

Abstract
The combined molecular dynamics (MD) and potential of mean force (PMF) simulations have been performed to determine the free energy profiles for the binding process of (−)-cocaine interacting with wild-type cocaine esterase (CocE) and its mutants (T172R/G173Q and L119A/L169K/G173Q). According to the MD simulations, the general protein-(−)-cocaine binding mode is not affected by the mutations, e.g. the benzoyl group of (−)-cocaine is always bound in a sub-site composed of aromatic residues W151, W166, F261, and F408 and hydrophobic residue L407, while the carbonyl oxygen on the benzoyl group of (−)-cocaine is hydrogen-bonded with the oxyanion-hole residues Y44 and Y118. According to the PMF-calculated free energy profiles for the binding process, the binding free energies for (−)-cocaine with the wild-type, T172R/G173Q, and L119A/L169K/G173Q CocEs are predicted to be −6.4, −6.2, and −5.0 kcal/mol, respectively. The computational predictions are supported by experimental kinetic data, as the calculated binding free energies are in good agreement with the experimentally-derived binding free energies, i.e. −7.2, −6.7, and −4.8 kcal/mol for the wild-type, T172R/G173Q, and L119A/L169K/G173Q, respectively. The reasonable agreement between the computational and experimental data suggests that the PMF simulations may be used as a valuable tool in new CocE mutant design that aims to decrease the Michaelis-Menten constant of the enzyme for (−)-cocaine.
Introduction
The naturally occurring and biologically active (−)-cocaine is considered to be the most addictive substance abused by millions of people worldwide.1,2,3,4 The disastrous medical and social consequences of cocaine addiction have made the development of an effective pharmacological treatment a high priority.5,6 It has been demonstrated that cocaine exerts its effects on central nervous system (CNS) through blocking the reuptake of neurotransmitter dopamine, thus potentiating the effects of dopamine in the synapse.7, 8, 9 The traditional pharmacodynamic approach has failed to produce a therapeutically useful small-molecule drug due to the difficulties inherent in blocking a blocker like cocaine.3,7,9 As an alternative, pharmacokinetic approach using an efficient cocaine-metabolizing enzyme has become a promising strategy for treatment of cocaine overdose and abuse. The enzyme strategy aims at accelerating the hydrolysis of cocaine and, therefore, eliminating cocaine quickly from the peripheral circulation.10,11,12,13,14 For this purpose, bacterial cocaine esterase (CocE)11,12,15,16,17,18,19,20,21,22,23,24 is a promising choice as a potential anti-cocaine agent for therapeutic treatment of cocaine overdose and abuse, because CocE is the most efficient natural enzyme against (−)-cocaine. Native CocE and the designed thermostable mutants are capable of protecting against cocaine-induced lethality.19,20,22,23 Thus, it is interesting to develop CocE mutants that are more efficient for the catalytic hydrolysis of (−)-cocaine.
Generally speaking, rational design of a highly efficient enzyme mutant is extremely challenging, particularly when the enzymatic reaction process consists of multiple steps.19,21,22,23,24 The catalytic efficiency (kcat/KM) of an enzyme for a substrate is determined by both the reaction rate constant (kcat) and the Michaelis-Menten constant (KM). The later is associated with the binding affinity of substrate with the enzyme. In order to design a mutant enzyme with an improved catalytic efficiency for a given substrate, one needs to design possible amino-acid mutations that can increase the binding affinity (associated with a smaller KM value) of the substrate and/or increase the corresponding kcat value. The general reaction pathway for CocE-catalyzed hydrolysis of (−)-cocaine has been uncovered by extensive molecular dynamics (MD) simulations and reaction-coordinate calculations using first-principles quantum mechanics/molecular mechanics (QM/MM) methods.21 The computational study have revealed that (−)-cocaine is bound in a site located on the interface of three domains of CocE,11,19 and the rate-determining step is the nucleophilic attack on the carbonyl carbon at the benzoyl group of (−)-cocaine by a water molecule. In our previous studies,19,21,24 we have designed and discovered several thermostable mutants of CocE through computational design, followed by in vitro and in vivo studies. The designed CocE mutants, i.e. T172R, G173Q, T172RG173Q, and L169K, have significantly increased thermostability of the enzyme in vitro and in vivo.19,22,23 Results obtained from previous studies19,20,22,23,24 have revealed that the enzyme can be stabilized by enhanced intra-molecular interactions resulted from these specific mutations. However, it is unclear about how these enhanced intra-molecular interactions affect the CocE-(−)-cocaine binding. Answer to this question is essential for us to better understand the binding mechanism of CocE, and such understanding will help us to rationally design novel mutants of this enzyme with higher catalytic efficiency against (−)-cocaine.
In the present study, we first performed MD simulations and potential of mean force (PMF) simulations to determine the free energy profiles for the substrate binding process of wild-type CocE and T172R/G173Q mutant. The combined MD and PMF simulations have revealed that these two enzyme-substrate (ES) complexes have similar free energy profiles, and the calculated binding affinity for (−)-cocaine with the thermostable mutant is lower than that of the wild-type CocE-(−)-cocaine binding. Based on the analysis of the simulated ES complexes and the calculated binding free energies, the L119A/L169K/G173Q mutant is predicted to have a significantly lower binding affinity with (−)-cocaine compared to the wild-type or the T172R/G173Q mutant. The computational prediction has been confirmed by wet experimental tests. The agreement between the computational and experimental data suggests that the PMF simulation is a reliable protocol to predict the binding free energy of new CocE mutant binding with (−)-cocaine. The novel insights obtained from the MD and PMF simulations should be helpful for future design of CocE mutants with an improved catalytic efficiency against (−)-cocaine.
