

Abstract
Loperamide is an FDA-approved antidiarrhea drug which acts on the μ-opioid receptors in the mesenteric plexus of large intestine and exhibits limited side effects. We hypothesized that loperamide might reverse the multidrug resistance (MDR) of human cancer cells to chemotherapeutic agents. MCF-7/MDR1 cells express high level of MDR1 and are resistant to doxorubicin. We found that loperamide significantly enhanced the cytotoxicity of doxorubicin to MCF-7/MDR1 cells in a dose-dependent manner. In conclusion, loperamide reversed the resistance of MCF-7/MDR1 cells to doxorubicin, suggesting that chemotherapy in combination with loperamide may benefit patients with MDR tumors once applied in clinic.
Keywords: Multidrug resistance, Breast cancer, Loperamide, Doxorubicin
INTRODUCTION
Multidrug resistance (MDR) of tumor cells is a major problem in cancer chemotherapy (1, 2). Several mechanisms have been established for the MDR development including activation of the ATP binding cassette transporters, expression changes of genes involved in apoptosis, and increased enzyme activity of the glutathione-mediated detoxification pathways (1, 2). The classical mechanism of MDR development involves the overexpression of P-glycoprotein (P-gp) that facilitates the efflux of chemotherapeutic agents from tumor cells (3–5). Chemotherapeutic agents kill most tumor cells but also select for surviving cells that overexpress P-gp. These surviving cells are resistant to the drugs used to treat patients and other structurally unrelated drugs that are MDR substrates as well (6–8). Compounds that compete for P-gp may have the potential to enhance the efficacy of anticancer drugs that are MDR substrates. Significant effort has been aimed at the development of MDR reversers (1, 2). A large number of compounds have been tested for their reversal activities on MDR tumor cells (9–12). These agents antagonize MDR through competitive inhibition of the P-gp-mediated transport of antitumor drugs (2). Generally, the majority of tested compounds are either insufficient in efficacy or exhibit unacceptable toxicity or unpredictable pharmacokinetic interactions (13). There is a need for effective and safe agents that reverse MDR of tumor cells.
Loperamide, an FDA-approved antidiarrhea drug, acts on the μ-opioid receptors in the mesenteric plexus of large intestines (14). Loperamide is widely used in the clinic to control diarrhea induced by digestive disorders, chemotherapy, and radiotherapy. In humans, many key organs including brain, liver, and heart express P-gp, and P-gp serves to minimize retention of toxic substances in normal cells. Recent studies have shown that loperamide acts as a P-gp substrate in the blood–brain barrier (BBB) (15, 16). Loperamide molecules normally do not cross the BBB to any significant extent, but those that cross the BBB are quickly exported from the brain by the P-gp in the BBB. However, the amount of loperamide in the central nervous system (CNS) is sevenfold higher in MDR1 gene knockout mice than in wild-type mice (17). In addition, evidence has shown that loperamide can block high-voltage-activated calcium channels and N-methyl-D-aspartate-evoked responses in cultured hippocampal pyramidal and dorsal root ganglion neurons of rats and mice (14). Calcium is an important intracellular second messenger involved in the regulation of many cellular processes. A number of tested MDR reversers are known to be calcium channel blockers (2). Having these findings, we hypothesized that loperamide might act as a reverser of MDR and benefit patients receiving chemotherapy. In the present study, we investigated the reversal effect of loperamide on MDR tumor cells using the doxorubicin-sensitive MCF-7 and doxorubicin-resistant MCF-7/MDR1 human breast cancer cells. Doxorubicin is a well-known P-gp substrate and frequently used to treat cancers.
