

Summary
Darier’s Disease (DD), caused by mutations in the endoplasmic reticulum (ER) Ca2+ ATPase ATP2A2 (SERCA2b), is a skin disease that exhibits impaired epidermal cell-to-cell adhesion and altered differentiation. Although previous studies have shown that keratinocyte Ca2+ sequestration and fluxes are controlled by sphingolipid signaling, the role of this signaling pathway in DD previously has not been investigated. We show here that sphingosine levels increase and sphingosine kinase (SPHK1) expression decreases after inactivating SERCA2b with the specific SERCA2 inhibitors thapsigargin (TG) or siRNA to SERCA2b. Conversely, inhibiting sphingosine lyase rescues the defects in keratinocyte differentiation, E-cadherin localization, Desmoplakin (DP) translocation, and ER Ca2+ sequestration seen in TG-treated keratinocytes. To our knowledge, it was previously unreported that the keratinocyte sphingolipid and Ca2+ signaling pathways intersect in ATP2A2- controlled ER Ca2+ sequestration, E-cadherin and desmoplakin localization and Ca2+ - controlled differentiation, and thus may be important mediators in DD.
Keywords: Darier’s Disease, keratinocyte, calcium, sphingolipid, acantholysis, apoptosis, E-cadherin, desmoplakin
Introduction
Darier’s disease (DD) (Darier, 2000,White, 1889) is an inherited skin disease characterized by keratotic papules and plaques found mostly in skin flexures and on the chest, upper back, face and scalp. Histopathology of DD skin reveals impaired differentiation, acantholysis and pathologic apoptosis. Patients with DD also have been reported to suffer from neuropsychiatric disease (Gordon-Smith, et al., 2010). Mutations in the endoplasmic reticulum (ER) Ca2+ ATPase (gene ATP2A2; protein SERCA2b), which sequesters Ca2+ in the ER, cause DD (Sakuntabhai, et al., 1999a, Sakuntabhai, et al., 1999b). The disease is thought to be caused by haploinsufficiency (Foggia, et al., 2006). Treatment for DD largely has been empiric, and is only partially effective in some patients, although a variety of pharmacologic and physical agents have been proposed (Kragballe, 1995; Reuter, et al., 2007; Stewart and Yell, 2008; Cooper and Burge, 2003).
Ca2+ signaling is implicated in keratinocyte and epidermal differentiation, cell-to-cell adhesion and apoptosis. Normal Ca2+ signaling requires both IP3-mediated ER and Golgi Ca2+ release and Ca2+ influx though Ca2+ -permeable ion channels, leading to transcription of differentiation-specific genes (Bikle, et al., 2001) and stimulating formation of adherens junctions containing E-cadherin (Green, et al., 2010). Well known as a structural protein, E-cadherin also recently has been shown to have important signaling functions, as two kinases contained within the E-cadherin complex (Xie and Bikle, 2007; Xie, et al., 2009) maintain the prolonged Ca2+ increase essential for calcium induced differentiation.
Sphingolipid metabolism (fig 1A) constitutes a complementary Ca2+ signaling system in keratinocytes. Heightened levels of intracellular Sphingosine-1-phosphate (S1P) increase cytosolic Ca2+ levels by direct mobilization of Ca2+ from Thapsigargin (TG) sensitive stores (Meyer zu Heringdorf, et al., 2003), while increases in extracellular S1P concentrations activate Store Operated Calcium Entry (SOCE), through activation of G-protein coupled receptors (Mehta, et al., 2005). In keratinocytes, increased intracellular S1P enhances keratinocyte growth and differentiation, and blocks apoptosis (Hong, et al., 2008; Lichte, et al., 2008). Activating sphingosine kinase (SPHK1) increases intracellular S1P; thus, agents that activate this enzyme enhance expression of the differentiation markers filaggrin and involucrin in HaCAT keratinocytes (Hong, et al., 2008). In contrast, blocking SPHK1 increases apoptosis in UVB-treated keratinocytes (Uchida, et al., 2010). S1P levels also can be increased by preventing S1P breakdown (Figure 1A) via Sphingosine phosphate lyase (SGPL1) (Reviewed by Kumar and Saba, 2009).
Figure 1. Sphingosine Metabolism is Controlled by SERCA2.