Methods
MD Simulations
The initial structures of the ES complexes were prepared based on the published X-ray crystal structures of CocE22 and the results of our previous molecular docking and MD simulations.19,21,24 The PDB code is 3I2J at resolution of 2.01 Å for wild-type CocE, 3I2G at resolution of 2.50 Å for G173Q, 3I2F at resolution of 2.50 Å for the T172R/G173Q mutant, and 3I2H at resolution of 1.65 Å for the L169K mutant.22 The initial structure of the L119A/L169K/G173Q mutant was prepared based on the X-ray crystal structures of the L169K and G173Q mutants. By superimposing the Cα atoms of the G173Q mutant with the corresponding Cα atoms of the L169K mutant, the atomic positions of the Q173 side chain were copied and the structure of the L169K/G173Q mutant was generated. Starting from the structure of the L169K/G173Q mutant, the structure of the L119A/L169K/G173Q mutant was generated through the L119A mutational modeling by using the X-leap module of Amber 9 program.25 The coordinates of the backbone atoms plus the Cβ atom of L119 were used for the corresponding atoms of A119. The redundant atoms on L119 side chain were deleted and the hydrogen atoms of A119 were added automatically. The generated structure of the L119A/L169K/G173Q mutant was then energy-minimized in order to optimize the interactions between the mutated residues and the surrounding residues. The energy minimization was performed by using the Sander module of Amber 9 program, via a combined use of the steepest descent/conjugate gradient algorithms, with a convergence criterion of 0.01 kcal mol−1 Å−1, and the non-bonded cutoff distance was set to 10.0 Å. The energy minimization was performed first on the mutated residues, and then on the residues within 5 Å around any of the mutated residues. The structure of the L119A/L169K/G173Q CocE-(−)-cocaine complex was constructed in a similar way as that for the other ES complexes. In order to further relax each constructed ES structure, MD simulations were performed by using the Sander module of Amber 9 program package.25 The general procedure of the MD simulations was similar to that used in our previously reported other computational studies.19,21,24 In particular, the molecular mechanics force field parameters and the partial charges of (−)-cocaine atoms were adopted directly from those developed in our previous studies.6,13,14,19,21,24 Briefly, the partial charges of (−)-cocaine atoms were calculated by using the restrained electrostatic potential-fitting (RESP) protocol implemented in the Antechamber module of Amber 9 program, following the electrostatic potential (ESP) calculation at ab initio HF/6-31G* level using Gaussian 03 program.26 Each of the ES complex structures was solvated in a rectangular box of TIP3P water molecules27 with a minimum solvent-wall distance of 10 Å. Sodium counter ions (Na+) were added to neutralize the solvated system. The solvated system was gradually heated to 298.15 K by weak-coupling method28 and equilibrated for 400 ps. During the MD simulations, a 10.0 Å non-bonded interaction cutoff was used and the non-bonded list was updated every 25 steps. The motion for the mass center of the system was removed every 1,000 steps. The particle-mesh Ewald (PME) method29,30 was applied to treat long-range electrostatic interactions. The lengths of covalent bonds involving hydrogen atoms were fixed with the SHAKE algorithm,31 enabling the use of a 2-fs time step to numerically integrate the equations of motion. Finally, the production MD was kept running for ~2.0 ns with a periodic boundary condition in the NTP ensemble at T = 298.15 K with Berendsen temperature coupling, and at P = 1 atm with isotropic molecule-based scaling.28,32
Potential of Mean Force (PMF) Simulations
In order to explore the free energy profiles for the process of (−)-cocaine binding with wild-type CocE and its mutants, PMF simulations were carried out by using umbrella-sampling33 MD simulations. The classic PMF definition34 can be represented by a function of reaction coordinate as
| (1) |
In Eq.(1), ρ(χ) is the probability density along the reaction coordinate χ, R is the gas constant, T is the simulation temperature, U(χ) is the biasing potential applied in the umbrella-sampling MD simulations, and F is the normalization constant. According to this approach, the reaction coordinate is usually divided into different regions, i.e., windows, and each of which is sampled separately. A biasing (umbrella) potential, i.e. U(χ), is applied for each window in order to obtain nearly uniform sampling of the potential energy surface. In the present study, the reaction coordinate was defined as the distance from the mass center of the non-hydrogen atoms of (−)-cocaine to the mass center of the non-hydrogen atoms on the side chains of residues H87, V121, and L146 of the enzyme. The total number of windows for each complex structure was about 70, depending on the starting structure of each system. Each window was separated by 0.3 Å, covering the reaction coordinate from ~11.43 Å to 32.93 Å. The biasing force constant applied in different windows of umbrella-sampling was 10.0 kcal/(mol·Å2). For each umbrella-sampling window, the initial complex structure was selected from the last snapshot of the PMF simulations of the previous window. The selected structure for each window was first equilibrated for 200 ps and then kept running for 800 ps for production sampling. The frequency for data collection was set to 1 fs, which was the same as that of the time step of umbrella-sampling MD.
After all the umbrella-sampling MD simulations were finished for each system, the data collected from separate simulation windows were combined along the reaction coordinate. These data were then used to calculate the PMF for the whole binding process with the weighed histogram analysis method (WHAM)35,36 using the code developed by Alan Grossfield (http://membrane.urmc.rochester.edu/Software/WHAM/WHAM.html).
Most of the MD and umbrella-sampling MD simulations were performed on a supercomputer (e.g. DELL Cluster with 388 nodes or 4,816 processors) at University of Kentucky's Computer Center. Some other modeling and computations were carried out on SGI Fuel workstations in our own lab.
