

Abstract
Background/Aims
Considerable evidence has indicated that apoptosis plays an important role in hepatocyte death in chronic liver disease. However, the cellular and molecular mechanisms underlying liver regeneration in the disease are largely unknown. Plausibly, certain molecules expressed to counteract apoptosis might provide survival advantage of certain liver cells. Therefore, we investigated a possible expression of decoy receptor 3 (DcR3) of the tumor necrosis factor (TNF) receptor family in chronic liver diseases since DcR3 is known to inhibit apoptosis mediated by pro-apoptotic TNF family ligands including FasL.
Methods
A series of liver biopsies from patients with different stages of fibrosis were subjected to immunohistochemistry and in situ hybridization.
Results
Both DcR3 protein and mRNA were mainly expressed in biliary epithelial cells and infiltrating lymphocytes in the diseased livers. Most noticeably, intense DcR3 expression was observed in newly developing biliary ductules in regenerative nodules as well as dysplastic nodules of cirrhotic livers. In addition, DcR3 secretion in hepatocellular carcinoma (HCC) cells in culture was via the activation of mitogen-activated protein kinases.
Conclusion
DcR3 was specifically expressed in chronic liver diseases and HCC cells, and DcR3 might facilitate the survival of liver cells by exerting its anti-apoptotic activity during the progression of liver cirrhosis and hepatocarcinogenesis.
Keywords: Apoptosis, FasL, cirrhosis, HCC, HPC
Introduction
Apoptosis (programmed cell death) is a normal cellular process that is critical for maintaining a balance between cell loss and cell gain. Dysregulation of apoptosis has been implicated in the pathogenesis of cancer and other diseases of various organs, including the liver (1). The TNF family (TNFSF) cytokines TNF and Fas ligand (FasL) can induce apoptosis in a variety of cell types expressing their cognate receptors, TNF-RI (p55) and Fas (CD95), respectively. The activation of these receptors triggers the recruitment of TRADD (TNF receptor -associated death domain) or FADD (Fas-associated death domain) that bind to intracellular domains of TNF-RI and Fas receptors, respectively, which leads to apoptosis through the activation of caspases, a family of cysteine proteases. Both TNF/TNF-R1 and FasL/Fas systems are considered to play major roles in the injury of liver cells (2, 3).
Apoptosis in virus infected hepatocytes can contribute to viral clearance, but the activation of apoptotic pathways can also cause unnecessary destruction of liver cells that can lead to formation of excessive scar tissue and cirrhosis. A significant increase of apoptotic liver cells was found in chronic viral hepatitis and cirrhosis (4).
Little is known about how certain liver cells acquire survival advantage to proliferate alongside cell death in chronically damaged livers, particularly in regenerative nodules of cirrhotic livers. Speculatively, pro-survival molecules expressed to counteract increased cell death during the progression of chronic liver diseases might enable certain liver cells to survive and proliferate. Soluble decoy receptor 3 (DcR3) which is already known to inhibit the activity of FasL might possess such potential.
DcR3 expression was first discovered in colorectal and lung carcinomas and thus it was hypothesized that DcR3 might aid cancer cells to evade FasL-mediated cytotoxic attack by host immune cells (5). DcR3 belongs to the decoy receptor group in the TNF receptor (TNFR) family that also includes DcR1, DcR2, and osteoprotegerin (OPG). DcR3 exhibits closest homology to OPG, also a soluble receptor in the family. The DcR3 gene is mapped at 20q13.3 on the human chromosome (6) and the cDNA comprises 903 nucleotides corresponding to 300 amino acids with a putative signal peptide. Due to the lack of transmembrane motif in the cDNA sequence, DcR3 is exclusively secreted as a soluble protein (7).
Soluble recombinant human DcR3 was shown to bind to three different ligands of the TNF family namely, FasL, LIGHT and TL1A (5, 8, 9). As a result, DcR3 is believed to inhibit diverse cellular effects that are mediated by the interaction between these ligands and their membrane-bound cognate receptors: FasL and TL1A bind to death-domain containing receptors Fas and DR3 (death receptor 3), respectively, and LIGHT interacts with either herpes virus entry mediator (HVEM) or lymphotoxin beta receptor (LTbetaR).
As expected, DcR3 can prevent most observed cellular responses that are mediated by its three ligands via their cognate receptors. DcR3 can prevent apoptosis mediated by FasL (5, 10, 11) and LIGHT (12), and inhibit TL1A mediated caspase induction in cells (9). Furthermore, DcR3 was shown to provide survival benefits in animal models of fulminant hepatic apoptosis (13) and acute pulmonary inflammation (14) that are induced by administering exogenous FasL. These results signified that DcR3 might have a therapeutic value to treat Fas/FasL mediated pathological conditions.
In an effort to identify molecules that may be important for hepatic cell survival and regeneration in chronic liver disease, we investigated the expression of DcR3 in liver tissues from the patients with chronic hepatitis C infection as well as the possible secretion of soluble DcR3 in hepatocellular carcinoma (HCC) cells in culture. The results demonstrated that DcR3 is noticeably expressed in developing nodules in cirrhotic livers and that the MAP kinases ERK1/2 and JNK are responsible for DcR3 secretion in HCC cells.
Materials and Methods
Reagents and cell lines
Kinase inhibitors, PD98059, U0126, and SP600125 were obtained from LC Laboratories (Woburn, MA). SB203580, SB202190, and pyrrolidine dithiocarbamate (PDTC) were purchased from Sigma (St Louis, MO). Hepatocellular carcinoma (HCC) cell lines, SK-Hep1, HepG2, and Hep3B were purchased from the American Type Culture Collection (ATCC, Manassas, VA). DMEM was obtained from Cellgro (Herndon, VA) and heat-inactivated FBS was from Invitrogen (Carlsbad, CA).