A. Sphingosine metabolism in keratinocytes. B. SERCA2 Inactivation Increases Sphingosine. Normal human keratinocytes were treated with TG 100 nM for 2 hours. Sphingosine levels were measured with HPLC (see Materials and Methods) 24 and 48 hours after TG treatment. Data are presented as the mean ± SD. N=3 for each data point. * = p<0.05 C. SPHK1 Expression is Decreased After SERCA2 Inhibition. Normal human keratinocytes were treated with 10 and 100 nM TG for 2 hours, washed, recultured and harvested at 24 (grey bars) and 48 (black bars) hours. In separate experiments, normal human keratinocytes were treated with siRNA to SERCA2b, and then harvested at 48 hours (marble bars). Control keratinocytes were treated with TG vehicle or scrambled siRNA (marble bar) and normalized to 1. SPHK1 expression was assessed with RT-PCR (see Materials and Methods).
We show here that the sphingolipid and Ca2+ signaling pathways intersect in controlling SERCA2- ER Ca2+ sequestration. SERCA2b inhibition decreases SPHK1 expression and reproduces the defects in cell growth, adhesion and differentiation seen in DD. In contrast, inhibiting SGPL1 compensates for decreased SPHK1 and rescues or ameliorates defects in differentiation, E-cadherin processing, desmoplakin translocation, and ER Ca2+ sequestration. Currently, SGPL1 is being targeted as possible therapeutic target for rheumatologic disease and multiple sclerosis. These therapeutic agents could also prove valuable as therapy for DD.
Results and Discussion
Increased sphingosine can enhance cell death/cell cycle arrest, previously reported in a canine model of DD (Muller, et al., 2006), while increased levels of S1P promote cell growth and viability. The relative amounts of these lipids are determined by the activity of SPHK1, which metabolizes sphingosine to S1P; and SGPL1, which degrades S1P to phosphoethanolamine and hexadecanol (Fig. 1A). Because abnormal sphingolipid signaling and DD share impaired cell differentiation, cell survival and calcium signaling, we first examined whether pharmacologically inactivating the ER Ca2+ ATPase SERCA2b could alter sphingolipid metabolism. We found that low concentrations of TG (100nM) increased sphingosine concentrations (Fig. 1B). We were able to detect decreased sphingosine kinase mRNA expression after treatment with even lower TG concentrations (10-100nM) (Fig. 1C). A similar decrease in SPHK1 expression was seen after inactivating SERCA2b with siRNA (Fig. 1C). Both siRNA and TG were used to impair SERCA2 function in subsequent experiments. The dose of SERCA2b siRNA used for these experiments was chosen to reproduce the average 50% reduction in SERCA2b expression seen in DD patients (Foggia, et al., 2006) (figure S1). In the same paper, the authors report an average basal level of ER calcium for DD cells that is 60% lower than control as observed by transient cytosolic calcium increase after TG exposure. We observed that a similar decrease in ER calcium was obtained by treating cells with 10nM TG (Celli, et al., 2010, and figure 7c) and we therefore used this concentration for the functional studies presented in this report.
Figure 7. Defects in ER Ca2+ Sequestration are Induced by ER SERCA2 Inhibition, and are Normalized by Inhibiting SGPL1.

Primary normal human keratinocytes were transfected with the ER-targeted Ca2+ sensor D1ER (see Methods). Cells also were transfected with SGPL1 siRNA or scrambled siRNA as a control. 8 hours after siRNA transfection, cells were treated with 10 nM thapsigargin for two hours. The cells then were washed, and incubated in high calcium media for an additional 24 hours. A) average FRET ratio; B) average ER Ca2+ concentration; C) Relative Ca2+ changes between control (black bars) and TG treated cells (grey bars) in untreated, scrambled siRNA, and SGPL1 siRNA treated samples. Data are presented as the mean +/− s.e.m. N=15–29 cells from two independent sets of experiments in each group. Significance was calculated using a one-way ANOVA. Distributions with p<0.05 were assumed as statistically different.