Experimental Procedure
Site-directed mutagenesis was generated by using QuickChange (Stratagene) and CocE cDNA cloned in the bacterial expression vector, pET-22b (+). The enzyme (the L119A/L169K/G173Q mutant of CocE) was expressed as 6×His-tagged proteins in E. coli BL-21 (DE3) cells grown at 37°C. Protein expression was induced with 1 mM isopropyl-β-thiogalactopyranoside (Sigma Aldrich) for ~15 h at 18°C. Cells were pelleted, resuspended in 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, and a protease inhibitor cocktail (34 μg/ml each of L-tosylamido-2-phenylethyl chloromethyl ketone, 1-chloro-3-tosylamido-7-amino-2-heptanone, and phenylmethylsulfonyl fluoride, and 3 μg/ml each of leupeptin and lima bean trypsin inhibitor) and lysed using a French Press (Thermo Fisher Scientific, Waltham, MA). The 6×His-tagged enzyme was enriched using HisPur™ Cobalt Resin (Thermo Fisher Scientific, Waltham, MA) storage buffers containing 20 mM HEPES, pH 8.0, 2 mM MgCl2, 1 mM EDTA, and 1 mM dithiothreitol. The fractions were concentrated by using an Amicon Ultra-50K centrifuge (Millipore, Billerica, MA). The enzyme concentration was determined using CB-Protein Assay™ Kit (from CALBIOCHEM) with bovine serum albumin as a standard.
To determine the catalytic activity of the enzyme against (−)-cocaine, the initial rates of the enzymatic hydrolysis of (−)-cocaine at various concentrations were determined by using a sensitive radiometric assay based on toluene extraction of [3H](−)-cocaine labeled on its benzene ring, as we did for the catalytic activity of BChE mutants against (−)-cocaine.13,14,37,38,39,40,41 Briefly, to initiate the enzymatic reaction, 100 nCi of [3H](−)-cocaine was mixed with 100 μl of culture medium. The enzymatic reaction proceeded at room temperature (25°C) with varying concentration of (−)-cocaine. The reaction was stopped by adding 200 μl of 0.05 M HCl, which neutralized the liberated benzoic acid while ensuring a positive charge on the residual (−)-cocaine. [3H]benzoic acid (one of the reaction products) was extracted by 1 ml of toluene and measured by scintillation counting. Finally, the measured (−)-cocaine concentration-dependent radiometric data were analyzed by using Prism 5 (GraphPad Software Inc., San Diego, CA).
Results and Discussion
MD-simulated ES Structures
As suggested in our previous study on CocE-catalyzed reaction mechanism,21 the (−)-cocaine binding site is located on the interface of the three domains of CocE. Figure 1 depicts the most important distances tracked from the MD simulations and the typical ES structure of wild-type CocE-(−)-cocaine complex derived from the last snapshot of the MD simulations. The plots for the tracked positional root-mean square deviation (RMSD) of all non-hydrogen atoms versus the simulation time are provided as Supporting Information (Figure S2). The performed MD simulations were also served to obtain stable ES structure used as the starting structure in subsequent PMF simulations (discussed below). As shown in Figure 1A, the distance from the carbonyl carbon at the benzoyl group of (−)-cocaine to the hydroxyl oxygen at the side chain of residue S117 fluctuates around 3.2 Å. Such a distance is suitable for the nucleophilic attack by the hydroxyl oxygen on the side chain of residue S117, which initiates the first chemical reaction step of the catalytic hydrolysis. The carbonyl oxygen on the benzoyl group of (−)-cocaine interacts with the oxyanion hole residues Y44 and Y118 through hydrogen-bonding interactions. In the typical CocE-(−)-cocaine complex (Figure 1B), the benzoyl group of (−)-cocaine is located in a sub-binding site composed of hydrophobic residues W151, W166, L169, L407, F408, F261, and P150 of CocE, packing in parallel with the aromatic side chain of W166, and in perpendicular with the aromatic side chain of F261. The mode of binding for the benzoyl group of (−)-cocaine is supported indirectly by the observations from the X-ray crystal structures of CocE in complex with either the benzoic acid (PDB code 1JU4 at resolution of 1.63 Å) or phenyl boronic acid (PDB code 1JU3 at resolution of 1.58 Å).11 As revealed in these X-ray structures, the phenyl ring of either the benzoic acid or the phenyl boronic acid was bound in the similar sub-site as that of the benzoyl group of (−)-cocaine (Figure 1B) and interacted with several hydrophobic residues including W166 and F261 of CocE. The binding mode of the benzoyl group of (−)-cocaine is also consistent with earlier results from site-directed mutagenesis of CocE.12 As reported, each of the mutations W151A, W166A, F261A, L407A, and F408A had some negative impact on the substrate binding, ca. 2~80-fold increase in the experimentally measured KM value, indicating a dramatic decrease in the substrate binding affinity. According to our modeled wild-type CocE-(−)-cocaine complex structure (Figure 1B), the hydrophobic packing between the benzoyl group of (−)-cocaine and the surrounding residues would be dramatically weakened by mutating any of these residues into Alanine residue which has a much smaller side chain.
Figure 1.

(A) Plots of important distances tracked through MD simulations on the binding structure of wild-type CocE-(−)-cocaine complex. C32---S117OG represents the distance between the carbonyl carbon on the benzoyl group of (−)-cocaine and the hydroxyl oxygen on the side chain of residue S117 of CocE; O33---Y44HH refers to the distance between the carbonyl oxygen on the benzoyl group of (−)-cocaine and the hydroxyl hydrogen on the side chain of residue Y44 of CocE; O33---Y118H represents the distance between the carbonyl oxygen on the benzoyl group of (−)-cocaine and the backbone hydrogen of residue Y118; Y44OH---W166NE refers to the hydrogen bond distance between the side chain of Y44 and the side chain of W166; S117HG---H287NE and H287HD---D259OD1/2 represent the distances related to the hydrogen bonds within the catalytic triad S117-H287-D259. (B) MD-simulated binding structure for wild-type CocE-(−)-cocaine complex. CocE is shown as gold ribbons and (−)-cocaine is in ball-and-stick style. Residues of CocE within 5 Å around (−)-cocaine molecule are shown in stick style and colored by atom type. Important distances are indicated with dashed lines and labeled.
