risk or risk-neutral haplotypes and five with the protective or protective-neutral haplotypes were recruited from the local SLE collections through the ORDRCC for further study. These included two SLE patients and three controls in the risk groups, and three SLE patients and two controls with protective and protective-neutral haplotypes. The risk group was enriched for controls so that the effects of the IRF5 haplotypes could be better studied in the absence of potential confounding genetic influences or factors related to lupus.
Table 1.
Frequency of IRF5 haplotypes in lupus patients and healthy controls. Haplotype frequencies observed in controls and systemic lupus erythematosus (SLE) patient cohort at single-nucleotide polymorphisms rs2004640 (T : risk, G: protective) and rs10954213 (A: risk, G: protective).
| Haplotype | Allele 1 | Allele 2 | Frequency (patients) n = 1390 | Frequency (controls) n = 2039 | SLE risk |
|---|---|---|---|---|---|
| 1 | TA | TA | 0.2576 | 0.1947 | Risk/risk |
| 2 | TA | TG | 0.4460 | <0.0001 | Risk/neutral |
| 3 | GA | GG | 0.2108 | 0.2737 | Protective/protective |
| 4 | GG | TG | 0.0165 | 0.1810 | Protective/neutral |
In all, 368 genes were found to be significantly (P < 0.05) differentially expressed between the risk and the nonrisk individuals. Gene set enrichment analysis was used to look for gene pathways overrepresented when comparing expression data from the risk haplotype and the protective haplotype phenotypic groups in the case or control groups separately. This analysis examines 6,769 a priori defined functional gene sets [46, 48]. In the unaffected controls with the risk haplotype, nine gene sets were enriched with a false discovery rate (FDR) <25% (Table 2), and 19 were enriched with a nominal P value <0.01, but an FDR >25% (Supplemental Table 2 which available online at doi:10.1155/2011/594056). In the lupus patients with the risk haplotype five gene sets were enriched with an FDR <25%. However, four of these were different versions of interferon-alpha gene sets. The fifth was the lupus-related interferon response signature (Table 2). Seven gene sets were enriched with a nominal P value <0.01 but a FDR >25% in the SLE risk haplotype cells, including the Toll-like receptor (TLR) gene set (Supplemental Table 1).
Table 2.
Effect of the IRF5 risk haplotype on the expression of gene sets. Gene set enrichment analysis showed gene sets enriched in the risk haplotypes of either SLE-unaffected controls or SLE patients without EBV infection. Genes shown have a false discovery rate (FDR) of <25%.
| Gene set name | Affected pathways or cellular conditions | No. of genes | Normalized enrichment score | P value | FDR q-value |
|---|---|---|---|---|---|
| Unaffected controls | |||||
|
| |||||
| CROONQUIST_IL6_STROMA_UP | IL-6 exposure | 37 | −2.026 | 0.0018 | 0.038 |
| PASSERINI_INFLAMMATION | Inflammation | 23 | −1.751 | 0.0112 | 0.212 |
| PASSERINI_PROLIFERATION | Proliferation | 62 | −1.782 | <0.0001 | 0.224 |
| ADIP_DIFF_CLUSTER2 | Differentiation | 41 | −1.752 | 0.004 | 0.225 |
| CROONQUIST_RAS_STROMA_DN | Ras activation | 21 | −1.757 | 0.0038 | 0.229 |
| UVB_NHEK3_C6 | UV light exposure | 27 | −1.763 | 0.0039 | 0.234 |
| HOHENKIRK_MONOCYTE_DEND_DN | Dendritic cell maturation | 121 | −1.724 | 0.0348 | 0.239 |
| LEE_DENA_UP | Murine liver cancer | 59 | −1.769 | <0.0001 | 0.241 |
| ZUCCHI_EPITHELIAL_DN | Breast cancer metastasis | 44 | −1.785 | 0.0117 | 0.245 |
|
| |||||
| SLE patients | |||||
|
| |||||
| IFNALPHA_HCC_UP | IFNα | 29 | −1.968 | 0.0038 | 0.042 |
| IFNALPHA_NL_HCC_UP | IFNα | 18 | −1.878 | 0.0096 | 0.063 |
| RADAEVA_IFNA_UP | IFNα | 49 | −1.897 | 0.0059 | 0.07 |
| IFNALPHA_NL_UP | IFNα | 27 | −1.847 | 0.0099 | 0.075 |
| BENNETT_SLE_UP | SLE | 28 | −1.785 | 0.0082 | 0.138 |
Fewer gene sets were as highly enriched in the protective haplotype cells. No gene sets were significantly enriched at an FDR level <25% in either the unaffected controls or the lupus patients with the protective haplotype. However, 39 gene sets were enriched at P < 0.01 in the SLE patients with the protective haplotype, and 35 gene sets were enriched at P < 0.01 level in the controls with the protective haplotype (Supplemental Tables 3 and 4).
3.2. Identification of Lupus-Related Pathways Differentially Affected by IRF5 Haplotype
Genes from the enriched gene sets described above which also demonstrated differences in expression in the previous analysis were included in a global pathway analysis using the Ingenuity Pathway Analysis system. This analysis uses the curated Ingenuity Knowledge Base to associate sets of genes and expression data with established gene pathways. Fisher's exact test was used to quantify the degree of association with these pathways. When the cells with the risk haplotype (both stimulated and unstimulated) were compared to those with the non-risk haplotypes, three canonical pathways were found with statistically significant (P < 0.01) association: the interferon (Figure 1), Toll-like receptor (Figure 2), and B-cell receptor (Figure 3) pathways. Interestingly, all three of these pathways have significant implications for lupus. These three canonical pathways and relative changes in expression following EBV exposure are represented in Figures 1–3 and Table 3.
Figure 1.