Tissues
Paraffin-embedded needle liver biopsy specimens were selected from the files of the Department of Pathology of the Aristotle University Medical School, and used according to the Declaration of Helsinki principles and Institutional Review Board policies. The biopsies were derived from 20 patients with chronic hepatitis C (CHC) infection. Patient demographics are provided in Table 1. These 20 cases were representative of the different grades of fibrosis usually seen in CHC (5 cases each with no or mild fibrosis, moderate fibrosis, severe fibrosis, and cirrhosis). Selection was otherwise random. In addition, 5 archival surgical resection specimens with HCV cirrhosis were also included. Disease activity was graded according to the histological activity index (HAI) (15) and fibrosis was staged from 0 to 4 (0 = no fibrosis to 4 = cirrhosis). Four histologically normal looking liver biopsies were used as control specimens.
Table 1.
Biopsy specimens
| case # | Age/Gender | HAI | Fibrosis | Biliary-ductule | Ductular-reaction | Lymphocytes | Stroma |
|---|---|---|---|---|---|---|---|
| 312/00 | 35 M | mild | 0 | yes | n.a. | very few | absent |
| 2988/03 | F | mild | 1 | yes | n.a. | very few | absent |
| 4397/03 | M | mild | 1 | yes | n.a. | few | absent |
| 4089/03 | M | mild | 1 | yes | n.a. | few | absent |
| 4518/03 | 32 M | mild | 1 | yes | n.a. | very few | absent |
| 3352/03 | 43 M | mild | 2 | yes | n.a. | none | absent |
| 3020/03 | 56 F | moderate | 2 | yes | n.a. | very few | absent |
| 1592/03 | M | mild | 2 | none | n.a. | very few | absent |
| 2718/03 | 60 F | mild | 2 | yes | n.a. | few | absent |
| 2987/03 | M | moderate | 2 | yes | n.a. | very few | absent |
| 2802/03 | 47 M | moderate | 3 | yes | n.a. | few | absent |
| 807/03 | M | moderate | 3 | yes | n.a. | very few | absent |
| 4398/03 | M | moderate | 3 | yes | n.a. | none | absent |
| 2158/94 | mild | 3 | very few | n.a. | very few | absent | |
| 3523/03 | 69 F | mild | 4 | yes | n.a. | few | present |
| 419/03 | 60 M | moderate | 4 | yes | yes | many | present |
| 4740/02 | 64 F | moderate | 4 | yes | yes | many | present |
| 1791/04 | 68 F | severe | 4 | yes | yes | many | present |
| 4239/03 | F | severe | 4 | yes | yes | many | present |
| 3334/03 | 51 M | severe | 4 | yes | yes | many | present |
Immunohistochemistry (IHC)
Liver tissue sections were prepared from archived paraffin blocks of liver biopsy and resection specimens. For DcR3 IHC, the monoclonal antibody (mAb) MD3F4 specific to human DcR3 was employed (16). Tissue sections were cut at 2 μm thickness and mounted on positively charged slides. Paraffin slides were then incubated overnight at 58 °C. After dewaxing in multiple fresh xylenes, the slides were rehydrated in descending ethanol changes. Endogenous peroxidase was blocked for 20 min with 3% H2O2 in methanol and subsequently slides were rinsed in distilled water. No epitope unmasking was used since this step was determined to be unnecessary for human DcR3 reactivity with MD3F4. To abrogate non-specific binding, sections were incubated with a commercially available blocking solution, Powerblock (Innogenex, San Ramon, CA), at 37 °C for 30 min. Slides were incubated with MD3F4 (2.5 μg/ml) diluted in Powerblock at 37 °C for 1 hr. After washing in PBS, a secondary biotinylated anti-mouse antibody (DAKO, Glostroup, Denmark), diluted at 1:200 in Powerblock, was applied for 1 hr at room temperature (RT). Color development was achieved with streptavidin-peroxidase and diaminobenzidine (DAB) (both from DAKO). For counterstain, haematoxylin was used.
In situ hybridization (ISH)
Paraffin sections from the resection specimens were also used for ISH. The SP6/T7-Polymerase Digoxigenin (DIG)-Labeling and Transcription Kit (Roche, Indianapolis, IN) was used to synthesize sense (negative control) and anti-sense DIG-labeled DcR3 riboprobes using the template plasmid that has the human DcR3 cDNA insert flanked by SP6 and T7 sites in pGM-T vector (Promega, Madison, WI). The cDNA (275 bps between nts 329~603; Genbank # AF1044419) was designed to span exon 1 through exon 2 to increase specific hybridization of DcR3 mRNA from the gene. Tissue slides were dewaxed, rehydrated, digested with pepsin in 0.1 N HCl for 30 min at 37°C, acetylated in freshly prepared 1:10 triethanolamine with MgCl2, pH 8, with 0.25% acetic anhydrite, and prehybridized for 2 hr at 48°C in a hybridization buffer containing 50% formamide, 5X SSC, RNase inhibitor and 10 μg/ml sssDNA. The riboprobes were then added on the slides in new hybridization buffer, denatured for 15 min at 70°C and incubated overnight at 45°C. Washings included 20 min in 2X SSC, 15 min in 1X SSC (twice), 15 min in 0.2X SSC at 56°C and 15 min in 0.2X SSC at RT. After blocking for 1 hr, slides were then incubated for 2 hrs with an anti-digoxigenin antibody conjugated with alkaline phosphatase (Roche) and subsequently washed in a Tris pH 7.5-NaCl-Tween buffer. Color was developed with NBT/BCIP (Roche) with the addition of 2 drops of levamizole (Sigma). Sense and antisense probes were run in parallel, and also reactions were performed without adding the probes as controls. Dark blue perinuclear signals were considered as positive for the presence of DcR3 mRNA.