The methyl ester group of (−)-cocaine stays just above residue H287 of CocE with a distance of ~3.8 Å between the methyl carbon and the center of the side chain of residue H287 (Figure 1B). The methyl ester group is also in close packing with residues V116, M141, and L290 of domain I (residues from #1 to #144 and from #241 to #354) of CocE. The cationic head group of (−)-cocaine is partly exposed to the surrounding solvent, and is surrounded by the side chains of residues Y44, A51, Q55, and L169. As residue Q55 is within 5 Å around the cationic head group of (−)-cocaine, it can be expected that the Q55D or Q55E mutation will increase the binding affinity (−)-cocaine, as the Q55D or Q55E will enhance the electrostatic interactions between the mutated residue and the cationic head group of (−)-cocaine. This explains why the experimentally measured KM value for the Q55E mutant decreased 2.5 fold.12
The tracked important distances from MD trajectories and the MD-simulated ES structures are depicted in Figure 2 for (−)-cocaine binding with the T172R/G173Q mutant, and in Figure 3 for (−)-cocaine binding with the L119K/L169K/G173Q mutant. In general, the binding mode of (−)-cocaine with each of these CocE mutants is similar to that of (−)-cocaine with wild-type CocE. For example, the benzoyl group of (−)-cocaine is stabilized in the binding site by hydrogen-bonding interactions between its carbonyl oxygen (O33) and the oxyanion-hole residues Y44 and Y118 as seen from the tracked O33---Y44HH and O33---Y118H distances in Figures 2 and 3, respectively. However, the tracked O33---Y44HH distance in each of the mutant-(−)-cocaine complexes is longer than that in the wild-type CocE-(−)-cocaine complex as shown in Figures 2A and 3A, and as listed in Table 1 for the simulated average values (based on the MD trajectories). As tracked through the MD simulations, the average O33---Y118H distance in each mutant-(−)-cocaine complex is much longer than that in the wild-type-(−)-cocaine complex (Table 1). According to our previous studies on other receptor-ligand binding systems,42,43 the contribution of hydrogen bonding to the total binding free energy between a protein and a ligand can be calculated as the following equation:
| (1) |
in which Ri is the H…O distance for the ith hydrogen bond between the protein and ligand, and the calibrated parameters β12 = 5.571 and β10 = 668.580. Using Eq.(1), the difference in binding free energy contributed from these hydrogen bonding interactions between the mutant-(−)-cocaine complex and the wild-type-(−)-cocaine complex can be conveniently calculated, i.e. ΔΔGHB = ΔGHB(mutant) − ΔGHB(wild-type), and the calculated results are also listed in Table 1. As shown in Table 1, the calculated ΔΔGHB values for the mutant-(−)-cocaine complexes are all positive. The data for the tracked hydrogen-bond distances and the calculated ΔΔGHB values indicate that the binding free energy for (−)-cocaine binding with each of these mutants should be higher than that of the wild-type CocE-(−)-cocaine binding.
Figure 2.

(A) Important distances tracked through MD simulations on the binding structure of the T172R/G173Q mutant-(−)-cocaine complex. The definitions for these distances (C32---S117OG, O33---Y44HH, O33---Y118H, Y44OH---W166NE, S117HG---H287NE, and H287HD---D259OD1/2) are the same as those in Figure 1A. (B) MD-simulated binding structure for the T172R/G173Q mutant-(−)-cocaine complex.
Figure 3.

(A) Important distances tracked through MD simulations for the binding structure of the L119A/L169K/G173Q mutant-(−)-cocaine complex. The definitions for the distances shown in up-panel (C32---S117OG, O33---Y44HH, O33---Y118H, Y44OH---W166NE, S117HG---H287NE, and H287HD---D259OD1/2) are the same as those in Figure 1A, except the N20---K169NZ for the distance between the nitrogen atom on the cationic head of (−)-cocaine and the positive charged head of the side chain of residue K169. (B) The tracked distance between the carbonyl oxygen on the benzoyl group of (−)-cocaine and the backbone Cα atom of residue #119 of the L119A/L169K/G173Q mutant-(−)-cocaine complex (red curve) in comparison with that of the wild-type CocE-(−)-cocaine complex (black curve). (C) MD-simulated binding structure for the L119A/L169K/G173Q mutant-(−)-cocaine complex.
Table 1.
Simulated average distances (with the root-mean square fluctuations) related to the hydrogen bonding between the carbonyl oxygen (O33) on the benzoyl group of (-)-cocaine and the oxyanion-hole residues Y44 (hydroxyl hydrogen on the side chain, Y44HH) and Y118 (backbone hydrogen, Y118H) for each CocE-(-)-cocaine complex averaged from the MD trajectories, and the calculated contributions from the hydrogen bonding to the binding free energy differences (ΔΔGHB).
| Average distance | Wild-type | T172R/G173Q | L119A/L169K/G173Q |
|---|---|---|---|
| O33---Y44HH (Å) | 1.95 ± 0.20 | 2.05 ± 0.23 | 2.08 ± 0.22 |
| O33---Y118H(Å) | 2.52 ± 0.25 | 2.90 ± 0.43 | 2.83 ± 0.34 |
| ΔΔGHB (kcal/mol)a | 0.00 | 0.29 ± 0.14 | 0.50 ± 0.06 |
ΔΔGHB = ΔGHB(mutant) − ΔGHB(wild-type).
As the residues #172 and #173 are not directly involved in the formation of the substrate-binding site of CocE, the T172R/G173Q mutations must affect the substrate binding indirectly. As shown in Figure 2 and listed in Table 1, the hydrogen bonding between the oxyanion-hole residues (Y44 and Y118) and the benzoyl group of (−)-cocaine in the T172R/G173Q mutant-(−)-cocaine complex is weakened compared to that in the wild-type CocE-(−)-cocaine complex (Figure 1), but the weakening effect on the substrate binding is not dramatic.