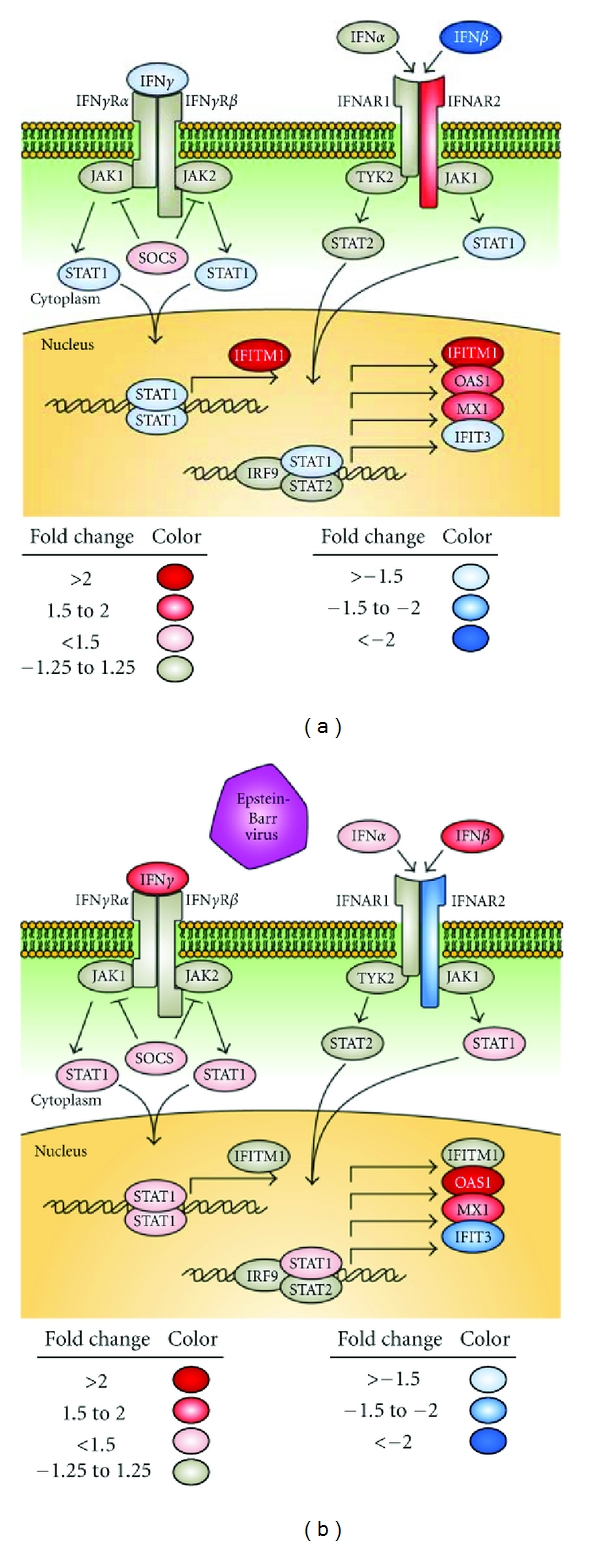
Association of the canonical interferon pathway with the IRF5 haplotype. Naïve B cells were either exposed to EBV for 16 hours or left unstimulated. Whole-genome expression levels were compared between cells with the risk and protective haplotypes using the Illumina platform. Significant association of the data set with the canonical IFN pathway was discovered using Ingenuity Pathway Analysis (P < 0.01). Interactions between genes in the interferon pathway based on Ingenuity Pathway Analysis are shown for both the unstimulated (a) and the EBV-infected conditions (b). Blue gene symbols represent genes with relatively lower expression in the cells with the risk haplotype, while genes with red shading are upregulated in the risk cells. IFIT3: IFN-induced protein with tetratricopeptide repeats 3, SOCS: suppressor of cytokine signaling.
Figure 2.
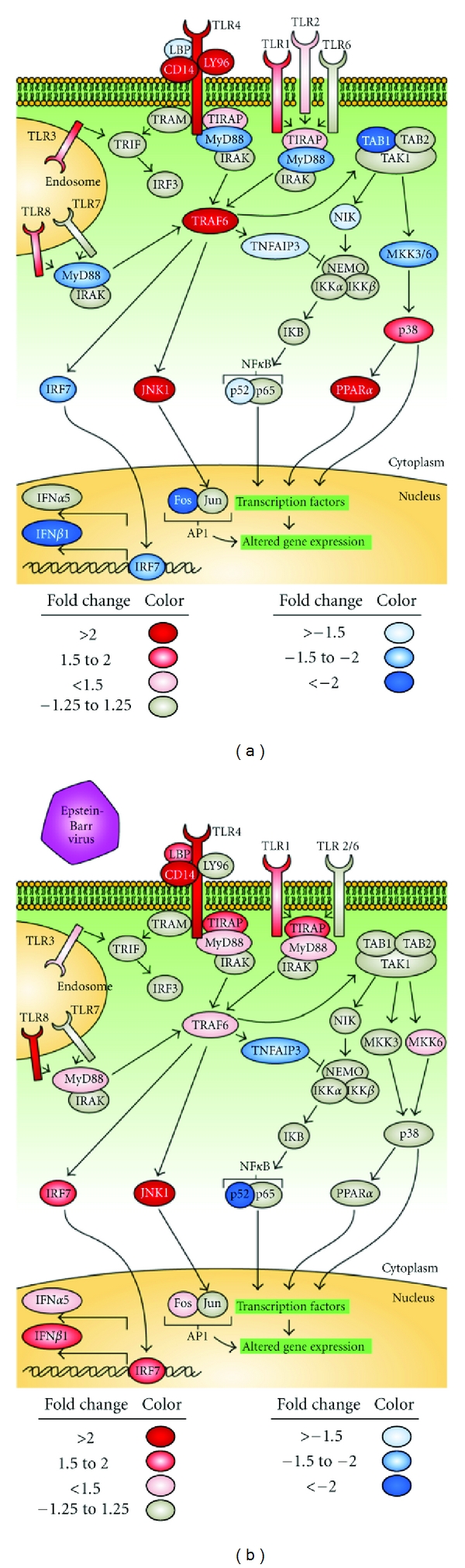
Association of the canonical Toll-like receptor pathway with IRF5 haplotype. Differential regulation of the TLR pathway was seen using Ingenuity analysis (P < 0.01). Unstimulated conditions are shown on the left, and EBV-exposed conditions on the right. Genes with altered expression based on haplotype are shown as either red or blue. IKB: inhibitor of NFκB, IKK: IKB kinase, JNK: c-Jun N-terminal kinase, LBP: lipopolysaccharide-binding protein, MKK: mitogen activated protein kinase kinase, NIK: NFκB inducing kinase, PPARα: peroxisome proliferator-activated receptor α, TAB1: transforming growth factor β-activated kinase 1, TIRAP: Toll/IL-1 receptor domain containing adaptor protein, and TRAF6: tumor necrosis factor receptor-associated factor 6.
Figure 3.