The ER Ca2+ store is important for normal keratinocyte signaling and differentiation (Callewaert, et al., 2003; Foggia, et al., 2006). Previous experiments demonstrated that early differentiation protein expression is inhibited by disabling the ER Ca2+ ATPase (Li, et al., 1995b; Li, et al., 1995a). Early differentiation proteins also are decreased in SERCA2 knockout mouse keratinocytes (Hong, et al., 2010), and defects in E-cadherin localization have been reported in human DD (Tada and Hashimoto, 1998). Decreased E-cadherin levels and/or trafficking may directly lead to decreased cell-to-cell adhesion via impaired formation of adherens junctions and may additionally impair IP3 mediated keratinocyte Ca2+ signaling because the E-cadherin complex formation in the membrane is essential for PLCγ1 activity (Xie and Bikle, 2007; Xie, et al., 2009). We therefore assessed whether modulating the sphingolipid pathway could reproduce or rescue defects in keratinocyte differentiation and E-cadherin expression or localization. We observed abnormal keratinocyte morphology and growth 24 hours (data not shown) and 48 hours after TG treatment (Fig. 2D). Inhibition of SGPL1 with siRNA rescued these defects (Fig 2F). Western blotting showed that SERCA2 inhibition with TG also led to defects in involucrin protein synthesis and E-cadherin protein synthesis and processing. These defects could be normalized or ameliorated by inhibiting SGPL1 synthesis (Fig. 3). We found that SGPL1 inhibition was more effective in normalizing the defects in involucrin and E-cadherin expression caused by lower doses of TG (10 nM) than after higher (100nM) TG (data not shown), likely because the effects of SERCA2 inhibition became irreversible at higher doses. Treatment with exogenous S1P had no effect (Fig. S3). Vitamin D and cannabinoid analogues also were ineffective (Fig. S3).
Figure 2. Defects in Keratinocyte Morphology, Induced by SERCA2 Inhibition, are Normalized by Inhibiting SGPL1.

Human Keratinocytes were cultured in growth media containing 0.06 mM Ca2+. When 80% confluent, some of the cells were transfected with siRNA against SGPL1 for 8 hours, then 10 nM (final concentration) TG was added. After two hours exposure to TG cells were washed and fed with growth media containing 1.2 mM Ca2+. Images were acquired 48 hours after raising extracellular Ca2+. A. Vehicle treated cells showing normal morphology: cells are tightly packed and flattened after 48 hours in high calcium; B and C) cells treated with scrambled siRNA or siRNA against SGPL1 show no morphological difference from untreated cells. D) 10 nM TG treatment produces larger keratinocytes that have not flattened. E) Treatment with scrambled siRNA has no effect on TG treated cells F) cells treated with both TG and siRNA to SGPL1 demonstrate a morphology similar to control keratinocytes. All images were acquired at the same magnification. The scale bar in panel A indicates 50μm.
Figure 3. Defects in Involucrin and E-cadherin synthesis and Processing Are Normalized by Inhibiting SGPL1.
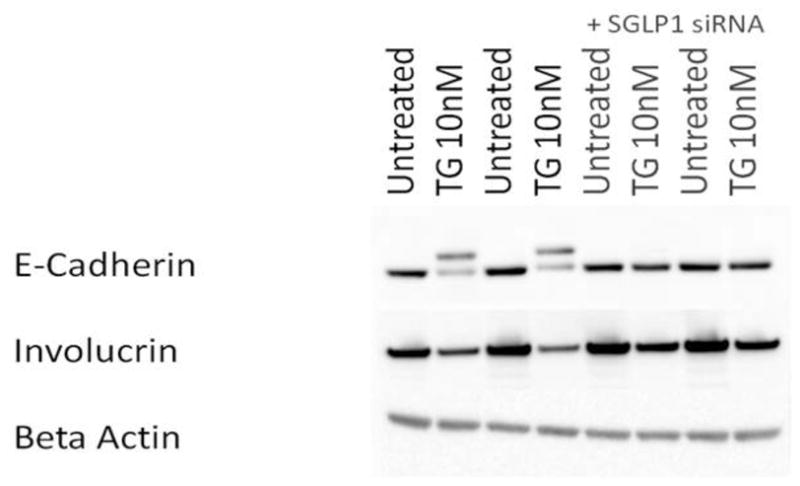
Human Keratinocytes were cultured and treated as in Figure 2, above. Cells were harvested 48 hours after raising extracellular Ca2+. E-cadherin and involucrin protein levels were assessed using Western blotting (see Methods).