Concerning the L119A/L169K/G173Q mutant binding with (−)-cocaine, the residue #119 stays behind the catalytic residue S117 and the oxyanion-hole residue Y118 from the same α-helix, and does not directly contact with substrate (−)-cocaine. We tested L119A mutation as we initially expected this mutation to add more free space of the substrate binding site so that the (−)-cocaine binding could be improved. However, as shown in Figure 3, the tracked distance from the carbonyl oxygen at the benzoyl group of (−)-cocaine to the Cα atom of residue #119 in the L119A/L169K/G173Q mutant-(−)-cocaine complex (red curve in Figure 3B) is longer than that in the wild-type CocE-(−)-cocaine complex (black curve in Figure 3B). As listed in Table 1, the average distances for the hydrogen bonding between the benzoyl group of (−)-cocaine and the oxyanion-hole residues Y44 and Y118 in the L119A/L169K/G173Q mutant-(−)-cocaine complex are also longer than the corresponding hydrogen-bonding distances in the wild-type-(−)- cocaine complex. The data from the MD simulations on the L119A/L169K/G173Q mutant-(−)-cocaine complex suggest that the L119A/L169K/G173Q mutation will lead to a considerable decrease in the affinity for the mutant enzyme binding with (−)-cocaine.
Free Energy Profiles and Experimental Kinetic Data
In order to predict the binding free energy of (−)-cocaine with wild-type CocE and its mutants, the PMF simulations were performed starting from the MD-simulated ES complex structures. Based on the data collected from the umbrella-sampling MD simulations, the PMF for each of the ES structures was determined. Figure 4 depicts the PMF-calculated free energy profiles. The distance between the mass center of the non-hydrogen atoms of (−)-cocaine and the mass center of the non-hydrogen atoms on the side chains of residues H87, V121, and L146 of the enzyme was used as the reaction coordinate for the PMF calculations. Such selection of the reaction coordinate was based on the structural features of our modeled CocE-(−)-cocaine binding structures in the present study, as we found that the direction of reaction coordinate roughly went through the central point of the active site of CocE to reach the molecular surface of the enzyme.
Figure 4.
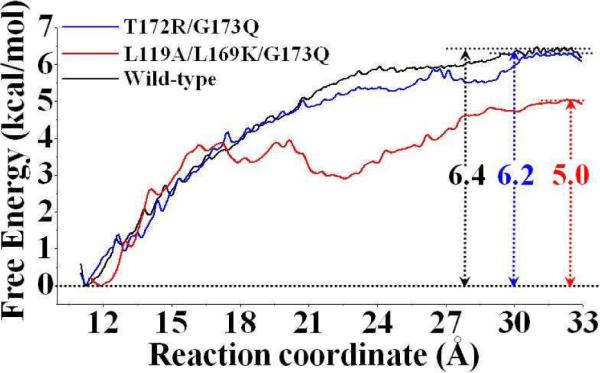
Free energy profiles determined for the binding of (−)-cocaine with wild-type CocE (black curve), T172R/G173Q mutant (blue curve), and L119A/L169K/G173Q mutant (red curve). The reaction coordinate was defined as the distance between the mass center of the non-hydrogen atoms of (−)-cocaine and the mass center of the non-hydrogen atoms on the side chains of residues H87, V121, and L146 of the enzyme.
To test whether the PMF simulations reached convergence, we calculated the binding free energy for the T172R/G173Q CocE-(−)-cocaine binding by using different lengths of the MD trajectory. As shown in Figure S1 of Supporting Information, the PMF for the T172R/G173Q CocE-(−)-cocaine binding was determined by using two different lengths of the MD trajectory for each window, i.e. 0.2−1.0 ns and 0.2−0.8 ns. There was no significant difference between the free energy profiles for the T172R/G173Q CocE-(−)-cocaine binding determined based on the 0.2−1.0 ns and 0.2−0.8 ns of the MD trajectory; the curve of the free energy profile corresponding to the 0.2−0.8 ns almost perfectly overlaps with that (corresponding to 0.2−1.0 ns) shown in Figure 4 (the blue), as seen in Supporting Information (Figure S2). These data suggest that 1.0 ns for each window of the PMF simulations should be sufficient for obtaining the converged results from the PMF simulations.
Based on the PMF-calculated free energy profiles (Figure 4), we cannot identify an obvious free energy barrier along the reaction coordinate in the simulated wild-type CocE-(−)-cocaine or T172R/G173Q mutant-(−)-cocaine binding process. According to the simulated free energy profile (black curve in Figure 4) for the process of wild-type CocE- (−)-cocaine binding and the MD-simulated ES structure, (−)-cocaine molecule can diffuse smoothly from external solvent to the active site of CocE, and its benzoyl group slides down to its sub-binding site around aromatic side chains of W151, W166, and F261. For the T172R/G173Q mutant-(−)-cocaine binding, the simulated free energy profile (blue curve in Figure 4) is also similar to that of the wild-type CocE-(−)-cocaine binding, except for the different starting point of the reaction coordinate. In order to check possible structural adaptation of the enzyme during the process of binding with (−)-cocaine, we tracked the size of the sub-binding site for the benzoyl group of (−)-cocaine along the reaction coordinate of the PMF simulations. Let us use the T172R/G173Q CocE-(−)-cocaine binding structure as an example for discussion here. We selected the distance from the center of mass of W166 side chain to the center of mass of F261 side chain as the criterion according to the structural features of the MD simulated T172R/G173Q CocE-(−)-cocaine complex (Figure 2). As shown in Supporting Information (Figure S2), the distance from the center of mass of W166 side chain to the center of mass of F261 side chain fluctuated at 11.5 Å ± 1.0 Å. Such small fluctuation suggests that the size of the sub-binding site for the benzoyl group of (−)-cocaine had no significant change along the binding process. For the L119A/L169K/G173Q mutant-(−)-cocaine binding, the simulated free energy profile (red curve in Figure 4) shows a local minimum when the reaction coordinate has a value around 23 Å. A detailed check of the umbrella-sampling MD simulations on the L119A/L169K/G173Q mutant-(−)-cocaine binding revealed that the carbonyl oxygen atom on the benzoyl group of (−)-cocaine was hydrogen-bonded with the positively charged head group on the side chain of residue K169 when the reaction coordinate was around 20 Å (see Supporting Information, Figure S5). The (−)-cocaine molecule starts to leave away from residue K169 as the value of reaction coordinate becomes smaller than 20 Å, and it reaches the edge of active site of CocE when the value of reaction coordinate becomes smaller than 16 Å. Further binding process for the L119A/L169K/G173Q mutant with (−)-cocaine starting from this local minimum has a local free energy barrier of 1.5 kcal/mol.