MATERIAL AND METHODS
MCF-7 and MCF-7/MDR1 human breast cancer cells
The parental drug-sensitive MCF-7 and multidrug resistant MCF-7/MDR1 cell lines were obtained from Dr. M.M. Gottesman, National Institutes of Health. MCF-7/MDR1 cell line is a subclone of MCF-7 cells stably transfected with MDR1 gene. Both cell lines were routinely maintained in exponential growth in 5% carbon dioxide at 37°C. DMEM fortified with 10% fetal bovine serum (FBS), glutamine (5 mM), sodium pyruvate (1 mM), and antibiotics (100 units/mL of penicillin and 100 μg/ml of streptomycin) were used for MCF-7 cell culture. Same medium with addition of 60 ng/mL of colchicine was used for MCF-7/MDR1 cell culture. In the experiments, the MCF-7/MDR1 cells were subcultured with removal of colchicine. The culture medium, FBS, sodium pyruvate, and antibiotics were all purchased from Invitrogen (Carlsbad, CA). Loperamide, doxorubicin, colchicine, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma (St. Louis, MO).
MTT assay of cell viability
The viability of MCF-7 and MCF-7/MDR1 cells after treatment with doxorubicin in the presence or absence of loperamide was analyzed with MTT assay. Briefly, cells were seeded into 96-well microplates (1 × 104 cells/well) and they were allowed to grow to 50%–70% confluence. The cells were then exposed to graded concentrations of doxorubicin alone or in combination with loperamide for different durations. After the old medium was removed, MTT solution (100 μL, 0.5 mg/mL) in phenol red-free DMEM was added to each well and the cells were further incubated for 3 hours (hr) at 37°C. The medium with MTT solution was then removed, followed by addition of 100 μL of dimethyl sulfoxide, and incubated for an extra 30 minutes (min). The absorbance was measured at the wavelength of 560 nm with a multiwell spectrophotometer (Bio-Rad, Hercules, CA). The half-maximal inhibitory concentrations (IC50) of doxorubicin in the presence or absence of loperamide were calculated. At least five replicates were performed in all experiments, and each experiment was repeated at least three times. The representative data are presented.
Confocal microscopic observation of intracellular accumulation of doxorubicin
Tumor cells were grown on 8-chamber glass slides. Twenty-four hours later, the cells at about 30% confluence were incubated with doxorubicin (10 and 20 μM, respectively) alone or in combination with loperamide (10 and 20 μM, respectively) for 3 hr. Cells were washed thrice with phosphate-buffered saline (PBS), and the slides were then mounted for confocal microscopy. To observe the efflux of intracellular doxorubicin, another set of samples were further incubated for 2 hr in fresh medium without doxorubicin and loperamide. Images were acquired with an Olympus FV300 confocal microscope (Olympus, Center Valley, PA) (18). All samples were analyzed under the same condition.
Flow cytometric quantification of net uptake and eflux of doxorubicin
Cell net uptake and efflux of doxorubicin in the presence or absence of loperamide were quantified with a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Briefly, MCF-7/MDR1 cells were harvested from cultures in the exponential phase of growth, seeded into 10-cm culture dishes, and allowed to grow overnight. The cells were then incubated with doxorubicin alone or in combination with loperamide (10 μM and 20 μM, respectively) for 3 hr. One set of the cell samples was washed twice with PBS and harvested, and the fluorescence intensity was determined. These samples also served as the baseline controls for doxorubicin efflux analysis. To determine the efflux of doxorubicin from cells, all other samples were further incubated in fresh medium without doxorubicin and loperamide or with loperamide alone for extra 30 min and 2 hr, separately. Cells without any drugs were used to assess the background fluorescence in the FL2-H channel. A minimum of 10,000 events were collected for each sample. The data were analyzed with CELLQuest 3.2.1.f1 software (BD Biosciences). The fluorescence intensity was expressed as GeoMean. The net uptake of doxorubicin was expressed as the fluorescence signal intensity of the treated cells minus that of the cells without exposure to any drugs. The efflux rate was expressed as percentage loss of intracellular net fluorescence intensity relative to the baseline with the formula of (baseline intensity − intensity at 30 min or 2 hr after drug removal)/baseline intensity × 100%. The data from 3 replicates are shown.