The additional, higher molecular weight band seen on immunoblots after TG treatment suggested that E-cadherin intracellular processing and localization might also be disrupted when ER Ca2+ stores were depleted, similar to what has previously been reported for DD (Hakuno, et al., 2000). To test this, we localized E-cadherin first using an antibody that selectively binds to the extracellular epitope (Fig. 4A-D). E-cadherin normally responds to raised extracellular Ca2+ by forming continuous, fine intercellular strands (Fig. 4A). TG treatment lead to a dose-dependent disruption in these cell-to-cell contacts, with E-cadherin localization becoming coarse and irregular at very low (10nM) TG concentrations (Fig. 4B), and discontinuous at 100 nM TG concentrations (data not shown). SGPL1 inhibition normalized E-cadherin localization at 10 nM TG (Fig. 2D), but only partially rescued TG-induced E-cadherin disruption at the 100 nM dose (not shown). Treatment with scrambled siRNA had no significant effect (Fig. 4C). These data suggest that E-cadherin is not properly localized to the plasma membrane after SERCA2 inhibition.
Figure 4. Defects in E-cadherin localization Are Normalized by Inhibiting SGPL1.

A-D) Keratinocytes cultured in 1.2 mM Ca2+ for 48 hours were stained with an E-cadherin antibody that recognizes an extracellular epitope. E-H) Keratinocytes cultured in 1.2 mM Ca2+ for 48 hours stained with an E-cadherin antibody that recognizes an intracellular epitope. (A and E) Control keratinocytes. (B and F) After treatment with 10 nM TG, extracellular E-cadherin staining (B) is discontinuous, with filamentous strands (arrows). (F) Perinuclear accumulation of E-cadherin is seen (arrows). (C and G) Scrambled siRNA does not rescue intracellular or extracellular E-cadherin localization (arrows). (D and H) SGPL1 inhibition with siRNA restores continuous, fine extracellular E-cadherin localization (D) and reverses the E-cadherin intracellular retention (H) seen after SERCA2 inhibition with TG.
To test whether SERCA2 inhibition lead to defective E-cadherin, we next localized E-cadherin using an antibody that binds to an intracellular E-cadherin epitope. After raising extracellular Ca2+, the vast majority of E-cadherin was found at cell-to-cell junctions in normal keratinocytes (Fig. 4E) with only punctuate foci of intracellular E-cadherin seen. After treatment with 10 nM TG, extracellular E-cadherin again appeared irregular and beaded (Fig. 4F). In addition, perinuclear aggregates of E-cadherin also were seen (Fig. 4F), suggesting that ER Ca2+ depletion leads to intracellular E-cadherin accumulation. This process also was dose-dependent, and the vast majority of E-cadherin was localized intracellularly after treatment with 100 nM TG (not shown). As above, inhibition of SGPL1 was able to normalize E-cadherin localization in keratinocytes treated with 10 nM TG (Fig.4H), but only partially rescued keratinocytes treated with the higher 100 nM TG (not shown). Treatment with scrambled siRNA did not rescue the defects caused by TG treatment (Fig. 4G).
Downregulation of SERCA2b expression by siRNA resulted in a similar pattern of intracellular and extracellular E-cadherin staining (Fig. 5D and J respectively) that could be normalized by SGPL1 siRNA treatment (Fig. 5E and K). Treatment with a scrambled siRNA sequence had no effect (Fig. 5F and L).
Figure 5. Defects in E-cadherin localization following downregulation of SERCA2b expression with siRNA Are Normalized by Inhibiting SGPL1.

A-F) Extracellular E-cadherin staining. A-C) Untreated, SGPL1 siRNA, and scrambled siRNA treated control cells show linear, smooth staining at the cell-to-cell borders; D) Treatment with SERCA2 siRNA results in rough, discontinuous, filamentous borders, and larger cells similar to TG treated cells (compare to Figure 4, above); E) SGPL1 inhibition with siRNA restores normal E-cadherin localization; F) Scrambled siRNA has no effect.
G-L) Intracellular E-cadherin staining; G-I) Untreated, SGPL1 siRNA, and scrambled siRNA treated control cells show normal, minimal intracellular retention of E-cadherin; J) Cells treated with siRNA to SERCA2b demonstrate more diffuse intracellular staining, irregular cell borders and larger cells; K) SGPL1 siRNA treatment restores normal E-cadherin localization; L) scrambled siRNA has no effect.