Cell culture and ELISA
SK-Hep1 cells were cultured in DMEM supplemented with 10% heat-inactivated FBS. One day before inhibitor treatment, cells were plated at 2 × 105 cells/well in 24-well tissue culture plates. After aspirating the medium, fresh culture medium with or without inhibitors was added to the wells (0.5 ml/well). The culture plates were then incubated for 22 hr at a 37°C, 5% CO2 culture incubator. At the end of inhibitor treatment, cell viability was confirmed by MTT assay according to the manufacture’s instruction (R&D systems, Minneapolis, MN). Cells were incubated with the tetrazolium compound MTT which was reduced by metabolically active cells to insoluble purple formazan dye crystals. After solubilizing crystals by detergent, the absorbance was read by at 570 nm.
DcR3 releases in culture supernatant were measured by the human DcR3 specific quantitative ELISA as we described (7). Briefly, 100 μl of culture supernatants were added to wells of 96-well ELISA plates that were coated with the capture mAb (MD3E2) and blocked with 3% BSA. The standard DcR3 protein and test samples were added to the wells and the plates were incubated overnight at 4°C. After washing, a biotinylated detection mAb (MD3B1) was incubated to wells for 2 hr at RT. After washing, peroxidase-conjugated streptavidin (Vector Lab., Burlingame, CA) was added to the wells. Color was developed using TMB substrate solution (KPL, Gaithersburg, MD). After stopping the color reaction with 1 N H2SO4, plates were read at absorbance 450 nm in an Emax ELISA plate reader (Molecular Device Co., Sunnyvale, CA).
Results
Localization of DcR3 immunoreactivity in CHC
In most diseased tissue sections, DcR3 immunoreactivity was primarily detected in certain biliary duct epithelial cells (Fig. 1). DcR3 immunostain was largely absent in hepatocytes, hepatic stellate cells, and kupffer cells. Inflammatory cells infiltrating the liver were generally negative for DcR3 with few positive lymphocytes present in cases with mild to severe fibrosis (Fig. 1). As the severity of fibrosis increases, there were more infiltrating lymphocytes showing positive DcR3 expression as they were more noticeable in cirrhosis (Fig. 2). Similarly to other normal organs, DcR3 immunoreactivity was not detected in normal liver tissue controls, and no staining was observed in the diseased tissues stained with an isotype-matching negative control mAb (data not shown).
Fig. 1. Representative examples of DcR3 localization in CHC with fibrosis.

In upper case (panels A & B), DcR3 immunoreactivity (dark brown indicated by arrows) was detected in certain bile duct epithelial cells, and infiltrating lymphocytes were largely negative for DcR3. In lower case stained with DAB without counterstain, DcR3 immunostaining (red-brown area marked by arrows) was also seen in biliary ducts and some endothelial cells were DcR3 positive (marked by e in panel C). Both DcR3 positive (dark brown indicated by p) and negative (light blue indicated by n) hepatocytes’ nuclei were detected in panel B (DAB, haematoxylin counterstain). Also, DcR3 positive hepatocytes’ nuclei were readily observed in panel D without counterstain. Original magnification: A, x100; B & D, x400; C, x40.
Fig. 2. Representative examples of DcR3 localization in CHC with cirrhosis.
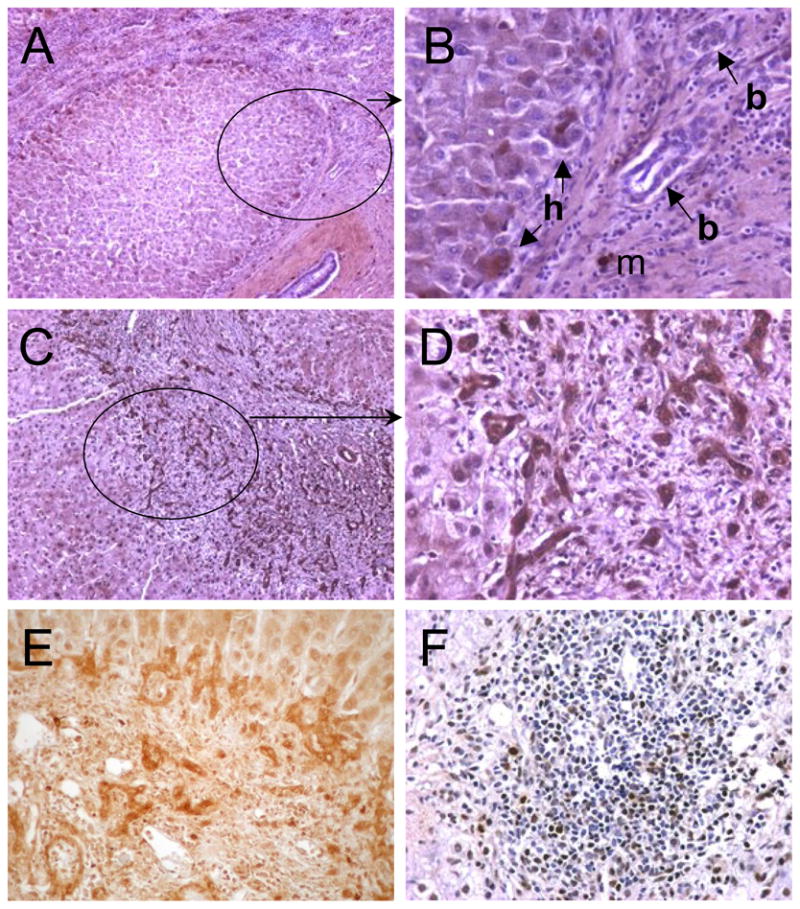
In the upper panels (panels A & B), the pattern with positive developing biliary ductules were practically absent and existing bile ducts were negative for DcR3 immunostain (panel B, b-arrows). Relatively few hepatocytes in the periphery of the nodules were DcR3 immunoreactive (panel B, h-arrows) and mast cells (m) showed immunoreactivity. In the lower panels (panels C & D), the circled area (panel C) shows growing part of the cirrhotic nodule that is magnified again in panel D. In panel D, intense immunostains were detected in structures called hepatobiliary ductules, regarded as an intermediary between hepatocytes and biliary duct epithelial cells. Panel E shows another cirrhosis specimen showing positively stained biliary duct and ductules (DAB, no counterstain). Panel F shows DcR3 immunoreactive infiltrating lymphocytes (dark brown). Original magnification: A & C, x100; B, D, E, & F, x400; (A ~ D & F, DAB, haematoxylin counterstain).