Semiquantitative reverse transcriptase polymerase chain reaction (RT-PCR)
Total RNA was extracted from cells with TRIzol reagents (Invitrogen). PCR was set up according to the instructions of SuperScript One-Step RT-PCR with Platinum Taq Kit (Invitrogen). Two sets of primers purchased from Sigma were used to amplify β-actin (383 base pairs, used as an internal control) and MDR1 gene (157 base pairs). PCR was performed using GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA). After a formation of cDNA step at 50°C for 40 min, 25 cycles of PCR amplification were performed. Each cycle consisted of a denaturing step at 94°C for 15 seconds (s), annealing at 55°C for 30 s, and extension at 72°C for 30 s, followed by a final step at 72°C for 7 min. The amplified fragments were separated via 2% (W/V) agarose gel electrophoresis and detected after ethidium bromide staining. The intensity of each band was determined with the image analysis system CHEMI DOC XRS (Bio-Rad). The specific gene expression level was expressed as the ratio of densitometric values from MDR1 in relation to the internal control, β-actin (MDR1 gene expression/β-actin expression).
Western blot analysis
Cells were washed twice with PBS and collected in protein lysis buffer (Sigma). The mixture was placed on ice for 30 min. Following centrifugation at 15,000 rpm for 15 min at 4°C, the supernatant was collected and the protein concentration was determined with the Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad). Whole cell lysate (30 μg protein) was resolved in 8% SDS-PAGE gel, transferred to polyvinylidene difluoride membrane (Immobilon; Amersham Corp., Arlington Heights, IL), and probed with monoclonal antibodies against MDR1 (Santa Cruz Biotech, Santa Cruz, CA) and β-actin (Santa Cruz Biotech), separately, at 4°C overnight. Blots were washed thrice (10 min each) with PBS + 0.1% Tween 20 and incubated with horseradish peroxidase-conjugated anti-mouse antibody (Santa Cruz Biotech) for 2 hr at room temperature. Blots were developed with ECL detection system (Bio-Rad).
Statistical analysis
The results were analyzed with the statistical software OriginPro 7.0 (OriginLab, Northampton, MA). A p value of < .05 was considered to be significantly different between any two sets of data.
RESULTS
Loperamide-mediated reversal of MCF-7/MDR1 cell resistance to doxorubicin
MCF-7 cells were sensitive to doxorubicin. After treatment with 3 μM doxorubicin for 72 hr, the cell viability was only 11% of the viability of untreated control cells. The IC50 of doxorubicin was ~1.2 μM against MCF-7 cells [see Figure 1(A)]. In contrast, MCF-7/MDR1 cells were highly resistant to doxorubicin. The IC50 of doxorubicin against MCF-7/MDR1 cells increased to 11 μM, 9.2-fold more resistant than the parental MCF-7 cells [see Figure 1(B)]. After treatment with 10 and 20 μM of doxorubicin for 72 hr, the viabilities of MCF-7/MDR1 cells were 52% and 36% relative to the viability of untreated control cells, respectively.
Figure 1.

Cell viability of MCF-7 and MCF-7/MDR1 cells. Cells treated with doxorubicin alone for 72 hr, indicating (A) the sensitivity of the MCF-7 cells and (B) the resistance of MCF-7/MDR1 cells to doxorubicin. (C) Loperamide-mediated enhancement of doxorubicin cytotoxicity to MCF-7/MDR1 cells, showing a dose-dependent manner of the enhancement.
Loperamide significantly enhanced the doxorubicin cytotoxicity to MCF-7/MDR1 cells and the enhancement was dose-dependent [see Figure 1(C)]. In the presence of 10 μM loperamide and doxorubicin for 72 hr, the cell viability was 56% and 24% of the viability of the cells treated with 10 and 20 μM of doxorubicin alone, respectively (both p < .05). In the presence of 20 μM loperamide and doxorubicin, the cell viability was only 34% and 18% of the viability of the cells treated with 10 and 20 μM of doxorubicin alone, respectively (both p < .05). The IC50 of doxorubicin decreased from 11 to 3 μM and 1 μM in the presence of 10 and 20 μM of loperamide, respectively. Loperamide did not significantly enhance the doxorubicin cytotoxicity to MCF-7 cells (data not shown). Only mild cytotoxicity (<7%) to MCF-7 and MCF-7/MDR1 cells was observed when the cells were exposed to loperamide alone at the levels up to 20 μM.