As shown in Figure 4, the calculated binding free energy (ΔGbind) is −6.4 kcal/mol for wild-type CocE, −6.2 kcal/mol for the T172R/G173Q mutant, and −5.0 kcal/mol for the L119A/L169K/G173Q mutant. The difference in the binding free energy between the T172R/G173Q mutant and wild-type CocE, i.e. ΔΔGbind = ΔGbind(mutant) − ΔGbind(wild-type), is 0.2 kcal/mol. The calculated ΔΔGbind for the difference between the L119A/L169K/G173Q mutant and the wild-type CocE is 1.4 kcal/mol. According to the calculated relative binding free energies, (−)-cocaine should have a much lower binding affinity with the L119A/L169K/G173Q mutant than that with wild-type CocE or the T172R/G173Q mutant, and the order of the binding affinity is wild-type CocE > T172R/G173Q mutant > L119A/L169K/G173Q mutant.
In order to know how well the calculated binding free energies are, we estimated the corresponding experimental binding free energies from available experimental data, i.e. the experimental values of Michaelist-Menten constant KM under the well-known rapid equilibration assumption as KM ≈ Kd (dissociation constant). Under the rapid equilibration assumption, we may have
| (2) |
Our recently reported experimental kinetic analysis revealed that KM = 13 μM for the T172R/G173Q mutant against (−)-cocaine.44 Experimental determination of the KM value for wild-type CocE against (−)-cocaine has been a challenge due to the thermal instability of the wild-type enzyme. Nevertheless, according to the most recently reported kinetic analysis,22 the KM = 5.7 μM for wild-type CocE against (−)-cocaine. Thus, when T = 298.15 K, we may have ΔGbind(expt) = −7.2 kcal/mol for wild-type CocE binding with (−)-cocaine, ΔGbind(expt) = −6.7 kcal/mol for the T172R/G173Q mutant binding with (−)-cocaine. The experimentally-derived binding free energies are reasonably close to the corresponding PMF-calculated binding free energies.
Further, in order to examine the computational prediction on the L119A/L169K/G173Q mutant, we carried out site-directed mutagenesis to make the L119A/L169K/G173Q mutant and performed in vitro kinetic analysis. The kinetic analysis revealed that kcat = 2700 min−1 and KM = 0.3 mM for the L119A/L169K/G173Q mutant against (−)-cocaine in the room temperature. Compared to the T172R/G173Q mutant (kcat = 1082 min−1 and KM = 13 μM), the L119A/L169K/G173Q mutant has an increased kcat value of 2700 min−1 and an increased KM value of 0.3 mM. When KM = 0.3 mM, we have ΔGbind(expt) = −4.8 kcal/mol for the L119A/L169K/G173Q mutant binding with (−)-cocaine. The experimentally-derived binding free energy of −4.8 kcal/mol is close to the PMF-calculated binding free energy of −5.0 kcal/mol.
Conclusion
The combined molecular dynamics (MD) and potential of mean force (PMF) simulations have allowed us to determine the free energy profiles for the binding process of (−)-cocaine interacting with wild-type CocE and its mutants. The MD-simulated enzyme-substrate (ES) structures reveal that the binding mode for (−)-cocaine with each of the mutants (T172R/G173Q and L119A/L169K/G173Q) is generally similar to that of (−)-cocaine with wild-type CocE, e.g. the benzoyl group of (−)-cocaine is always bound in a sub-site composed of aromatic residues W151, W166, F261, and F408 and hydrophobic residue L407. The carbonyl oxygen on the benzoyl group of (−)-cocaine is hydrogen-bonded with the oxyanion-hole residues Y44 and Y118. The data obtained from the MD simulations indicate that the binding of (−)-cocaine with the L119A/L169K/G173Q mutant is less favorable compared to that of (−)-cocaine with wild-type CocE or the T172R/G173Q mutant.
The PMF simulations demonstrate that all of the three simulated ES structures have similar free energy profiles for the binding process, but with different starting points for the reaction coordinate. Based on the PMF simulations for the binding process, the calculated binding free energies for (−)-cocaine with the wild-type, T172R/G173Q, and L119A/L169K/G173Q CocEs are −6.4, −6.2, and −5.0 kcal/mol, respectively. The calculated relative binding free energies are reasonably close to the corresponding experimental values (−7.2 kcal/mol for the wild-type and −6.7 kcal/mol for the T172R/G173Q mutant) derived from the experimental KM values. The computational prediction for the L119A/L169K/G173Q mutant has been supported by experimental kinetic analysis showing KM = 0.3 mM (associated with ΔGbind = −4.8 kcal/mol) for the L119A/L169K/G173Q mutant against (−)-cocaine. The experimentally-derived binding free energy of −4.8 kcal/mol is in good agreement with the calculated binding free energy of −5.0 kcal/mol. The agreement between the computational and experimental data suggests that the PMF simulations may be used as a valuable tool in new CocE mutant design that aims to decrease the Michaelis-Menten constant and, thus, improve the catalytic efficiency of the enzyme for (−)-cocaine.
Supplementary Material
Acknowledgment
This work was supported by NIH (grants R01 DA025100, R01 DA021416, R01 DA032910, and R01 DA013930) and NSF (grant CHE-1111761). The authors also acknowledge the Computer Center at University of Kentucky for supercomputing time on a Dell Supercomputer Cluster consisting of 376 nodes or 4,512 processors.