In cases of CHC with cirrhosis, intense DcR3 immunoreactivity was observed in biliary duct and ductules (ductular reaction) in regenerative nodules (Fig. 2). All the liver specimens with cirrhosis (5 biopsy and 5 resection specimens) had ductular reactions that showed intense DcR3 immunostains. The staining in biliary epithelial cells of both duct and ductules in regenerative cirrhotic nodules was the most consistent observation and no such staining was observed in tissue sections with no presence of ductular reaction (Fig. 2).
Moreover, we found that in cirrhotic regions with marked atypia which could be regarded as early cancer, there were more DcR3 positive cells inside nodules than in pure cirrhosis with no atypia; inside the cirrhotic nodules the hepatocytes were practically negative for DcR3 (Fig. 3).
Fig. 3. Localization of DcR3 expression in dysplastic nodule.

The upper panels (A & B) show a case of pure cirrhosis with no atypia. Inside the cirrhotic nodules, the cells were practically negative for DcR3. The lower panels (C & D) present a case of cirrhosis with marked atypia which could be regarded as early cancer; cells inside the nodule were mostly DcR3 positive. Both DcR3 positive (dark brown) and negative (light blue) nuclei can be observed in panel D. Original magnification: A, C, x100; B, D, x400 (DAB, haematoxylin counterstain).
Curiously, DcR3 immunostaning was found in the nucleus of certain hepatocytes and biliary epithelial cells in most liver specimens (Figs. 1 and 3). The staining of liver cells including the nucleus staining was the same either with or without antigen retrieval. Such nucleus staining was not detected in our IHC on acute appendicitis specimens using the same antibody (16). Thus, it will be of interest to investigate whether the DcR3 reactivity in the nucleus is a unique feature in chronic liver diseases of viral etiology and whether DcR3 has any function as transcription activator/inhibitor being localized in the nucleus.
Detection of DcR3 mRNA expression in cirrhotic liver
Since DcR3 binds to FasL, LIGHT and TL1A, it is reasonable to speculate that some DcR3 might have bound to the cell surface via these membrane-bound ligands. All these TNF family ligands belong to type II transmembrane protein family which is present in both membrane bound and soluble forms. Therefore, the expression of DcR3 mRNA in cirrhotic liver sections was confirmed by In situ hybridization (ISH).
Similarly to IHC, there were noticeable hybridization signals in the periphery of cirrhotic nodules (Fig. 4). Signals were detected in small biliary ductules, hepatocytes located at nodular margins, as well as structures morphologically related to both hepatocytes and biliary epithelial cells. A prominent signal was also detected in clustered infiltrating lymphocytes (Fig. 4). According to electron microscopic studies, human hepatic progenitor cells (HPCs), which have been shown to form newly developing biliary ductules, were characterized by small size (10 μm), oval shape, and high nucleo/cytoplasm ratio. These HPCs were demonstrated to have bipotential capacity to become mature hepatocytes and biliary epithelial cells (17). Thus, the hybridization pattern also seemed to suggest that biliary ductules might be of HPC origin. Except for periportal regions, hepatocytes were largely negative for hybridization signals. There were no above the noise-level signals in control experiments using the sense cDNA probe (Fig. 4). Therefore, the localization of DcR3 mRNA in cirrhotic livers appeared to be in agreement with the results of DcR3 immunostaining.
Fig. 4. In situ hybridization of DcR3 transcripts in CHC with cirrhosis.