Observation of increased nuclear accumulation and decreased eflux
Confocal microscopy was used to observe the intracellular localization and accumulation of doxorubicin in MCF-7/MDR1 cells. Following treatment with doxorubicin alone for 3 hr, weak fluorescence was observed in the nuclei of the cells. The fluorescence was stronger in cells exposed to 20 μM than in cells exposed 10 μM of doxorubicin. Addition of loperamide increased doxorubicin accumulation in the nuclei of the cells as judged by more intense fluorescence (see Figure 2). The fluorescent intensity in the cell nuclei was dependent on the dosage of loperamide [see Figure 2(A)–(C)]. Dot-like pattern of fluorescence was evident in the cytoplasm of some cells exposed to 20 μM of each doxorubicin and loperamide [see Figure 2(C)]. Two hours after removal of doxorubicin and loperamide, the nuclear signal of doxorubicin was still evident [see Figure 2(E) and (F)]. On the contrary, only a weak cytoplasmic signal was observed in the control cells treated with doxorubicin alone after removal of doxorubicin [see Figure 2(D)]. The microscopic observation was consistent with the cell viability data and both indicated that loperamide decreased the efflux of doxorubicin from the cells.
Figure 2.

Confocal images of nuclear accumulation of doxorubicin (20 μM) in the presence or absence of loperamide in MCF-7/MDR1 cells. (A)–(C) show the images from the cells after treatment for 3 hr, demonstrating increased nuclear accumulation of doxorubicin in the presence of loperamide. (A) Cells treated doxorubicin alone. (B) Cells treated with doxorubicin and 10 μM loperamide. (C) Cells treated with doxorubicin and 20 μM loperamide. (D)–(F) are the images from the cells after the removal of both doxorubicin and loperamide. (D) Cells pretreated with doxorubicin alone. (E) Cells pretreated with doxorubicin and 10 μM loperamide. (F) Cells pretreated with doxorubicin and 20 μM loperamide. The original magnification of all images was 400×, and the images were taken under the same microscope settings.
Quantification of loperamide-mediated cellular uptake and eflux of doxorubicin
The loperamide-mediated net uptake and efflux of doxorubicin in the MCF-7/MDR1 cells were quantified with flow cytometric analysis. Similar to the findings of confocal microscopy, flow cytometric analysis also showed a significant increase of the net uptake and a decrease of the efflux of doxorubicin in the presence of loperamide [see Figure 3(A) and 3(B)].
Figure 3.

Flow cytometric analysis of the net uptake of doxorubicin and rhodamine 123 and eflux of doxorubicin. (a) and (b) Fluorescent intensity after treatment with doxorubicin (10 and 20 μM) alone or in combination with loperamide (10 and 20 μM) for 3 hr, showing loperamide-mediated increase of doxorubicin accumulation. (a) Representative of the flow cytometric analysis. (b) Fold change in the intracellular fluorescent signal intensity in the presence loperamide relative to doxorubicin alone. (c) and (d) Fluorescent intensity following removal of doxorubicin (10 and 20 μM), but not loperamide (10 and 20 μM) for 2 hr, showing decreased eflux of doxorubicin by loperamide. (c) Representative of the flow cytometric analysis. (d) Loss percentage of the intracellular fluorescent signal in the presence or absence of loperamide. Samples in (a) and (c): (1) untreated; (2) doxorubicin 10 μM; (3) doxorubicin 10 μM + loperamide 10 μM; (4) doxorubicin 10 μM + loperamide 20 μM; (5) doxorubicin 20 μM; (6) doxorubicin 20 μM + loperomide 10 μM; (7) doxorubicin 20 μM + loperomide 20 μM. (e) Fluorescent intensities in MCF-7/MDR1 cells were analyzed by flow cytometry after treatment with rhodamine 123 (100 μM) alone or in the combination with rhodamine 123 (100 or 200 μM) and loperamide (10 and 20 μM) for 3 hr.