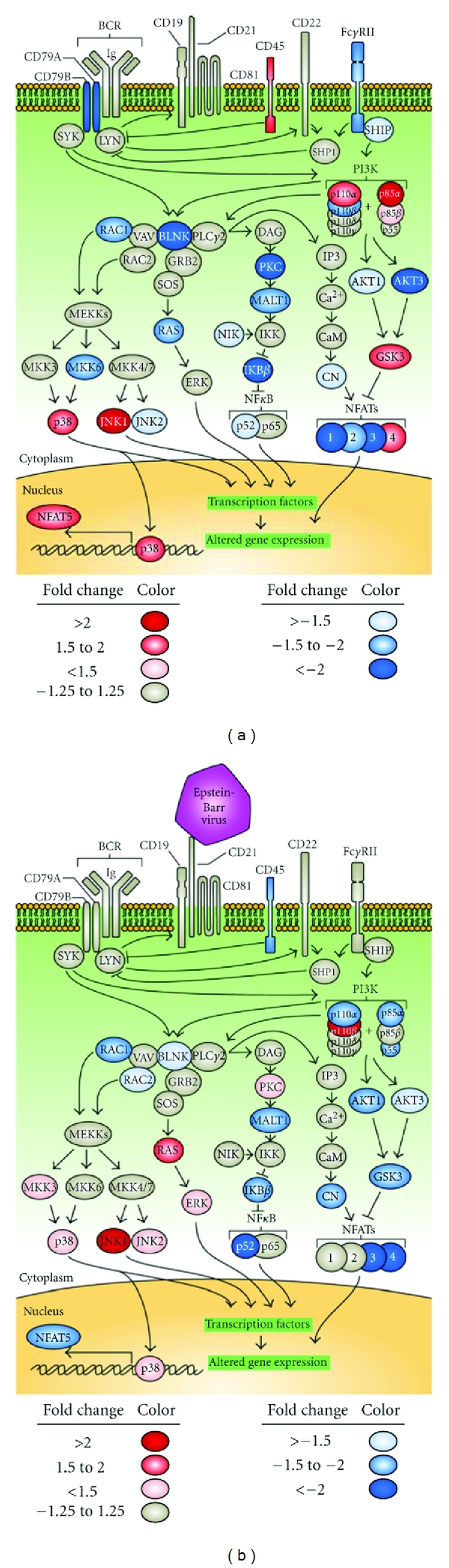
Association of the canonical B-cell receptor pathway with IRF5 haplotype. BCR pathway genes demonstrate a significant enrichment of changes in expression levels based on IRF5 haplytype, as determined by Ingenuity analysis (P < 0.01). The unstimulated condition is shown on the left, and the EBV-exposed condition is shown on the right. Genes with altered expression based on haplotype are shown as either blue or red. BLNK: B cell linker, CAM: Calmodulin, CN: Calcineurin, ERK: extracellular signal-regulated kinase, FCGR2B: fragment crystallizable γ receptor 2B, INPP5D: inositol polyphosphate-5-phosphatase D (SHIP), MALT1: mucosa associated lymphoid tissue lymphoma translocation gene 1, and PKC: protein kinase C.
Table 3.
Genes exhibiting differential expression between risk and nonrisk cells in the canonical pathways identified through ingenuity pathway analysis. Fold up/down column is positive in the case that the gene expression is higher in the risk haplotype cells, and negative in the case that gene expression is higher in the nonrisk cells. PIK3CA: phosphoinositide-3-kinase, catalytic, alpha; RAC1: Ras-related C3 botulinum toxin substrate 1. *The IFNB1 gene is found in both the interferon and Toll-like receptor pathways.
| Gene pathway | Gene symbol | Mock infected 16 hours | EBV live virus infected 16 hours | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Avg. nonrisk | Avg. risk | Ratio | Fold up/down | Avg. nonrisk | Avg. risk | Ratio | Fold up/down | ||
| Interferon | IFNB1* | 104.95 | 3.89 | 0.04 | −26.99 | 5.39 | 9.15 | 1.70 | 1.70 |
| STAT1 | 71.08 | 52.84 | 0.74 | −1.35 | 184.29 | 244.12 | 1.32 | 1.32 | |
| OAS1 | 56.04 | 84.55 | 1.51 | 1.51 | 49.87 | 107.78 | 2.16 | 2.16 | |
| MX1 | 3179.14 | 5161.02 | 1.62 | 1.62 | 3409.35 | 6199.20 | 1.82 | 1.82 | |
| IFNAR2 | 959.14 | 1637.11 | 1.71 | 1.71 | 1437.47 | 1094.97 | 0.76 | −1.31 | |
| IFITM1 | 439.88 | 944.41 | 2.15 | 2.15 | 2128.61 | 2602.68 | 1.22 | 1.22 | |
|
| |||||||||
| Toll-like receptor | IFNB1* | 104.95 | 3.89 | 0.04 | −26.99 | 5.39 | 9.15 | 1.70 | 1.70 |
| FOS | 116.48 | 53.84 | 0.46 | −2.16 | 90.05 | 120.60 | 1.34 | 1.34 | |
| MYD88 | 281.53 | 152.57 | 0.54 | −1.85 | 181.06 | 226.62 | 1.25 | 1.25 | |
| TNFAIP3 | 439.96 | 321.29 | 0.73 | −1.37 | 728.35 | 377.23 | 0.52 | −1.93 | |
| TLR1 | 26.50 | 46.76 | 1.76 | 1.76 | 67.59 | 116.37 | 1.72 | 1.72 | |
| LY96 (MD-2) | 560.79 | 1274.32 | 2.27 | 2.27 | 1128.47 | 1208.54 | 1.07 | 1.07 | |
| CD14 | 401.17 | 1272.53 | 3.17 | 3.17 | 139.53 | 450.09 | 3.23 | 3.23 | |
|
| |||||||||
| B-Cell receptor | CD79B | 2222.90 | 991.48 | 0.45 | −2.24 | 797.14 | 827.44 | 1.04 | 1.04 |
| CD79A | 41.32 | 19.08 | 0.46 | −2.17 | 6526.62 | 5973.72 | 0.92 | −1.09 | |
| RAC1 | 1407.29 | 930.84 | 0.66 | −1.51 | 4.36 | 2.64 | 0.61 | −1.65 | |
| MAPK9 | 105.87 | 71.02 | 0.67 | −1.49 | 62.50 | 80.35 | 1.29 | 1.29 | |
| AKT1 | 512.79 | 353.65 | 0.69 | −1.45 | 13.02 | 6.74 | 0.52 | −1.93 | |
| NFKB2 | 6.44 | 4.85 | 0.75 | −1.33 | 495.75 | 147.18 | 0.30 | −3.37 | |
| PIK3CA | 25.86 | 40.05 | 1.55 | 1.55 | 79.96 | 49.71 | 0.62 | −1.61 | |
| NFAT5 | 123.74 | 222.63 | 1.80 | 1.80 | 5.14 | 2.70 | 0.52 | −1.91 | |
| GSK3B | 68.58 | 123.65 | 1.80 | 1.80 | 177.23 | 91.07 | 0.51 | −1.95 | |
3.3. Identification of Individual Genes Differentially Expressed by IRF5 Haplotype
Several genes in the interferon pathway exhibited differential expression between either the risk and protective haplotypes or the EBV exposed and unexposed conditions. Genes with differential expression were selected based on inclusion in a significantly associated pathway, average expression values of at least thirty for one condition, and differential expression of at least 1.5-fold. Differential expression comparisons were done both with the unstimulated and the EBV-infected states (Table 3). Several genes were differentially expressed in the interferon pathway, including interferon-induced transmembrane protein 1 (IFITIM1), signal transducer and activator of transcription 1 (STAT1), IFNα receptor 2 (IFNAR2), 2′–5′-oligoadenylate synthetase 1 (OAS1), and MX1. The expression patterns of these genes varied based on IRF5 haplotype and EBV infection status (Figure 1).