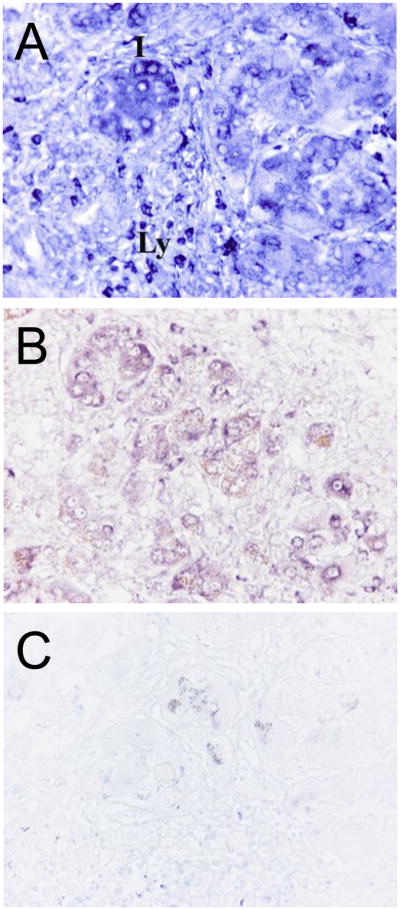
In the areas of active inflammation and architectural remodelling (panels A & B), there is intense staining in infiltrating lymphocytes (Ly), as well as in bile ductular and intermediate cells (I). Images in B and C (inset) were processed simultaneously to show the background level staining in CHC cirrhosis with sense-probe (negative control). Brown granules seen in panels B and C are hemosiderin. Original magnification: A, B, and C, x400.
DcR3 expression in HCC and SK-Hep1 cell line
It was previously reported that 60% of HCC express DcR3 protein and transcripts (18). Similarly, we also detected DcR3 protein and mRNA expression in some tissue sections of a dozen cases of HCC operative specimens (Fig. 5). As DcR3 is known to be exclusively released as soluble protein, we have further investigated whether certain HCC cell lines indeed secreted soluble DcR3 in culture supernatant. Among HCC cell lines, SK-Hep1 expressed noticeably high constitutive level of soluble DcR3 in culture supernatant and thus chosen for studying intracellular signaling pathways involved in DcR3 release.
Fig. 5. Localization of DcR3 expression in HCC.

The right panel (A) represents positive DcR3 immunoreactivity and the left panel (B) represents positive DcR3 mRNA expression (in HCC tissue section. Original magnification: A, x400 ((DAB, haematoxylin counterstain); B, x400.
During the initial screening of inhibitors in different signaling pathways, we found that mitogen-activated protein kinase (MAPK) but not protein kinase C (PKC) and PKA pathways were involved in DcR3 expression in SK-Hep1 cells. The MAPK pathway involves three major kinases, ERK1/2 (p44/p42), p38, and JNK, that are known to be responsible for numerous cellular behaviors and responses (19).
Among inhibitors of the MAPK pathway, PD98059 was selected since it was shown to selectively inhibit phosphorylation of MAPK activating kinase (MEK1/2) by the upstream kinase, MEK activating kinase (MEKK) (20). An inhibitor U0126 was also utilized since this compound particularly blocks MEK1/2, the kinases upstream of ERK1/2 (21, 22). A specific inhibitor of JNK, SP600125, was also tested since it selectively inhibits JNK-1, 2, 3 thus resulting in the inhibition of AP-1 phosphorylation (23). Also, widely used potent pyridinyl imidazole inhibitors of p38 MAPK, SB202190 and SB203580, were included. These inhibitors were chosen based on their well recognized specificity in the literature. Cells were treated with inhibitor concentrations around IC50 and the doses that are shown in Fig. 6 were confirmed not to adversely affect viability of SK-Hep1 during 22 hr incubation with the inhibitors.
Fig. 6. DcR3 release in SK-Hep1 was inhibited by inhibitors of ERK1/2 and JNK but not p38.

(A)Cells seeded in 24-well tissue culture plates were treated with the indicated inhibitors for 22 hr. DcR3 release in culture supernatants was quantified by ELISA. Experiments were conducted two independent times with triplicate samples and data are mean ± SD. PD98059 (2.5~20 μM), U0126 (200~400 nM), SP600125 (5~20 μM), and PDTC (50~100 μM) significantly inhibited DcR3 release in the cells compared to the cells with no inhibitor treatment. p values between untreated and each inhibitor (grouped effective doses) were less than 0.05 by unpaired t-test. Summary of DcR3 expression via the activation of MAPKs (B). Inhibitors of ERK, JNK, p38 MAPK that were used in Fig. 6A are indicated in parentheses. p38 MAPK inhibitors had no inhibitory effect in DcR3 release. The activation of NF-κB was likely via NIK (NF-κB inducing kinase pathways, not included in the diagram). The binding of transcription factors NF-κB and AP-1 (activated by JNK) to the promoter of DcR3 gene will eventually cause release of soluble DcR3 in SK-Hep1 cells.
The treatment of cells with ERK1/2 and JNK inhibitors (U0126, PD98059, and SP600125) resulted in significant inhibition of DcR3 expression in the cells by 50~70% (Fig. 6A). However, the two p38 MAPK inhibitors (SB202190 and SB203580) had no inhibitory effects on the cells. Instead, these inhibitors resulted in DcR3 release up to 25% in the culture supernatant. Previously, SB203580 was shown to activate Raf-1 (24), therefore the increased DcR3 release by these inhibitors might have been in part due to the activation of Raf-1, an upstream kinase of MEK.
Additionally, PDTC, a selective inhibitor of transcription factor NF-κB, was tested. NF-κB is widely known as a central transcriptional factor that controls transcriptional regulation of numerous genes involved in inflammation, infection and immunity. Furthermore, constitutive activation of NF-κB has been also reported in HCC (25). Similarly, SK-Hep1 cells also appeared to constitutively express activated NF-κB as PDTC inhibited DcR3 expression by 70% in the cells (Fig. 6A).