In the case of net uptake, after treatment of the MCF-7/MDR1 cells for 3 hr with doxorubicin alone, the fluorescence signal intensity (geometric mean) was 110.78 upon treatment with 10 μM and 212.66 upon treatment with 20 μM doxorubicin. Significantly, with addition of 10 μM of loperamide, the fluorescence signal intensity was 3.7-fold higher upon 10 μM and a 4.5-fold higher upon treatment with 20 μM doxorubicin. With addition with 20 μM of loperamide, the fluorescence signal intensity was 3.3-fold higher in 10 μM and 6.3-fold higher upon treatment with 20 μM doxorubicin [all, p < .05; see Figure 3(B)].
In the case of efflux of the intracellular doxorubicin after removal of the drugs from culture media, the intracellular signal loss was significantly lower in the cells treated with a combination of doxorubicin and loperamide compared with loss of fluorescent signal from treated with doxorubicin alone (p < .05). Relative to the signal before drug removal, the signal loss was 42.2% and 41.7% at 30 min and 49.9% and 57.6% at 2 hr in the cells with 3-hr treatment of doxorubicin alone at 10 and 20 μM, respectively. Differentially, the signal loss at 30 min was only 15.2% (with 10 μM of loperamide) and 7.4% (with 20 μM of loperamide) in the cells treated with the combination of loperamide and 10 μM of doxorubicin. This value was 19.6% (with 10 μM of loperamide) and 2.8% (with 20 μM of loperamide) in the cells treated with the combination of loperamide and 20 μM of doxorubicin. However, the signal loss rate (percentage) at 2 hr was similar in the cells regardless of presence or absence of loperamide, although the absolute residual signal intensity was still higher in cells treated with a combination of doxorubicin and loperamide than that with doxorubicin alone.
The efflux rate of the intracellular doxorubicin was further determined with continuous exposure of loperamide for 2 hr but removal of doxorubicin in the cells treated with both drugs. Consistently, continuous exposure of loperamide alone decreased the efflux of intracellular doxorubicin compared with efflux in the absence of loperamide. The signal loss was 25.5% and 12.6% for 10 μM of doxorubicin treatment and 21.3% and 10.9% for 20 μM of doxorubicin treatment in the presence of 10 and 20 μM of loperamide, respectively [all, p < .05; see Figure 3(C) and 3(D)].
Quantification of loperamide-mediated cellular uptake of rhodamine 123
A similar test was performed with rhodamine 123 in the MCF-7/MDR1 cells to validate the ability of loperamide-mediated cellular uptake and efflux of doxorubicin [see Figure 3(E)]. As expected, flow cytometric analysis also showed a significant increase of the net uptake of rhodamine 123 in the presence of loperamide [see Figure 3(E)]. Loperamide enhanced the uptake of rhodamine 123 in a dose-dependent fashion.
The mRNA and protein expression of MDR1 gene
To test whether loperamide changed the expression of MDR1 gene, the mRNA and protein levels were estimated with semi-quantitative RT-PCR and Western blot analysis. Following treatment with loperamide (10 and 20 μM) for 3 and 6 hr, no significant changes were detected at both mRNA and protein levels (Figure 4, upper panel), relative to the controls without loperamide exposure (Figure 4, lower panel).
Figure 4.