IFITM1 was more strongly expressed in the risk cells than in the nonrisk in the unstimulated condition (2.1-fold difference). When the cells were exposed to EBV, this difference disappeared, a result of a greater increase in expression (4.8-fold) in the nonrisk cells than the risk (2.8-fold). STAT1 was slightly underexpressed in the risk cells than in the nonrisk prior to EBV exposure (1.3-fold), but after EBV exposure it was more highly expressed in the risk cells (1.3-fold). IFNAR2 acted in the opposite manner; its expression was higher in the risk cells in the unstimulated condition (1.7-fold), but higher in the nonrisk cells after EBV infection (1.3-fold). IFNAR2 is an interferon receptor that contributes directly to the response to interferon, making this gene very interesting in the context of interferon regulation and responsiveness. OAS1 was overexpressed in the risk cells compared to the nonrisk cells in both the unstimulated and EBV-exposed conditions (1.5-fold and 2.2-fold, resp.), as was MX1 (1.6- and 1.8-fold).
The TLR pathway also contained several genes that were differentially expressed between the risk and protective haplotype-containing cells (Figure 2). Fos and myeloid differentiation primary response gene 88 (MyD88) are both under expressed in the unstimulated risk cells compared to the nonrisk (2.2- and 1.8-fold, resp.). Both of these genes switch from being downregulated in the risk cells before EBV exposure to upregulated in the risk cells after EBV exposure (1.3- and 1.2-fold, resp.). Another very interesting gene that was differentially expressed in the TLR pathway is tumor necrosis factor α-induced protein 3 (TNFAIP3). It is under expressed by 1.3-fold in the risk cells in the resting condition. After EBV exposure, expression is even more unbalanced, with 1.9-fold under expression in the risk cells. Genes of interest that are overexpressed in the risk cells in the TLR pathway without EBV exposure include CD14 (3.2-fold), lymphocyte antigen 96 (LY96, or MD-2) (2.3-fold), and TLR1 (1.7-fold).
The B-cell receptor (BCR) pathway exhibited differences in gene expression due to the IRF5 risk haplotype (Figure 3).
CD79A and CD79B, which together form part of the BCR, were both downregulated 2.2-fold in the uninfected risk cells, but this difference disappeared after EBV infection. Ras-related C3 botulinum toxin substrate 1 (RAC1), a gene involved in lymphocyte differentiation and survival [49], was overexpressed in risk cells under all conditions (1.5-fold in mock infected cells, 1.7-fold in EBV infected cells). Expression of the signaling protein AKT1 and the transcription factor NFκB2 were downregulated in EBV infected cells with the IRF5 risk haplotype (1.9- and 3.4-fold, resp.). In three other genes, phosphatidylinositol 3 kinase catalytic subunit α (PIK3CA), nuclear factor of activated T cells 5 (NFAT5), and glycogen synthase kinase 3β (GSK3B), the risk haplotype had a 1.5- to 1.8-fold increase in expression. However, when EBV was present, the risk haplotype showed a decrease of 1.6- to 1.9-fold of the same genes.
3.4. The Interferon Response Signature in Patients Depends on Haplotype
Lupus patients have a heightened interferon response signature in the peripheral blood [50–52]. This signature is heritable and is associated with the IRF5 risk haplotype [19, 26]. When we examined genes included in the interferon response signature, we found an interesting association with the IRF5 risk haplotype. Cells from the SLE patients with the risk haplotype had an interferon response signature under all conditions, whether exposed to EBV or not. However, the cells from SLE patients with the protective haplotype did not exhibit an interferon signature without EBV infection. The difference in expression of the interferon response genes between the unstimulated patient risk and the unstimulated patient protective cells was statistically significant (P = 0.011) (Figure 4). The risk haplotype cells derived from control individuals did not have heightened baseline expression of interferon response genes. After exposure to EBV, these cells developed an interferon response signature that was similar to that seen in the baseline and EBV-infected risk-haplotype lupus patients. Interestingly, the patients with the protective haplotype did not develop a strong interferon response signature even after exposure to EBV, indicating that the IRF5 protective haplotype is dampening the response to interferon compared to the risk haplotype (Figure 4).
Figure 4.
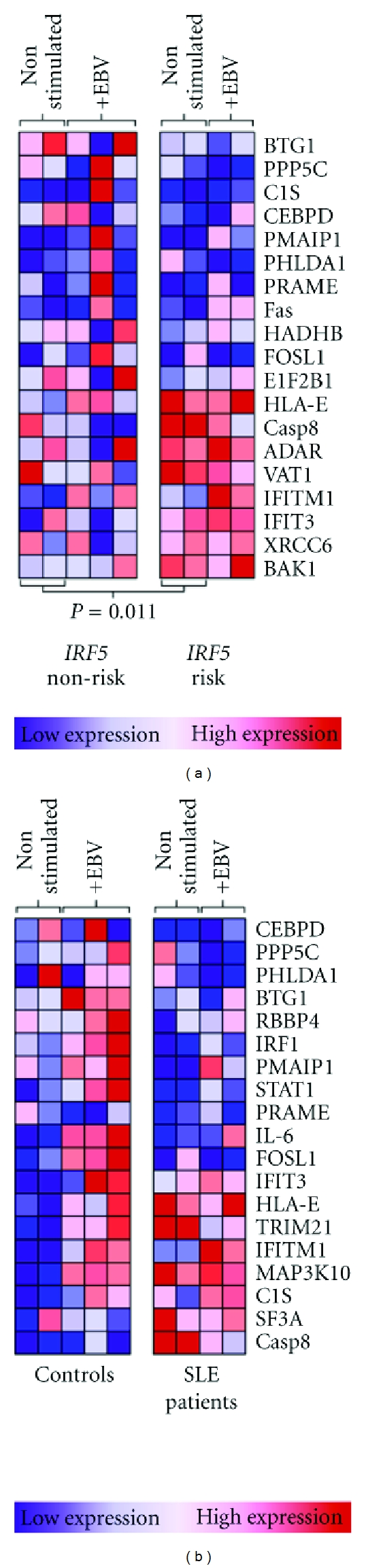
Dependence of the interferon response signature on IRF5 haplotype in SLE patients. Interferon signature genes identified in gene set enrichment analysis were compared between patients and controls, and between lupus patients with the IRF5 risk and nonrisk haplotypes. Each column represents expression of genes of interest in individual subjects. (a) Interferon signature genes of 3 IRF5 nonrisk and 2 IRF5 risk SLE patients. Gene expression comparisons are between IRF5 risk and nonrisk SLE patients, either nonstimulated or EBV infected. (b) Gene set enrichment of 3 control IRF5 risk haplotype individuals and 3 SLE patient IRF5 risk haplotype individuals. Gene expression comparisons between IRF5 risk patients and controls from nonstimulated and EBV-infected B cells. ADAR: adenosine deaminase, BAK: Bcl2-antagonist/killer, BTG: B cell translocation gene, C1S: complement component 1S, CASP: caspase, CEBPD: CCAAT/enhancer-binding protein δ, eIF2B: eukaryotic translation initiation factor 2B, FOSL: Fos-related antigen, HADHB: hydroxyacyl-Coenzyme A dehydrogenase/3-ketoacyl-Coenzyme A thiolase/enoyl-Coenzyme A hydratase β subunit, HLA: human leukocyte antigen, PHLDA: pleckstrin homology-like domain family A, PMAIP1: phorbol-12-myristate-13-acetate-induced protein 1 (Noxa), PRAME: melanoma antigen preferentially expressed in tumors, RBBP: retinoblastoma binding protein, SF3A: splicing factor 3A, TRIM: tripartite motif, VAT: vesicle amine transport, and XRCC: X-ray repair cross-complementing (Ku70).