Based on the results from the inhibitor studies, the signaling pathways that are most likely involved in DcR3 expression in liver cancer cells have been deduced (Fig. 6B). The figure shows that the signal transduction of DcR3 is mediated by ERK1/2/ and JNK but not p38 MAPK, and that the binding of transcriptional factors AP-1 and NF-κB in the promoter of human DcR3 gene induces DcR3 release in the liver cancer cells.
Discussion
The mechanism underlying liver regeneration in advanced chronic liver diseases is largely unknown. Therefore, the prominent expression of anti-apoptotic protein DcR3 in proliferating biliary epithelial cells in regenerative nodules of HCV-related cirrhosis is of great significance. We speculate that DcR3 release in chronic liver diseases might aid liver cells to resist apoptosis mediated by the interaction between pro-apoptotic ligands of DcR3 (FasL, LIGHT, TL1A) and their cognate receptors that are expressed on the surface of liver cells. The main source of these ligands might be infiltrating T-cells as all these ligands were shown to be expressed in activated T-cells, and the receptors of these ligands are usually expressed in a variety of cell types. The expression of LIGHT and TL1A in liver diseases has yet to be investigated but the expression of Fas and FasL has been demonstrated in chronic liver diseases.
In CHC, Fas expression was increased in hepatocytes in line with the severity of liver inflammation. FasL expression was also detected in infiltrating lymphocytes and thus it was indicated that Fas-positive hepatocytes were likely subjected to apoptosis mediated by the infiltrating lymphocytes (26, 27). The expression of Fas and FasL in hepatocytes was significantly higher in the chronic viral hepatitis group than in the cirrhosis group (28) thus suggesting that apoptotic cell death was likely less active in cirrhosis. FasL expression was also found in areas with lymphocytic infiltration in hepatitis B virus (HBV)-related cirrhosis (29).
Particularly in the liver, Fas/FasL and TNF/TNF-R1 extrinsic apoptosis pathways were reported to be coupled with the intrinsic mitochondrial apoptotic pathway which is regulated by pro-apototic (Bid, Bax, and Bak) and antiapoptotic (Bcl-2, and Bcl-xL) molecules of the Bcl-2 family (30). Bcl-2 expression was localized in bile ducts and mononuclear cells in CHC, but was not commonly present in hepatocytes (31). In CHC with cirrhosis, Bcl-2 expression was also found in bile ducts and mononuclear cells, but in contrast to CHC without cirrhosis, Bcl-2 was also expressed in hepatocytes. Therefore, it has been hypothesized that the increased antiapoptotic Bcl-2 might contribute to hepatocarcinogenesis and that cirrhosis is selection of Bcl-2 expressing cells (32).
Regardless, apoptosis can be primarily initiated by binding of FasL or TNF to their cell surface death receptors, and these extrinsic apoptotic pathways are sufficient to induce apoptosis without participation of the mitochondrial apoptotic pathway. Therefore, the expression of soluble DcR3 in chronic liver diseases might represent another mechanism that liver cells utilize to prevent extracellular pro-apoptotic ligands ever reaching to their cognate death receptors on the cell surface.
A variety of agents including proinflammatory cytokines TNF and IL-1, bacterial antigens and phorbol ester (PMA) can induce DcR3 release in a number of human primary cells and cancer cell lines. Previously, we had reported that p38 MAPK was not involved in DcR3 regulation in primary human monocytes and dendritic cells stimulated with endotoxin (LPS) (33). Also, both constitutive and LPS-induced DcR3 release in the colorectal cancer cells were mediated by ERK1/2 and JNK but not p38 MAPK (16). Thus, the herein reported lack of p38 MAPK appeared to be again a feature of DcR3 signaling in liver cancer cells. Therefore, all human cells expressing constitutive or induced DcR3 that we have so far checked, either primary culture or cancer cell lines, appeared to involve ERK1/2/ and JNK but not p38 MAPK. It is known that p38 MAPK plays a critical role in the regulation of pro-inflammatory and pro-apoptotic proteins such as TNF and FasL (34). Interestingly, reduced p38 MAPK activity was detected in human liver tumors compared with non-tumorous controls (35), and the loss of p38 MAPK activation was associated with increased tumorigenesis in liver cells (36). Therefore, the lack of p38 MAPK involvement might be an important feature in the regulation of DcR3 expression during the development of cirrhosis and carcinogenesis in the liver.
Human hepatic progenitor cells (HPCs), an equivalent of oval cells in rodents, are known to be activated to proliferate when the liver damage is extensive and chronic or if proliferation of hepatocytes is inhibited, such as in viral infection and cirrhosis (37). The presence of HPCs has been reported in diseased human livers including severe hepatic necrosis, focal nodular hyperplasia, primary biliary cirrhosis, as well as alcohol and hepatitis B and C virus associated cirrhosis (15, 17). Proliferating HPCs are undetectable in normal livers and HPC numbers in human liver rise with increasing severity of liver diseases. The involvement of HPCs in liver carcinogenesis has been hypothesized (38–40), however the mechanism underlying activation and proliferation of HPCs remains largely unknown. In rodents, it has been proposed that inflammatory growth factors may mediate the proliferation of oval cells (progenitor cells) during disease progression (41). Therefore, the presence of the antagonist of apoptosis, DcR3, in newly developing biliary ductules in regenerative cirrhotic nodules is of interest.