These studies demonstrate that E-cadherin requires normal ER Ca2+ stores to properly localize to the plasma membrane, and that the defects seen in cell-to-cell adhesion in DD may be due to defective E-cadherin localization.
To further investigate the effects of SERCA2 inactivation on cell to cell junctions, we assessed the effect of 10 nM TG on desmoplakin (DP) localization, which also is disrupted following SERCA2 inactivation (Hobbs, et al., 2011). In agreement with this previous report, we also observed DP localization at the cell to cell borders within 3 hours after raising extracellular Ca2+ in control cells (Fig. 6A-C). 48 hours after raising extracellular Ca2+, DP was almost entirely localized to cell-to-cell borders (Fig. 6G-I). Like E-cadherin, TG disrupted DP localization in a dose-dependent manner. Cells treated with 10 nM TG are shown in figure 6D and J, and cells treated with 100 nM TG are shown for comparison in Fig. S5. DP staining was mostly intracellular in TG-treated cells. Defects in DP staining and gaps between cells were noted at both the 3 and 48 hours time points (Fig. 6D and J respectively). Treatment with SGPL1 siRNA completely rescued DP localization 48 hours after calcium switch, but only partially at the earlier 3 hour time point (Fig. 6K and E respectively). Treatment with scrambled siRNA did not rescue the abnormal DP localization (figure 6C, F, I, L). SGPL1 siRNA also partially rescued DP localization in cells treated with 100nM TG (Fig. S5).
Figure 6. Defects in Desmoplakin (DP) localization, Caused by SERCA2 inactivation with TG, Are Normalized by Inhibiting SGPL1.

Desmoplakin staining of human keratinocytes treated with 10nM TG, 3hours (A-F) and 48 hours (G-L) after raising extracellular Ca2+. In control (A), SGPL1 (B) and scrambled siRNA-treated cells (C), DP accumulation at cell-to-cell borders is evident within 3 hours after raising extracellular Ca2+. (G-I) 48 hours after raising extracellular Ca2+, DP localizes exclusively at the cell to cell borders in control (G) , SGPL1 siRNA (H) and scrambled siRNA-treated (I) cells. Treatment with 10 nM TG reduces DP staining at the cell borders three hours after raising extracellular Ca2+ (D) and intracellular retention of DP is evident at 48 hours (J); SGPL1 inhibition with siRNA improves DP localization at 3 hrs (E), but complete normalization of DP localization is seen only at 48 hours (K). Scrambled siRNA had no effect (F and L).
We used the ER-targeted Ca2+ sensor D1ER to monitor ER Ca2+ depletion after SERCA2 inhibition, and to assess whether SGPL1 inhibition could ameliorate this loss. Figure 7A and figure 7B present the data as average FRET ratios and average calcium concentrations respectively. A decrease in FRET ratio indicates lower Ca2+ concentrations. FRET ratios were converted into calcium concentration following the protocol in (Rudolf, et al., 2006). siRNA transfection alone, with either a scrambled sequence or SGPL1 siRNA, leads to a small decrease in ER calcium levels. In agreement with a previous report (Celli, et al., 2010), treatment with 10nM thapsigargin caused a dramatic drop in ER calcium concentration in both untreated and scrambled siRNA transfected samples. However, treatment with 10nM TG had no significant effect on ER Ca2+ in cells pretreated with SGPL1 siRNA (Fig. 7A and B). TG treatment produced more than a 50% drop in ER Ca2+ concentration in both untreated keratinocytes and scrambled siRNA-transfected keratinocytes (Fig. 7C). In contrast, SGPL1 inhibition prevented this TG- induced Ca2+ loss (Fig. 7C). These data suggest that modulating sphingosine metabolism may normalize TG-induced abnormalities in keratinocyte differentiation and cell-to-cell adhesion by mitigating ER Ca2+ loss after SERCA2 inactivation. 100 nM TG almost completely emptied ER Ca2+ stores and could not be rescued by SGPL1 inhibition (data not shown). SGPL1 inhibition did not change SERCA2 protein levels or SPHK1 mRNA expression, suggesting that Ca2+ was normalized by a mechanism independent of SERCA2 or SPHK1 synthesis (Fig. S1 and S2).