4. Discussion
The IRF5 gene has been associated with risk for lupus. These findings demonstrate that the lupus-associated polymorphisms in the IRF5 gene have wide-reaching effects on B-cell responses to infection. The gene sets that were enriched in the risk haplotypes included interferon-related sets, which is encouraging considering that the genotype being examined is IRF5. Multiple gene sets that are related to lupus were enriched in the cells with the risk haplotypes, including IFNα sets, interleukin- (IL-) 6, inflammation, proliferation, and monocyte and dendritic cell genes, in addition to the SLE-related interferon gene set. The finding that these gene sets are the most strongly enriched in the risk haplotype indicates that the IRF5 risk haplotype has a strong influence on interferon signaling and inflammation, processes that are at the core of SLE. The finding that the most enriched gene sets were associated with interferon and lupus also indicates that these results are unlikely to be false positives obtained by chance, since the variable being studied is an interferon-affecting gene.
The gene set enrichment analysis techniques that were used are valuable because they identify not only individual genes, but also how strongly pathways that include those genes and the interactions between them are affected by the experimental conditions. This allows a much broader look into gene networks than looking only at individual genes. These studies point to the IRF5 risk haplotype having a wide influence on interferon and inflammation. The results identify targets for future investigation into the function of the IRF5 polymorphisms as well as other genetic influences on lupus.
As was the case with the gene set enrichment analysis, the identification of the interferon and Toll-like receptor pathways through Illumina pathway analysis suggests that the results are robust, as these are pathways that would be expected to be modulated by the underlying IRF5 haplotype of the donor. Interferon alpha is an extremely important cytokine in lupus [53]. These studies suggest that the interferon alpha pathway is strongly affected by genetic variation in the IRF5 gene, and show multiple genes that could potentially be targets for understanding interferon in lupus or potential therapeutic targets. Toll-like receptors are involved in response to infection through the recognition of pathogen-associated molecular patterns. Additionally, Toll-like receptors are important in the pathogenesis of lupus. They are capable of recognizing endogenous nucleic acids in the context of immune complexes found in lupus patient sera, thereby stimulating dendritic cell maturation and interferon alpha production [25, 54–60], a process which also involves IRF5 itself [25]. IRF5 is a very interesting transcriptional regulator in that it acts as both an activator when homodimerized and blocks activation when heterodimerized with IRF7 [61, 62]. This mechanism of action may help to explain how some of these pathways can exhibit relative upregulation or downregulation depending on the other conditions in the cell.
One of the more unexpected findings of this study was the modulation of the B-cell receptor pathway by the IRF5 haplotype. The B-cell receptor is important in the recognition of antigen and the survival, maturation, and proliferation of B cells. B cells produce the autoantibodies involved in lupus, as well as being important for antigen processing and presentation and T-cell activation. Differences in the activation threshold or other effects that may be seen with altered B-cell receptor gene expression may be very important to breaking self-tolerance or other aspects of B cell biology involved in SLE. Of particular interest in this respect is the recent finding that IRF5 controls antibody class switching to IgG2A, allowing lupus-like autoimmunity in mice [63]. The B-cell receptor and Toll-like receptor pathways are involved in antibody class switching, and the genes that were modulated by IRF5 variation in this study could represent mechanisms through which the IRF5 risk haplotype may contribute to class switching or other similar variations in humans.
The interferon response signature has been identified as a common feature in lupus. These studies examined how polymorphisms in the IRF5 gene affected the interferon response signature in both patients and controls. Interestingly, SLE patients with the risk haplotype demonstrated an interferon signature in both the infected and uninfected cells, while a strong interferon response was not found in the patients without the risk haplotype even when stimulated by EBV exposure. The controls with the risk haplotype lacked the interferon response signature in the basal state, but developed it after exposure to EBV, as would be expected. These findings suggest that the IRF5 risk haplotype is integral for the interferon response signature in both patients and controls. They also indicate that other factors contribute to a basal interferon response in lupus patients, since the IRF5 risk haplotype was not sufficient for the response signature to be present in the unstimulated control cells, as it was in the patients with the risk haplotype.
Cells were infected with EBV for two reasons. The first was to identify differences in gene expression patterns when cells were stimulated with a biologically relevant trigger for interferon production. The second reason was to identify areas that may start to explain the differences in EBV infection and response in lupus. Gene expression was examined for genes in the three pathways found to be significant by ingenuity analysis. In several cases, (IFITM1, IFNAR2, LY96, PIK3CA, NFAT5, and GSK3B) the baseline level of gene expression was higher in the risk cells, but after EBV infection, the gene expression was comparatively increased in the protective cells. In other genes, including CD79A, CD79B, STAT1, MyD88, and Fos, expression was lower in the risk cells but the difference diminished or reversed after EBV infection. Expression of one gene, TNFAIP3, was lower in the risk than in the protective haplotype subject unstimulated cells and was comparatively diminished further after EBV infection. These differences suggest several areas of investigation to understand differences in B cell biology in lupus and show that the IRF5 haplotype affects multiple genes related to EBV infection and response.