Footnotes
Supporting Information Available. Figure S1 for the experimental kinetic data obtained for (−)-cocaine hydrolysis catalyzed by the L119A/L169K/G173Q mutant of CocE, Figure S2 for the plots of the RMSD of the MD-simulated CocE-(−)-cocaine complexes, Figure S3 for the free energy profiles of the T172R/G173Q CocE binding with (−)-cocaine determined by using two different lengths of the MD trajectory, Figure S4 for the plot of a key distance in the simulated T172R/G173Q CocE-(−)-cocaine binding structure versus the reaction coordinate of the PMF simulations, and Figure S5 for typical snapshot structures of the simulated enzyme-(−)-cocaine complexes along the reaction coordinate. These materials are available free of charge via the Internet http://pubs.acs.org.
References
- 1.Mendelson JH, Mello NK. Management of cocaine abuse and dependence. New Eng. J. Med. 1996;334:965–972. doi: 10.1056/NEJM199604113341507. [DOI] [PubMed] [Google Scholar]
- 2.Sparenborg S, Vocci F, Zukin S. Peripheral cocaine-blocking agents: new medications for cocaine dependence. Drug Alcohol Depend. 1997;48:149–151. doi: 10.1016/s0376-8716(97)00072-0. [DOI] [PubMed] [Google Scholar]
- 3.Singh S. Chemistry, design, and structure-activity relationship of cocaine antagonists. Chem. Rev. 2000;100:925–1024. doi: 10.1021/cr9700538. [DOI] [PubMed] [Google Scholar]
- 4.Paula S, Tabet MR, Farr CD, Norman AB, Ball WJ., Jr. Three-dimensional quantitative structure-activity relationship modeling of cocaine binding by a novel human monoclonal antibody. J. Med. Chem. 2004;47:133–142. doi: 10.1021/jm030351z. [DOI] [PubMed] [Google Scholar]
- 5.Gorelick DA. Enhancing cocaine metabolism with butyrylcholinesterase as a treatment strategy. Drug Alcohol Depend. 1997;48:159–165. doi: 10.1016/s0376-8716(97)00119-1. [DOI] [PubMed] [Google Scholar]
- 6.Zhan C-G, Zheng F, Landry DW. Fundamental reaction mechanism for cocaine hydrolysis in human butyrylcholinesterase. J. Am. Chem. Soc. 2003;125:2462–2474. doi: 10.1021/ja020850+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaintdinov RR, Sotmikova TD, Caron MG. Monoamine transporter pharmacology and mutant mice. Trends Pharmacol. Sci. 2002;231:367–373. doi: 10.1016/s0165-6147(02)02044-8. [DOI] [PubMed] [Google Scholar]
- 8.Torres GE, Gainetdinov RR, Caron MG. Plasma membrane monoamine transporters: structure, regulation and function. Nat. Rev. Neurosci. 2003;4:13–25. doi: 10.1038/nrn1008. [DOI] [PubMed] [Google Scholar]
- 9.Chen R, Tilley MR, Wei H, Zhou F, Zhou F-M, Ching S, Quan N, Stephens RL, Hill ER, Nottoli T, Han DD, Gu HH. Abolished cocaine reward in mice with a cocaine-insensitive dopamine transporter. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9333–9338. doi: 10.1073/pnas.0600905103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Landry DW, Zhao K, Yang GX-Q, Glickman M, Georgiadis TM. Antibody-catalyzed degradation of cocaine. Science. 1993;259:1899–1901. doi: 10.1126/science.8456315. [DOI] [PubMed] [Google Scholar]
- 11.Larsen NA, Turner JM, Stevens J, Rosser SJ, Basran A, Lerner RA, Bruce NC, Wilson IA. Crystal structure of a bacterial cocaine esterase. Nature Struct. Biol. 2002;9:17–21. doi: 10.1038/nsb742. [DOI] [PubMed] [Google Scholar]
- 12.Turner JM, Larsen NA, Basran A, Barbas CF, III, Bruce NC, Wilson IA, Lerner RA. Biochemical characterization and structural analysis of a highly proficient cocaine esterase. Biochem. 2002;41:12297–12307. doi: 10.1021/bi026131p. [DOI] [PubMed] [Google Scholar]
- 13.Zheng F, Yang W, Ko M-C, Liu J, Cho H, Gao D, Tong M, Tai H-H, Woods JH, Zhan C-G. Most efficient cocaine hydrolase designed by virtual screening of transition states. J. Am. Chem. Soc. 2008;130:12148–12155. doi: 10.1021/ja803646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang W, Pan Y, Zheng F, Cho H, Tai H-H, Zhan C-G. Free-energy perturbation simulation on transition states and redesign of butyrylcholinesterase. Biophys. J. 2009;96:1931–1938. doi: 10.1016/j.bpj.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cooper ZD, Narasimhan D, Sunahara RK, Mierzejewski P, Jutkiewicz EM, Larsen NA, Wilson IA, Landry DW, Woods JH. Rapid and robust protection against cocaine-induced lethality in rats by the bacterial cocaine esterase. Mol. Pharmacol. 2006;70:1885–1891. doi: 10.1124/mol.106.025999. [DOI] [PubMed] [Google Scholar]
- 16.Ko M-C, Bowen LD, Narasimhan D, Berlin AA, Lukacs NW, Sunahara RK, Cooper ZD, Woods JH. Cocaine esterase: interactions with cocaine and immune responses in mice. J. Pharmacol. Exp. Ther. 2007;320:926–933. doi: 10.1124/jpet.106.114223. [DOI] [PubMed] [Google Scholar]