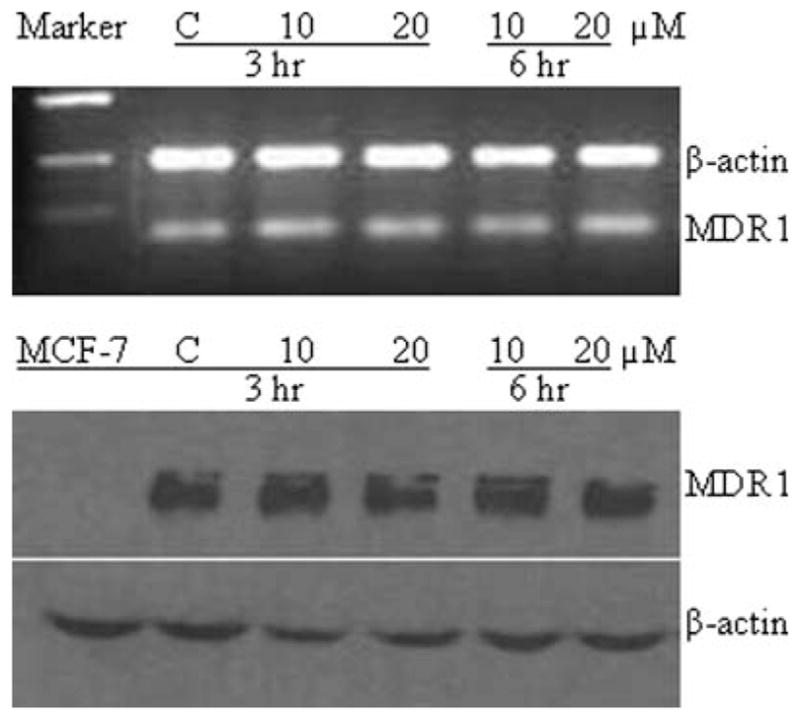
Semiquantitative RT-PCR and Western blot analysis of MDR1 gene expression following treatment with loperamide (20 μM) for 3 and 6 hr, showing no evident changes in the mRNA and protein levels. Upper panel: RT-PCR of MDR1 and control of β-actin; lower panel: Western blot analysis of MDR1 and β-actin (as control) protein expression.
DISCUSSION
Loperamide is an FDA-approved drug, well known for its effectiveness against diarrhea. Because of its high-affinity for P-gp, loperamide is also used as an indicator of permeability change in the BBB. We hypothesized that loperamide can effectively compete with chemotherapeutic agents for binding with P-gp and thus decrease their efflux from drug resistant cancer cells. Cell viability assays demonstrated that loperamide enhanced doxorubicin cytotoxicity against resistant cells in a dose-dependent manner. The findings were consistent with the confocal microscopic observations that the fluorescent signal of doxorubicin in the nuclei of resistant cells was stronger in the presence of loperamide. Quantification with flow cytometric analysis revealed that loperamide significantly increased the net cellular uptake and decreased the efflux of doxorubicin. Even when both doxorubicin and loperamide were removed from the media, the efflux rate of intracellular doxorubicin was still significantly low within 30 min. These results indicate that loperamide’s action is mediated by its ability to promote increased uptake and decreased efflux of doxorubicin in resistant cancer cells.
Studies designed to investigate cellular retention and efflux of doxorubicin in the absence or presence of loperamide may be complicated due to the cytotoxicity of doxorubicin. Therefore, we validated the findings by further investigation of the retention and efflux of rhodamine 123 in the presence of loperamide. Rhodamine 123 is a nontoxic fluorescent dye and a P-gp substrate (19, 20). Loperamide also enhanced the uptake of rhodamine 123 in a dose-dependent fashion [see Figure 3(E)]. The data with rhodamine 123 were consistent with the findings obtained with doxorubicin.