DcR3 has been considered to potentially have therapeutic value in treating fulminant hepatic failure, because recombinant soluble DcR3 could block exogenous FasL-mediated hepatocyte apoptosis in rodent hepatitis models (13). Hence, it is quite interesting to detect the expression of DcR3 in chronic liver disease. DcR3 might indeed have a therapeutic application in acute fulminant hepatitis in humans as DcR3 expression appeared to be associated with nodular regeneration in cirrhotic livers. However, in chronic liver disease, the inhibition of DcR3 release might retard the proliferation of liver cells thus resulting in slowing down hepatic regeneration, and possibly hepatocarcinogenesis. By targeting molecules responsible for fibrogenic cascade, liver fibrosis is increasingly considered to be a potentially treatable pathological complication (42). Therefore, understanding the cellular and molecular mechanism underlying the liver regeneration process in chronic liver disease might further provide better treatment modality in the future.
Acknowledgments
This study was in part supported by NIH grant DK07643 to S. Kim.
Footnotes
There if no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akazawa Y, Gores GJ. Death receptor-mediated liver injury. Semin Liver Dis. 2007;27:327–328. doi: 10.1055/s-2007-991510. [DOI] [PubMed] [Google Scholar]
- 2.Kondo T, Suda T, Fukuyama H, Adachi M, Nagata S. Essential roles of the Fas ligand in the development of hepatitis. Nat Med. 1997;3:409–413. doi: 10.1038/nm0497-409. [DOI] [PubMed] [Google Scholar]
- 3.Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med. 2004;8:445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papakyriakou P, Tzardi M, Valatas V, Kanavaros P, Karydi E, Notas G, et al. Apoptosis and apoptosis related proteins in chronic viral liver disease. Apoptosis. 2002;7:133–141. doi: 10.1023/a:1014472430976. [DOI] [PubMed] [Google Scholar]
- 5.Pitti RM, Marsters SA, Lawrence DA, Roy M, Kischkel FC, Dowd P, et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature. 1998;396:699–703. doi: 10.1038/25387. [DOI] [PubMed] [Google Scholar]
- 6.Fujita Y, Sakakura C, Shimomura K, Nakanishi M, Yasuoka R, Aragane H, et al. Chromosome arm 20q gains and other genomic alterations in esophageal squamous cell carcinoma, as analyzed by comparative genomic hybridization and fluorescence in situ hybridization. Hepatogastroenterology. 2003;50:1857–1863. [PubMed] [Google Scholar]
- 7.Chen J, Zhang L, Kim S. Quantification and detection of DcR3, a decoy receptor in TNFR family. J Immunol Methods. 2004;285:63–70. doi: 10.1016/j.jim.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Yu KY, Kwon B, Ni J, Zhai Y, Ebner R, Kwon BS. A newly identified member of tumor necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J Biol Chem. 1999;274:13733–13736. doi: 10.1074/jbc.274.20.13733. [DOI] [PubMed] [Google Scholar]
- 9.Migone TS, Zhang J, Luo X, Zhuang L, Chen C, Hu B, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 10.Bai C, Connolly B, Metzker ML, Hilliard CA, Liu X, Sandig V, et al. Overexpression of M68/DcR3 in human gastrointestinal tract tumors independent of gene amplification and its location in a four-gene cluster. Proc Natl Acad Sci U S A. 2000;97:1230–1235. doi: 10.1073/pnas.97.3.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roth W, Isenmann S, Nakamura M, Platten M, Wick W, Kleihues P, et al. Soluble decoy receptor 3 is expressed by malignant gliomas and suppresses CD95 ligand induced apoptosis and chemotaxis. Cancer Res. 2001;61:2759–2765. [PubMed] [Google Scholar]
- 12.Gill RM, Hunt JS. Soluble receptor (DcR3) and cellular inhibitor of apoptosis-2 (cIAP-2) protect human cytotrophoblast cells against LIGHT-mediated apoptosis. Am J Pathol. 2004;165:309–317. doi: 10.1016/S0002-9440(10)63298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connolly K, Cho YH, Duan R, Fikes J, Gregorio T, LaFleur DW, et al. In vivo inhibition of Fas ligand mediated killing by TR6, a Fas ligand decoy receptor. J Pharmacol Exp Ther. 2001;298:25–33. [PubMed] [Google Scholar]
- 14.Wortinger MA, Foley JW, Larocque P, Witcher DR, Lahn M, Jakubowski JA, et al. Fas ligand-induced murine pulmonary inflammation is reduced by a stable decoy receptor 3 analogue. Immunology. 2003;110:225–233. doi: 10.1046/j.1365-2567.2003.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1:431–435. doi: 10.1002/hep.1840010511. [DOI] [PubMed] [Google Scholar]
- 16.Kim S, Fotiadu A, Kotoula V. Increased expression of soluble decoy receptor 3 in acutely inflamed intestinal epithelia. Clin Immunol. 2005;115:286–294. doi: 10.1016/j.clim.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Xiao JC, Jin XL, Ruck P, Adam A, Kaiserling E. Hepatic progenitor cells in human liver cirrhosis: immunohistochemical, electron microscopic and immunofluorencence confocal microscopic findings. World J Gastroenterol. 2004;10:1208–1211. doi: 10.3748/wjg.v10.i8.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen HW, Gao SL, Wu YL, Peng SY. Overexpression of decoy receptor 3 in hepatocellular carcinoma and its association with resistance to Fas ligand-mediated apoptosis. World J Gastroenterol. 2005;11:5926–5930. doi: 10.3748/wjg.v11.i38.5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 20.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duncia JV, Santella JB, 3rd, Higley CA, Pitts WJ, Wityak J, Frietze WE, et al. MEK inhibitors: the chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg Med Chem Lett. 1998;8:2839–2844. doi: 10.1016/s0960-894x(98)00522-8. [DOI] [PubMed] [Google Scholar]