This report demonstrates that inhibiting SGPL1 rescues ER Ca2+ depletion-induced defects in keratinocyte differentiation and adhesion, and restores ER Ca2+ stores depleted by SERCA2 inhibition. How sphingosine metabolism and SERCA2 Ca2+ signaling interact in directing keratinocyte adhesion and differentiation is not yet well defined. Certainly, the actions of exogenous S1P in controlling intracellular Ca2+ have been studied extensively. However, we found that exogenous S1P was ineffective in rescuing adhesion and differentiation defects caused by SERCA2b inactivation (Fig. S3). These data suggest that generation of intracellular S1P is essential for rescuing the DD phenotype. Alternatively, extracellular S1P-stimulated Ca2+ release from the ER may be blunted when these stores already are depleted due to SERCA2 dysfunction. Exposure to increased intracellular S1P levels, on the other hand, has been shown to increase calcium levels in TG sensitive stores and to mobilize calcium from these stores into the cytosol, independently of the plasma membrane G-protein coupled S1P receptor (Claas, et al., 2010; Meyer zu Heringdorf, et al., 2003).
In a recent paper, Calloway et al (Calloway, et al., 2009) found that treatment of RBL mast cells with sphingosine and its positively charged analogues inhibited the Ca2+ influx via SOCE usually seen after TG treatment, by flipping to the inner leaflet of the plasma membrane and preventing Ora1/CRACM1 from interacting with STIM1. Thus, SERCA2b inactivation might cause not only ER Ca2+ depletion, but also lead to increased intracellular sphingosine, via SPHK1 inhibition. The increased sphingosine then might interfere with Ora/CRACM1/STIM interaction, exacerbating the low ER Ca2+ caused by SERCA2b loss.
We chose to study the effects of ER calcium depletion and SGPL1 down-regulation on E-cadherin because this protein has both a structural function and a role in calcium signaling (Xie and Bikle, 2007; Xie, et al., 2009). Moreover, calcium dependent E-cadherin aggregation at cell-to-cell adhesion points is known to be perturbed in DD (Tada and Hashimoto, 1998). In contrast to a previous study (Hobbs, et al., 2011) our experiments demonstrated that E-cadherin localization was disrupted after SERCA2 inhibition. This discrepancy may be explained by the longer timecourse (24–48 hours vs. 3 hours) used in our studies, based on previous findings that E-cadherin continues to aggregate and complex at cell-to-cell junctions for at least 7–20 hours (Vasioukhin, et al., 2000). Immunofluorescence micrographs of SERCA2 siRNA treated cells harvested 3 hours after calcium switch do not show significant defects in E-cadherin localization (Fig. S4) as compared to the longer time points, thus lending support to this hypothesis.
SGPL1 inhibition also normalized desmoplakin (DP) trafficking, which has been implicated in DD (Dhitavat, et al., 2003; Hobbs, et al., 2011). These studies suggest that modulating the sphingolipid pathway may ameliorate the defective cell-to-cell adhesion that is a hallmark of DD.
We studied cells treated with thapsigargin or siRNA against SERCA2 in lieu of DD keratinocytes, as previous studies conducted on DD keratinocytes revealed a wide heterogeneity in calcium mobilization in these cells (Foggia, et al., 2006). While further investigation on the effects of SGPL1 inhibition in DD cells is needed, we show here that reversing SERCA2 inhibition caused by thapsigargin or SERCA2 siRNA ameliorates the junctional and differentiation defects that are known to underlie DD pathology.
MATERIALS AND METHODS
Cell Culture
Normal human keratinocytes, passage 2-3, were plated in 0.03 mM Ca+2 154 CF media (Cascade, Life Sciences, NY). For experiments where TG was used, cells were treated with either scrambled siRNA (as a negative control) or siRNA against SGPL1 (Hs_SGPL1_2 Flexitube, Quiagen, Valencia, CA) when 80% confluent. 8 hours after siRNA treatment, thapsigargin at a final concentration of 10 or 100 nM, was added to some of the cells for 2 hours. The media was then replaced with 1.2 mM Ca+2 154 CF (Cascade, Life Sciences, NY), to induce differentiation. For experiments where siRNA against SERCA2 (silencing sequence: AAGCAGGACATCAATGAGCAA) was used, cells were transfected with siRNA against SERCA2, or a scrambled siRNA sequence for control, when cells were 60% confluent and treated with SGPL1 siRNA, or with a scrambled siRNA for control, 24 hours after. Hyperfect transfection reagent (Quiagen, Valencia, CA) was used for all siRNA transfections. Cells were harvested at time points noted in each figure after thapsigargin treatment. Vehicle (for TG) or scrambled siRNA were used as controls.