- 17.Jutkiewicz EM, Baladi MG, Cooper ZD, Narasimhan D, Sunahara RK, Woods JH. A bacterial cocaine esterase protects against cocaine-induced epileptogenic activity and lethality. Ann. Emerg. Med. 2009;54:409–420. doi: 10.1016/j.annemergmed.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ko MC, Narasimhan D, Berlin AA, Lukacs NW, Sunahara RK, Woods JH. Effects of cocaine esterase following its repeated administration with cocaine in mice. Drug Alcohol Depend. 2009;101:202–209. doi: 10.1016/j.drugalcdep.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao D, Narasimhan D, Macdonald J, Brim R, Ko M-C, Landry DW, Woods JH, Sunahara RK, Zhan C-G. Thermostable variants of cocaine esterase for long-time protection against cocaine toxicity. Mol. Pharmacol. 2009;75:318–323. doi: 10.1124/mol.108.049486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collines GT, Brim RL, Narasimhan D, Ko M-C, Sunahara RK, Zhan C-G, Woods JH. Cocaine esterase prevents cocaine-induced toxicity and the ongoing intravenous self-administration of cocaine in rats. J. Pharmacol. Exp. Ther. 2009;331(2):445–455. doi: 10.1124/jpet.108.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J, Hamza A, Zhan C-G. Fundamental reaction mechanism and free energy profile for (−)-cocaine hydrolysis catalyzed by cocaine esterase. J. Am. Chem. Soc. 2009;131:11964–11975. doi: 10.1021/ja903990p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narasimhan D, Nance M, Gao D, Ko M-C, Macdonald J, Yoon D, Landry D, Woods JH, Zhan C-G, Tesmer J, Sunahara RK. Thermostabilizing mutations of cocaine esterase. Protein Eng. Des. Sel. 2010;23(7):537–547. doi: 10.1093/protein/gzq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brim RL, Nance MR, Youngstrom DW, Narasimhan D, Zhan C-G, Tesmer JJ, Sunahara RK, Woods JH. A thermally stable form of cocaine esterase: a potential therapeutic agent for treatment of cocaine abuse. Mol. Pharmacol. 2010;77(4):593–600. doi: 10.1124/mol.109.060806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang X, Gao D, Zhan C-G. Computational design of a thermostable mutant of cocaine esterase via molecular dynamics simulations. Org. Biomol. Chem. 2011;9(11):4138–4143. doi: 10.1039/c0ob00972e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman PA. AMBER 9. University of California; San Francisco: 2006. [Google Scholar]
- 26.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery J,JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- 27.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 28.Morishita T. Fluctuation formulas in molecular dynamics simulations with the weak coupling heat bath. J. Chem. Phys. 2000;113:2976–2982. [Google Scholar]
- 29.Darden T, York D, Pedersen L. Particle mesh Ewald—an N_log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 30.Toukmaji A, Sagui C, Board J, Darden T. Efficient particle-mesh Ewald based approach to fixed and induced dipolar interactions. J. Chem. Phys. 2000;113:10913–10927. [Google Scholar]
- 31.Ryckaert J, Ciccotti PG, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 32.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 33.Torrie GM, Valleau JP. Nonphysical sampling distribution in monte carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 1977;23:187–199. [Google Scholar]
- 34.Kirkwood JG. Statistical mechanics of fluid mixtures. J. Chem. Phys. 1935;3:300–313. [Google Scholar]
- 35.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg J. The weighted histogram analysis method for free-energy calculations on biomolecules. I. Method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 36.Roux B. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commu. 1995;91:275–282. [Google Scholar]
- 37.Pan Y, Gao D, Yang W, Cho H, Yang G-F, Tai H-H, Zhan C-G. Computational redesign of human butyrylcholinesterase for anti-cocaine medication. Proc. Natl. Acad. Sci. USA. 2005;102:16656–16661. doi: 10.1073/pnas.0507332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao D, Cho H, Yang W, Pan Y, Yang G-F, Tai H-H, Zhan C-G. Computational design of a human butyrylcholinesterase mutant for accelerating cocaine hydrolysis based on the transition-state simulation. Angew. Chem. Int. Ed. 2006;45:653–657. doi: 10.1002/anie.200503025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan Y, Gao D, Yang W, Cho H, Zhan C-G. Free energy perturbation (FEP) simulation on the transition-states of cocaine hydrolysis catalyzed by human butyrylcholinesterase and its mutants. J. Am. Chem. Soc. 2007;129:13537–13543. doi: 10.1021/ja073724k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang W, Pan Y, Fang L, Gao D, Zheng F, Zhan C-G. Free-energy perturbation simulation on transition states and high-activity mutants of human butyrylcholinesterase for (−)-cocaine hydrolysis. J. Phys. Chem. B. 2010;114:10889–10896. doi: 10.1021/jp104989b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xue L, Ko M-C, Tong M, Yang W, Hou S, Zheng F, Woods JH, Tai H-H, Zhan C-G. Rational design, preparation and characterization of high-activity mutants of human butyrylcholinesterase against (−)-cocaine. Mol. Pharmacol. 2011;79:290–297. doi: 10.1124/mol.110.068494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang X, Zheng F, Chen X, Crooks PA, Dwoskin LP, Zhan C-G. Modeling subtype-selective agonists binding with α4β2 and α7 nicotinic acetylcholine receptors: effects of local binding and long-range electrostatic interactions. J. Med. Chem. 2006;49(26):7661–7674. doi: 10.1021/jm0606701. [DOI] [PubMed] [Google Scholar]
- 43.Huang X, Zheng F, Stokes C, Papke RL, Zhan C-G. Modeling binding modes of α7 nicotinic acetylcholine receptor with ligands: the roles of Gln117 and other residues of the receptor in agonist binding. J. Med. Chem. 2008;51:6293–6302. doi: 10.1021/jm800607u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu J, Zhao X, Yang W, Zhan C-G. Reaction mechanism for cocaine esterasecatalyzed hydrolyses of (+)- and (−)-cocaine: Unexpected common rate-determining step. J. Phys. Chem. B. 2011;115:5017–5025. doi: 10.1021/jp200975v. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.