The ability of loperamide to reverse MDR may be explained by its high affinity for P-gp, which allows it to more effectively bind P-gp than doxorubicin. Altered expression of MDR1 gene was not detected at both mRNA and protein levels in the cells treated with loperamide. P-gp inhibitors, such as verapamil, cyclosporine A, and PSC833, exhibit a similar competitive mechanism in reversing MDR (2, 10). However, it is unclear whether the binding of loperamide with P-gp causes conformational change in the P-gp protein. Notably, P-gp activity differs widely among individuals and a functionally relevant polymorphism in P-gp has been described in the literature (21). Loperamide is a substrate of P-gp in the BBB, but it is unclear whether loperamide acts as a substrate and/or inhibitor of P-gp in multidrug resistant cancer cells. In the studies by Wandel et al., loperamide has been shown to be a substrate with high dependence on P-gp in the basal–apical transport in the MDR1-transfected kidney-derived L-MDR1 cells (22). On the other hand, loperamide inhibits P-gp-mediated digoxin transport in Caco-2 human colon carcinoma cells which constitutively express P-gp. Loperamide treatment at 20 μM level resulted in greater than 50% inhibition of the P-gp activity in Caco-2 cells (22). Another mode of action of loperamide may be through its role in autophagy. Autophagy is a lysosome-dependent cellular catabolic mechanism for mediating the turnover of intracellular organelles and long-lived proteins. Reduction of autophagy results in accumulation of misfolded proteins in neurons. Loperamide has been shown to increase the degradation rate of long-lived cellular proteins. Another important feature of loperamide is that it blocks the action of voltage-dependent calcium channels (14). Many compounds recognized as MDR reversers are calcium channel blockers and calmodulin inhibitors (2). These features of loperamide may also contribute to its reversal effect on MDR. Elucidation of the mechanism of its action will facilitate the clinical application of loperamide in combination with chemotherapeutic drugs.
Loperamide is a high affinity P-gp substrate. With the system of multidrug resistant MCF-7/MDR1 cells and doxorubicin, we have confirmed the reversal effect of loperamide on the resistance of tumor cells to doxorubicin. Although further confirmation in animals is critical and studies are ongoing in our laboratory, the promising in vitro results suggest that loperamide may benefit patients with MDR tumors when used in combination with chemotherapeutic agents. Because loperamide has minimal side effects and is an over-the-counter drug, chemotherapy in combination with loperamide can be rapidly translated to clinical applications.
Acknowledgments
This work was supported in part by grants Howard/Hopkins Partnership grant, U54 CA091431 (YZ and SS) and P20 CA118770 (Y.Z) from the National Institutes of Health, and DAMD17-03-1-0759 from U.S. Army Medical Research and Materiel Command.
Footnotes
DECLARATION OF INTEREST
The authors report no declarations of interest.
References
- 1.Choi C. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005;5:30. doi: 10.1186/1475-2867-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clarke R, Currier S, Kaplan O, Lovelace E, Boulay V, Gottesman MM, Dickson RB. Effect of P-glycoprotein expression on sensitivity to hormones in MCF-7 human breast cancer cells. J Natl Cancer Inst. 1992;84:1506–1512. doi: 10.1093/jnci/84.19.1506. [DOI] [PubMed] [Google Scholar]
- 3.Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU, Duncan AM, Deeley RG. Overexpression of a transporter gene in a multidrug-resistant human lung AM cancer cell line. Science. 1992;258:1650–1654. doi: 10.1126/science.1360704. [DOI] [PubMed] [Google Scholar]
- 4.Coley HM. Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat Rev. 2008;34:378–390. doi: 10.1016/j.ctrv.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Cox DS, Scott KR, Gao H, Raje S, Eddington ND. Influence of multidrug resistance (MDR) proteins at the blood-brain barrier on the transport and brain distribution of enaminone anticonvulsants. J Pharm Sci. 2001;90:1540–1552. doi: 10.1002/jps.1104. [DOI] [PubMed] [Google Scholar]