ER calcium measurements
60% confluent cells were transfected with D1ER (gift of Prof. Tsien’s laboratory) using Mirrus Keratinocyte Transit lipid reagent, according to the manufacturer’s protocol. 24 hours after transfection, cells were treated with SGPL1 siRNA or scrambled siRNA and TG as described above. Dual channel fluorescence measurements were performed 24 hours later using a Zeiss LSM Meta confocal system (Carl Zeiss Inc., NY). Calibration of the Calcium sensitivity range of the probe and conversion of the FRET ratio into Calcium concentrations was performed following the protocol in (Palmer and Tsien, 2006; Rudolf, et al., 2006).
Sphingosine Analysis
Total lipids were extracted from keratinocytes and then sphingoid bases were converted to o-phthalaldehyde derivatives after dephosphorylation with alkaline phosphatase as previously reported (Min, et al., 2002). Sphingoid base o-phthalaldehyde derivatives were separated on C18 reverse phase HPLC column (Luna C18(2), 250 x 4.6 mm, 5μm, Phenomenex, Torrance, CA) using methanol:water (9:1) (v/v) with a 1 ml/min flow. Fluorescence intensity was monitored at Ex 360 nm and Em 430 nm. The sphingoid base content was reported as pmol per mg of protein.
Immunohistochemistry
Normal human keratinocytes were treated as described above (see Cell Culture). Samples were formaldehyde-fixed. Cells were stained with an antibody that stained both intracellular and extracellular E-cadherin (36/E-Cadherin BD Transduction Laboratories, Sparks, MD), or only the extracellular epitope of E-cadherin (HECD-1, abcam, Cambridge, MA). For desmoplakin immunofluorescence we used a mouse monoclonal antibody (1G4 gift of Dr. Kathy Green).
Photomicrographs shown in figure 4 were taken on a Zeiss 510 Meta confocal microscope (Carl Zeiss Inc. NY). Identical laser power and detector gain levels were used for all images. Photomicrographs in figures 5 and 6 were taken using a Zeiss observer/spinning disk confocal system (Carl Zeiss Inc. NY). Identical laser power and detector gain levels were used for all images.
Western Blotting
Cells were homogenized, and proteins were isolated using RIPA buffer (Sigma-Aldrich, Saint Louis, MO) containing protease inhibitors (Roche Applied Science, Indianapolis, IN). Protein quantification for equal loading was made with Thermo Scientific Pierce’s BCA assay kit. SDS-PAGE and transfer was performed using NuPAGE Novex 4-12% sodium dodecyl sulfate-polyacrylamide gels and nitrocellulose membranes according to Invitrogen’s NuPAGE protocol. Membranes were blocked in PBS with 5% non-fat dry milk and 0.05% Tween-20 and incubated with the following primary antibodies and dilutions: anti-Involucrin (Sigma Aldrich, Saint Louis, MO) at 1:100, anti-Ecadherin (Invitrogen, Life Science, NY) at 1:500, and anti-β-Actin-HRP (Sigma-Aldrich, Saint Louis, MO) at 1:30,000 anti-SERCA2. Chemiluminescent detection was performed with ECL Plus detection reagent (Thermo Scientific Pierce, Waltham, MA) using the Fujifilm LAS-3000 imaging system.
Quantitative PCR
All cDNA was generated using Roche Applied Science’s Transcriptor first strand cDNA synthesis kit. Quantitative PCR (QPCR) was performed using TaqMan fast universal PCR master mix and the ABI 7900HT real-time PCR machine (Applied Biosystems, Life sciences, NY) and target genes were normalized to GAPDH (primers and dual-labeled probes from Applied Biosystems). Human SPHK1 primers and probes used: CTGTCTGCTCCGAGGACTG forward, CGTGGTTCTTACACCACTGC reverse, TGACCTGCTACCTCGGCCGC probe.