- 22.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 23.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, et al. SP600125, an anthrapyrazolone inhibitor of Jun N terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalmes A, Deou J, Clowes AW, Daum G. Raf-1 is activated by the p38 mitogen-activated protein kinase inhibitor, SB203580. FEBS Lett. 1999;444:71–74. doi: 10.1016/s0014-5793(99)00034-4. [DOI] [PubMed] [Google Scholar]
- 25.Guo LL, Xiao S, Guo Y. Activation of transcription factors NF-kappaB and AP-1 and their relations with apoptosis associated-proteins in hepatocellular carcinoma. World J Gastroenterol. 2005;11:3860–3865. doi: 10.3748/wjg.v11.i25.3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayashi N, Mita E. Fas system and apoptosis in viral hepatitis. J Gastroenterol Hepatol. 1997;12:S223–S226. doi: 10.1111/j.1440-1746.1997.tb00504.x. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi N, Mita E. Involvement of Fas system-mediated apoptosis in pathogenesis of viral hepatitis. J Viral Hepat. 1999;6:357–365. doi: 10.1046/j.1365-2893.1999.00175.x. [DOI] [PubMed] [Google Scholar]
- 28.Chen NL, Bai L, Li L, Chen PL, Zhang C, Liu CY, et al. Apoptosis pathway of liver cells in chronic hepatitis. World J Gastroenterol. 2004;10:3201–3214. doi: 10.3748/wjg.v10.i21.3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galle PR, Hofmann WJ, Walczak H, Schaller H, Otto G, Stremmel W, et al. Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. J Exp Med. 1995;182:1223–1230. doi: 10.1084/jem.182.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Li S, Childs EE, Kuharsky DK, Yin XM. Activation of pro-death Bcl-2 family proteins and mitochondria apoptosis pathway in tumor necrosis factor-alpha-induced liver injury. J Biol Chem. 2001;276:27432–27440. doi: 10.1074/jbc.M102465200. [DOI] [PubMed] [Google Scholar]
- 31.Tsamandas AC, Thomopoulos K, Gogos C, Tepetes K, Kourelis T, Ravazoula P, et al. Expresssion of bcl-2 oncoprotein in cases of acute and chronic viral hepatitis type B and type C: a clinicopathologic study. Dig Dis Sci. 2002;47:1618–1624. doi: 10.1023/a:1015839807627. [DOI] [PubMed] [Google Scholar]
- 32.Frommel TO, Yong S, Zarling EJ. Immunohistochemical evaluation of Bcl-2 gene family expression in liver of hepatitis C and cirrhotic patients: a novel mechanism to explain the high incidence of hepatocarcinoma in cirrhotics. Am J Gastroenterol. 1999;94:178–182. doi: 10.1111/j.1572-0241.1999.00792.x. [DOI] [PubMed] [Google Scholar]
- 33.Kim S, McAuliffe WJ, Zaritskaya LS, Moore PA, Zhang L, Nardelli B. Selective induction of tumor necrosis receptor factor 6/decoy receptor 3 release by bacterial antigens in human monocytes and myeloid dendritic cells. Infect Immun. 2004;72:89–93. doi: 10.1128/IAI.72.1.89-93.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu SC, Gavrilin MA, Tsai MH, Han J, Lai MZ. p38 mitogen-activated protein kinase is involved in Fas ligand expression. J Biol Chem. 1999;274:25769–25776. doi: 10.1074/jbc.274.36.25769. [DOI] [PubMed] [Google Scholar]
- 35.Iyoda K, Sasaki Y, Horimoto M, Toyama T, Yakushijin T, Sakakibara M, et al. Involvement of the p38 mitogen-activated protein kinase cascade in hepatocellular carcinoma. Cancer. 2003;97:3017–3026. doi: 10.1002/cncr.11425. [DOI] [PubMed] [Google Scholar]
- 36.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, et al. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Theise ND, Saxena R, Portmann BC, Thung SN, Yee H, Chiriboga L, et al. The canals of Hering and hepatic stem cells in humans. Hepatology. 1999;30:1425–1433. doi: 10.1002/hep.510300614. [DOI] [PubMed] [Google Scholar]
- 38.Roskams TA, Libbrecht L, Desmet VJ. Progenitor cells in diseased human liver. Semin Liver Dis. 2003;23:385–396. doi: 10.1055/s-2004-815564. [DOI] [PubMed] [Google Scholar]
- 39.Lowes KN, Brennan BA, Yeoh GC, Olynyk JK. Oval cell numbers in human chronic liver diseases are directly related to disease severity. Am J Pathol. 1999;154:537–541. doi: 10.1016/S0002-9440(10)65299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Theise ND, Yao JL, Harada K, Hytiroglou P, Portmann B, Thung SN, et al. Hepatic ‘stem cell’ malignancies in adults: four cases. Histopathology. 2003;43:263–271. doi: 10.1046/j.1365-2559.2003.01707.x. [DOI] [PubMed] [Google Scholar]
- 41.Knight B, Matthews VB, Akhurst B, Croager EJ, Klinken E, Abraham LJ, et al. Liver inflammation and cytokine production, but not acute phase protein synthesis, accompany the adult liver progenitor (oval) cell response to chronic liver injury. Immunol Cell Biol. 2005;83:364–374. doi: 10.1111/j.1440-1711.2005.01346.x. [DOI] [PubMed] [Google Scholar]
- 42.Friedman SL. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nat Clin Pract Gastroenterol Hepatol. 2004;1:98–105. doi: 10.1038/ncpgasthep0055. [DOI] [PubMed] [Google Scholar]