Although a detailed analysis of each gene involved in these pathways is beyond the scope of this paper, the genes with expression differences between the risk and protective haplotypes are suggestive in several instances. One of the genes identified with promise to affect lupus is TNFAIP3. This gene is a transcription factor that is produced in response to inflammation. It has been shown to be critical to limiting inflammation by terminating NFκB responses [64].Variants have recently been associated with risk for lupus and other autoimmune and inflammatory diseases [65–71], and it is often suppressed in tumors, especially lymphomas [66, 72, 73]. Other promising genes identified by these experiments include STAT4, IFITM1, and IFNAR2, which are all involved in the response to interferon, and several B cell signaling genes, including NFAT5, GSK3B, and NFκB2.
Although EBV was used in part to simulate an infected state in B cells, EBV itself could be involved in the etiology of lupus by affecting several pathways. The three pathways identified here are all involved in EBV infection. EBV may stimulate these pathways through several mechanisms, including both infection and binding of virions to the receptors involved in these pathways. Although the effect of EBV infection on differential gene expression was somewhat variable, for many of the genes examined in this study there was overexpression in the risk cells, which subsequently diminished after EBV infection. This pattern, as well as that seen with the interferon response signature, suggests that the IRF5 risk haplotype makes these cells appear more activated in the resting state. Because of this heightened activation state, there is less difference in the response to EBV infection in the risk cells, with the nonrisk cells often catching up to or passing the risk cells in expression of several genes following viral infection. An activated basal state would be likely to promote inappropriate cellular responses and possibly heightened sensitivity to self-antigens, including those recognized by TLRs.
These findings identify several key pathways that are affected by the IRF5 risk haplotype and are involved in the B cell response to antigen stimulation and viral infection. Many of the genes involved in these pathways have definite potential to alter the response to EBV infection and affect the development of lupus. These merit further investigation. Since all of these pathways are likely to be involved in the development of lupus, further comparison of these pathways in other cell types such as plasmacytoid dendritic cells will be beneficial to understanding the origins and pathogenesis of lupus. It will also be beneficial to examine more closely the role of EBV in regulating expression of these genes, through the use of EBV mutants, and to dissect the role of IRF5 in each pathway and gene set identified.
Supplementary Material
The supplemental data contains gene sets for SLE risk or protective allele groups that were significantly enriched at p < 0.01, but did not cross the threshold of 25% FDR (Tables 1 and 2). Tables 3 and 4 include the gene sets that were enriched in the non–risk cells compared to the risk cells. Also included in the supplemental tables are all of the gene sets that were significantly enriched for each group after EBV infection (Table 5).
Acknowledgments
This work was funded by the National Institutes of Health Grants P30 AR053483 (J. James and J. Guthridge), P20 RR015577 (J. James), N01 AI50026 (J. James and J. Guthridge), R01 AI063274 and R01 AR056360 (P. Gaffney), R3724717 (J. Harley), AR042460 (J. Harley), P01 AI083194 (J. Harley), P20 RR020143 (P. Gaffney), R01 DE018209 (J. Harley), and the Office for Human Research Studies award no. HR08–037 from the Oklahoma Center for the Advancement of Science & Technology (J. Guthridge).
References
- 1.Deapen D, Escalante A, Weinrib L, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis & Rheumatism. 1992;35(3):311–318. doi: 10.1002/art.1780350310. [DOI] [PubMed] [Google Scholar]
- 2.Block SR. A brief history of twins. Lupus. 2006;15(2):61–64. doi: 10.1191/0961203306lu2263ed. [DOI] [PubMed] [Google Scholar]
- 3.Guo L, Deshmukh H, Lu R, et al. Replication of the BANK1 genetic association with systemic lupus erythematosus in a European-derived population. Genes & Immunity. 2009;10(5):531–538. doi: 10.1038/gene.2009.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han S, Kim-Howard X, Deshmukh H, et al. Evaluation of imputation-based association in and around the integrin-α-M (ITGAM) gene and replication of robust association between a non-synonymous functional variant within ITGAM and systemic lupus erythematosus (SLE) Human Molecular Genetics. 2009;18(6):1171–1180. doi: 10.1093/hmg/ddp007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu K, Li QZ, Delgado-Vega AM, et al. Kallikrein genes are associated with lupus and glomerular basement membrane-specific antibody-induced nephritis in mice and humans. The Journal of Clinical Investigation. 2009;119(4):911–923. doi: 10.1172/JCI36728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu R, Vidal GS, Kelly JA, et al. Genetic associations of LYN with systemic lupus erythematosus. Genes & Immunity. 2009;10(5):397–403. doi: 10.1038/gene.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Namjou B, Sestak AL, Armstrong DL, et al. High-density genotyping of STAT4 reveals multiple haplotypic associations with systemic lupus erythematosus in different racial groups. Arthritis & Rheumatism. 2009;60(4):1085–1095. doi: 10.1002/art.24387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozyrev SV, Abelson AK, Wojcik J, et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nature Genetics. 2008;40(2):211–216. doi: 10.1038/ng.79. [DOI] [PubMed] [Google Scholar]
- 9.Demirci FY, Manzi S, Ramsey-Goldman R, et al. Association of a Common Interferon Regulatory Factor 5 (IRF5) variant with increased risk of Systemic Lupus Erythematosus (SLE) Annals of Human Genetics. 2007;71(part 3):308–311. doi: 10.1111/j.1469-1809.2006.00336.x. [DOI] [PubMed] [Google Scholar]
- 10.Graham RR, Kyogoku C, Sigurdsson S, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(16):6758–6763. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nature Genetics. 2008;40(2):204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufman KM, Kelly JA, Herring BJ, et al. Evaluation of the genetic association of the PTPN22 R620W polymorphism in familial and sporadic systemic lupus erythematosus. Arthritis & Rheumatism. 2006;54(8):2533–2540. doi: 10.1002/art.21963. [DOI] [PubMed] [Google Scholar]
- 13.Kawasaki A, Kyogoku C, Ohashi J, et al. Association of IRF5 polymorphisms with systemic lupus erythematosus in a Japanese population: support for a crucial role of intron 1 polymorphisms. Arthritis & Rheumatism. 2008;58(3):826–834. doi: 10.1002/art.23216. [DOI] [PubMed] [Google Scholar]