- 6.Damle BD, Sridhar R, Desai PB. Dipyridamole modulates multidrug resistance and intracellular as well as nuclear levels of doxorubicin in B16 melanoma cells. Int J Cancer. 1994;56:113–118. doi: 10.1002/ijc.2910560120. [DOI] [PubMed] [Google Scholar]
- 7.Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007–1017. [PubMed] [Google Scholar]
- 8.Fojo T, Coley HM. The role of eflux pumps in drug-resistant metastatic breast cancer: new insights and treatment strategies. Clin Breast Cancer. 2007;7:749–756. doi: 10.3816/CBC.2007.n.035. [DOI] [PubMed] [Google Scholar]
- 9.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 10.Gottesman MM, Ling V. The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett. 2006;580:998–1009. doi: 10.1016/j.febslet.2005.12.060. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmöller J, Johne A, Cascorbi I, Gerloff T, Roots I, Eichelbaum M, Brinkmann U. Functional polymorphisms of the human multiresistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA. 2000;97:3473–3478. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamel AM, El-Sharkawy N, Yassin D, Shaaban K, Hussein H, Sidhom I, Abo El-Naga S, Ameen M, El-Hattab O, Aly El-Din NH. P-gp expression and Rh 123 eflux assay have no impact on survival in Egyptian pediatric acute lymphoblastic leukemia patients. J Egypt Natl Canc Inst. 2005;17:165–172. [PubMed] [Google Scholar]
- 13.Li J, Xu LZ, He KL, Guo WJ, Zheng YH, Xia P, Chen Y. Reversal effects of nomegestrol acetate on multidrug resistance in adriamycin-resistant MCF7 breast cancer cell line. Breast Cancer Res. 2001;3:253–263. doi: 10.1186/bcr303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loetchutinat C, Saengkhae C, Marbeuf-Gueye C, Garnier-Suillerot A. New insights into the P-glycoprotein-mediated effluxes of rhodamines. Eur J Biochem. 2003;270:476–485. doi: 10.1046/j.1432-1033.2003.03403.x. [DOI] [PubMed] [Google Scholar]
- 15.Ludwig JA, Szakács G, Martin SE, Chu BF, Cardarelli C, Sauna ZE, Caplen NJ, Fales HM, Ambudkar SV, Weinstein JN, Gottesman MM. Selective toxicity of NSC73306 in MDR1-positive cells as a new strategy to circumvent multidrug resistance in cancer. Cancer Res. 2006;66:4808–4815. doi: 10.1158/0008-5472.CAN-05-3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nobili S, Landini I, Giglioni B, Mini E. Pharmacological strategies for overcoming multidrug resistance. Curr Drug Targets. 2006;7:861–879. doi: 10.2174/138945006777709593. [DOI] [PubMed] [Google Scholar]
- 17.Pauli-Magnus C, Feiner J, Brett C, Lin E, Kroetz DL. No effect of MDR1 C3435T variant on loperamide disposition and central nervous system effects. Clin Pharmacol Ther. 2003;74:487–498. doi: 10.1016/S0009-9236(03)00234-0. [DOI] [PubMed] [Google Scholar]
- 18.Weaver JL, Pine PS, Aszalos A, Schoenlein PV, Currier SJ, Padmanabhan R, Gottesman MM. Laser scanning and confocal microscopy of daunorubicin, doxorubicin, and rhodamine 123 in multidrug-resistant cells. Exp Cell Res. 1991;196:323–329. doi: 10.1016/0014-4827(91)90267-x. [DOI] [PubMed] [Google Scholar]
- 19.Schinkel AH, Wagenaar E, Mol CA, van Deemter L. P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest. 1996;97:2517–2524. doi: 10.1172/JCI118699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skarke C, Jarrar M, Schmidt H, Kauert G, Langer M, Geisslinger G, Lötsch J. Effects of ABCB1 (multidrug resistance transporter) gene mutations on disposition and central nervous effects of loperamide in healthy volunteers. Pharmacogenetics. 2003;13:651–660. doi: 10.1097/00008571-200311000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Wu J, Lu Y, Lee A, Pan X, Yang X, Zhao X, Lee RJ. Reversal of multidrug resistance by transferrin-conjugated liposomes co-encapsulating doxorubicin and verapamil. J Pharm Pharm Sci. 2007;10:350–357. [PubMed] [Google Scholar]
- 22.Wandel C, Kim R, Wood M, Wood A. Interaction of morphine, fentanyl, sufentanil, alfentanil, and loperamide with the eflux drug transporter P-glycoprotein. Anesthesiology. 2002;96:913–920. doi: 10.1097/00000542-200204000-00019. [DOI] [PubMed] [Google Scholar]