- 14.Kelly JA, Kelley JM, Kaufman KM, et al. Interferon regulatory factor-5 is genetically associated with systemic lupus erythematosus in African Americans. Genes & Immunity. 2008;9(3):187–194. doi: 10.1038/gene.2008.4. [DOI] [PubMed] [Google Scholar]
- 15.Kozyrev SV, Alarcon-Riquelme ME. The genetics and biology of Irf5-mediated signaling in lupus. Autoimmunity. 2007;40(8):591–601. doi: 10.1080/08916930701510905. [DOI] [PubMed] [Google Scholar]
- 16.Moser KL, Neas BR, Salmon JE, et al. Genome scan of human systemic lupus erythematosus: evidence for linkage on chromosome 1q in African-American pedigrees. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(25):14869–14874. doi: 10.1073/pnas.95.25.14869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moser KL, Kelly JA, Lessard CJ, Harley JB. Recent insights into the genetic basis of systemic lupus erythematosus. Genes & Immunity. 2009;10(5):373–379. doi: 10.1038/gene.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harley ITW, Kaufman KM, Langefeld CD, Harley JB, Kelly JA. Genetic susceptibility to SLE: new insights from fine mapping and genome-wide association studies. Nature Reviews Genetics. 2009;10(5):285–290. doi: 10.1038/nrg2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-α activity in systemic lupus erythematosus patients. Arthritis & Rheumatism. 2008;58(8):2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reddy MV, Velázquez-Cruz R, Baca V, et al. Genetic association of IRF5 with SLE in Mexicans: higher frequency of the risk haplotype and its homozygozity than Europeans. Human Genetics. 2007;121(6):721–727. doi: 10.1007/s00439-007-0367-6. [DOI] [PubMed] [Google Scholar]
- 21.Graham RR, Kozyrev SV, Baechler EC, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nature Genetics. 2006;38(5):550–555. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 22.Sigurdsson S, Nordmark G, Garnier S, et al. A risk haplotype of STAT4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Human Molecular Genetics. 2008;17(18):2868–2876. doi: 10.1093/hmg/ddn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin HJ, Jae JM, Walls D, Hayward SD. Manipulation of the toll-like receptor 7 signaling pathway by Epstein-Barr virus. Journal of Virology. 2007;81(18):9748–9758. doi: 10.1128/JVI.01122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnes BJ, Richards J, Mancl M, Hanash S, Beretta L, Pitha PM. Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. The Journal of Biological Chemistry. 2004;279(43):45194–45207. doi: 10.1074/jbc.M400726200. [DOI] [PubMed] [Google Scholar]
- 25.Yasuda K, Richez C, Maciaszek JW, et al. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes Is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. The Journal of Immunology. 2007;178(11):6876–6885. doi: 10.4049/jimmunol.178.11.6876. [DOI] [PubMed] [Google Scholar]
- 26.Niewold TB, Hua J, Lehman TJA, Harley JB, Crow MK. High serum IFN-α activity is a heritable risk factor for systemic lupus erythematosus. Genes & Immunity. 2007;8(6):492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harley BJ, Harley ITW, Guthridge JM, James JA. The curiously suspicious: a role for Epstein-Barr virus in lupus. Lupus. 2006;15(11):768–777. doi: 10.1177/0961203306070009. [DOI] [PubMed] [Google Scholar]
- 28.Harley JB, James JA. Epstein-Barr virus infection induces lupus autoimmunity. Bulletin of the NYU Hospital for Joint Diseases. 2006;64(1-2):45–50. [PubMed] [Google Scholar]
- 29.James JA, Harley JB. Epstein-Barr virus infection may be an environmental risk factor for SLE in children and teenagers. Arthritis & Rheumatism. 1999;42:1122–1126. doi: 10.1002/1529-0131(199908)42:8<1782::aid-anr36>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 30.James JA, Harley JB, Scofield RH. Epstein-Barr virus and systemic lupus erythematosus. Current Opinion in Rheumatology. 2006;18(5):462–467. doi: 10.1097/01.bor.0000240355.37927.94. [DOI] [PubMed] [Google Scholar]
- 31.James JA, Kaufman KM, Farris AD, Taylor-Albert E, Lehman TJA, Harley JB. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. The Journal of Clinical Investigation. 1997;100(12):3019–3026. doi: 10.1172/JCI119856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.James JA, Neas BR, Moser KL, et al. Systemic lupus erythematosus in adults is associated with previous Epstein-Barr virus exposure. Arthritis & Rheumatism. 2001;44(5):1122–1126. doi: 10.1002/1529-0131(200105)44:5<1122::AID-ANR193>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 33.Lu JJY, Chen DY, Hsieh CW, Lan JL, Lin FJ, Lin SH. Association of Epstein-Barr virus infection with systemic lupus erythematosus in Taiwan. Lupus. 2007;16(3):168–175. doi: 10.1177/0961203306075800. [DOI] [PubMed] [Google Scholar]
- 34.James JA, Scofield RH, Harley JB. Lupus humoral autoimmunity after short peptide immunization. Annals of the New York Academy of Sciences. 1997;815:124–127. doi: 10.1111/j.1749-6632.1997.tb52054.x. [DOI] [PubMed] [Google Scholar]
- 35.Poole BD, Scofield RH, Harley JB, James JA. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity. 2006;39(1):63–70. doi: 10.1080/08916930500484849. [DOI] [PubMed] [Google Scholar]
- 36.Sabbatini A, Bombardieri S, Migliorini P. Autoantibodies from patients with systemic lupus erythematosus bind a shared sequence of SmD and Epstein-Barr virus-encoded nuclear antigen EBNA I. European Journal of Immunology. 1993;23(5):1146–1152. doi: 10.1002/eji.1830230525. [DOI] [PubMed] [Google Scholar]
- 37.Sabbatini A, Dolcher MP, Marchini B, Bombardieri S, Migliorini P. Mapping of epitopes on the SmD molecule: the use of multiple antigen peptides to measure autoantibodies in systemic lupus erythematosus. The Journal of Rheumatology. 1993;20(10):1679–1683. [PubMed] [Google Scholar]
- 38.Sundar K, Jacques S, Gottlieb P, et al. Expression of the Epstein-Barr virus nuclear antigen-1 (EBNA-1) in the mouse can elicit the production of anti-dsDNA and anti-Sm antibodies. Journal of Autoimmunity. 2004;23(2):127–140. doi: 10.1016/j.jaut.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Poole BD, Gross T, Maier S, Harley JB, James JA. Lupus-like autoantibody development in rabbits and mice after immunization with EBNA-1 fragments. Journal of Autoimmunity. 2008;31(4):362–371. doi: 10.1016/j.jaut.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McClain MT, Poole BD, Bruner BF, Kaufman KM, Harley JB, James JA. An altered immune response to Epstein-Barr nuclear antigen 1 in pediatric systemic lupus erythematosus. Arthritis & Rheumatism. 2006;54(1):360–368. doi: 10.1002/art.21682. [DOI] [PubMed] [Google Scholar]
- 41.Kang I, Quan T, Nolasco H, et al. Defective control of latent Epstein-Barr virus infection in systemic lupus erythematosus. The Journal of Immunology. 2004;172(2):1287–1294. doi: 10.4049/jimmunol.172.2.1287. [DOI] [PubMed] [Google Scholar]
- 42.Moon UY, Park SJ, Oh ST, et al. Patients with systemic lupus erythematosus have abnormally elevated Epstein-Barr virus load in blood. Arthritis Research & Therapy. 2004;6(4):R295–302. doi: 10.1186/ar1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Babcock GJ, Hochberg D, Thorley-Lawson AD. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity. 2000;13(4):497–506. doi: 10.1016/s1074-7613(00)00049-2. [DOI] [PubMed] [Google Scholar]
- 44.Knowlton N, Dozmorov IM, Centola M. Microarray data analysis toolbox (MDAT): for normalization, adjustment and analysis of gene expression data. Bioinformatics. 2004;20(18):3687–3690. doi: 10.1093/bioinformatics/bth424. [DOI] [PubMed] [Google Scholar]
- 45.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 46.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kozyrev SV, Lewén S, Reddy PMVL, et al. Structural insertion/deletion variation in IRF5 is associated with a risk haplotype and defines the precise IRF5 isoforms expressed in systemic lupus erythematosus. Arthritis & Rheumatism. 2007;56(4):1234–1241. doi: 10.1002/art.22497. [DOI] [PubMed] [Google Scholar]
- 48.Subramanian S, Tus K, Li QZ, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(26):9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dumont C, Henderson R, Tybulewicz VL. Characterization of the roles of Rac1 and Rac2 GTPases in lymphocyte development. Methods in Enzymology. 2008;439:235–254. doi: 10.1016/S0076-6879(07)00418-1. [DOI] [PubMed] [Google Scholar]
- 50.Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. The Journal of Experimental Medicine. 2003;197(6):711–723. doi: 10.1084/jem.20021553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crow MK. Interferon-α: a new target for therapy in systemic lupus erythematosus? Arthritis & Rheumatism. 2003;48(9):2396–2401. doi: 10.1002/art.11226. [DOI] [PubMed] [Google Scholar]
- 53.Niewold TB, Clark DN, Salloum R, Poole BD. Interferon alpha in systemic lupus erythematosus. Journal of Biomedicine and Biotechnology. 2010;2010:8 pages. doi: 10.1155/2010/948364. Article ID 948364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Batteux F, Palmer P, Daëron M, Weill B, Lebon P. FCγRII (CD32)-dependent induction of interferon-alpha by serum from patients with lupus erythematosus. European Cytokine Network. 1999;10(4):509–514. [PubMed] [Google Scholar]
- 55.Bave U, Magnusson M, Eloranta ML, Perers A, Alm GV, Rönnblom L. FcγRIIa is expressed on natural IFN-α-producing cells (plasmacytoid dendritic cells) and is required for the IFN-α production induced by apoptotic cells combined with lupus IgG. The Journal of Immunology. 2003;171(6):3296–3302. doi: 10.4049/jimmunol.171.6.3296. [DOI] [PubMed] [Google Scholar]
- 56.Lovgren T, Eloranta M-L, Bave U, Alm GV, Rönnblom L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis & Rheumatism. 2004;50(6):1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 57.Lovgren T, Eloranta M-L, Kastner B, Wahren-Herlenius M, Alm GV, Rönnblom L. Induction of interferon-α by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjögren's syndrome auto antigen—associated RNA. Arthritis & Rheumatism. 2006;54(6):1917–1927. doi: 10.1002/art.21893. [DOI] [PubMed] [Google Scholar]
- 58.Rönnblom L, Alm GV. Systemic lupus erythematosus and the type I interferon system. Arthritis Research & Therapy. 2003;5(2):68–75. doi: 10.1186/ar625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rönnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008;17(5):394–399. doi: 10.1177/0961203308090020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nature Immunology. 2007;8(5):487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 61.Barnes BJ, Field AE, Pitha-Rowe PM. Virus-induced heterodimer formation between IRF-5 and IRF-7 modulates assembly of the IFNA enhanceosome in vivo and transcriptional activity of IFNA genes. The Journal of Biological Chemistry. 2003;278(19):16630–16641. doi: 10.1074/jbc.M212609200. [DOI] [PubMed] [Google Scholar]
- 62.Barnes BJ, Moore PA, Pitha PM. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon α genes. The Journal of Biological Chemistry. 2001;276(26):23382–23390. doi: 10.1074/jbc.M101216200. [DOI] [PubMed] [Google Scholar]
- 63.Savitsky DA, Yanai H, Tamura T, Taniguchi T, Honda K. Contribution of IRF5 in B cells to the development of murine SLE-like disease through its transcriptional control of the IgG2a locus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(22):10154–10159. doi: 10.1073/pnas.1005599107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Werner SL, Kearns JD, Zadorozhnaya V, et al. Encoding NF-κB temporal control in response to TNF: distinct roles for the negative regulators IκBα and A20. Genes & Development. 2008;22(15):2093–2101. doi: 10.1101/gad.1680708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Graham RR, Cotsapas C, Davies L, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nature Genetics. 2008;40(9):1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang NI, Yoon HY, Lee YR, et al. A20 attenuates allergic airway inflammation in mice. The Journal of Immunology. 2009;183(2):1488–1495. doi: 10.4049/jimmunol.0900163. [DOI] [PubMed] [Google Scholar]
- 67.Trynka G, Zhernakova A, Romanos J, et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-κB signalling. Gut. 2009;58(8):1078–1083. doi: 10.1136/gut.2008.169052. [DOI] [PubMed] [Google Scholar]
- 68.Bates JS, Lessard CJ, Leon JM, et al. Meta-analysis and imputation identifies a 109kb risk haplotype spanning TNFAIP3 associated with lupus nephritis and hematologic manifestations. Genes & Immunity. 2009;10(5):470–477. doi: 10.1038/gene.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gateva V, Sandling JK, Hom G, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nature Genetics. 2009;41(11):1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han JW, Zheng HF, Cui Y, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nature Genetics. 2009;41(11):1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 71.Musone SL, Taylor KE, Lu TT, et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nature Genetics. 2008;40(9):1062–1064. doi: 10.1038/ng.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Honma K, Tsuzuki S, Nakagawa M, et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood. 2009;114(12):2467–2475. doi: 10.1182/blood-2008-12-194852. [DOI] [PubMed] [Google Scholar]
- 73.Kato M, Sanada M, Kato I, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459(7247):712–716. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The supplemental data contains gene sets for SLE risk or protective allele groups that were significantly enriched at p < 0.01, but did not cross the threshold of 25% FDR (Tables 1 and 2). Tables 3 and 4 include the gene sets that were enriched in the non–risk cells compared to the risk cells. Also included in the supplemental tables are all of the gene sets that were significantly enriched for each group after EBV infection (Table 5).