Statistical Analysis
Data were analyzed using the paired, two-tailed Students T test and one-way ANOVA.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants AR051930 and the San Francisco Veterans Administration Medical Service.
Abbreviations
- DD
Darier Disease
- S1P
Sphingosine 1 Phosphate
- TG
Thapsigargin
- SPKH1
Sphingosine Kinase 1
- SGPL1
Sphingosine Lyase
- SERCA2
Sarco-Endoplasmic Reticulum Calcium ATPase
- DP
Desmoplakin
Footnotes
Conflict of interest: The authors have no conflict of interest.
References
- Bikle DD, Ng D, Tu CL, et al. Calcium- and vitamin D-regulated keratinocyte differentiation. Mol Cell Endocrinol. 2001;177:161–171. doi: 10.1016/s0303-7207(01)00452-x. [DOI] [PubMed] [Google Scholar]
- Callewaert G, Parys JB, De Smedt H, et al. Similar Ca(2+)-signaling properties in keratinocytes and in COS-1 cells overexpressing the secretory-pathway Ca(2+)-ATPase SPCA1. Cell Calcium. 2003;34:157–162. doi: 10.1016/s0143-4160(03)00070-8. [DOI] [PubMed] [Google Scholar]
- Calloway N, Vig M, Kinet JP, et al. Molecular clustering of STIM1 with Orai1/CRACM1 at the plasma membrane depends dynamically on depletion of Ca2+ stores and on electrostatic interactions. Mol Biol Cell. 2009;20:389–399. doi: 10.1091/mbc.E07-11-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli A, Mackenzie DS, Crumrine DS, et al. Endoplasmic reticulum Ca2+ depletion activates XBP1 and controls terminal differentiation in keratinocytes and epidermis. Br J Dermatol. 2010;164:16–25. doi: 10.1111/j.1365-2133.2010.10046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claas RF, ter Braak M, Hegen B, et al. Enhanced Ca2+ storage in sphingosine-1-phosphate lyase-deficient fibroblasts. Cell Signal. 2010;22:476–483. doi: 10.1016/j.cellsig.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Cooper SM, Burge SM. Darier's disease: epidemiology, pathophysiology, and management. Am J Clin Dermatol. 2003;4:97–105. doi: 10.2165/00128071-200304020-00003. [DOI] [PubMed] [Google Scholar]
- Darier J. Jean Darier: "The three planes of dermatological diagnosis". 1923. Ann Dermatol Venereol. 2000;127:981–986. [PubMed] [Google Scholar]
- Dhitavat J, Cobbold C, Leslie N, et al. Impaired trafficking of the desmoplakins in cultured Darier's disease keratinocytes. J Invest Dermatol. 2003;121:1349–1355. doi: 10.1046/j.1523-1747.2003.12557.x. [DOI] [PubMed] [Google Scholar]
- Foggia L, Aronchik I, Aberg K, et al. Activity of the hSPCA1 Golgi Ca2+ pump is essential for Ca2+-mediated Ca2+ response and cell viability in Darier disease. J Cell Sci. 2006;119:671–679. doi: 10.1242/jcs.02781. [DOI] [PubMed] [Google Scholar]
- Gordon-Smith K, Jones LA, Burge SM, Munro CS, Tavadia S, Craddock N. The neuropsychiatric phenotype in Darier disease. Br J Dermatol. 2010;163:515–522. doi: 10.1111/j.1365-2133.2010.09834.x. [DOI] [PubMed] [Google Scholar]
- Green KJ, Getsios S, Troyanovsky S, et al. Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb Perspect Biol. 2010;2:a000125. doi: 10.1101/cshperspect.a000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakuno M, Shimizu H, Akiyama M, et al. Dissociation of intra- and extracellular domains of desmosomal cadherins and E-cadherin in Hailey-Hailey disease and Darier's disease. Br J Dermatol. 2000;142:702–711. doi: 10.1046/j.1365-2133.2000.03415.x. [DOI] [PubMed] [Google Scholar]
- Hobbs RP, Amargo EV, Somasundaram A, et al. The calcium ATPase SERCA2 regulates desmoplakin dynamics and intercellular adhesive strength through modulation of PKC{alpha} signaling. Faseb J. 2011 doi: 10.1096/fj.10-163261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JH, Yang YM, Kim HS, et al. Markers of squamous cell carcinoma in sarco/endoplasmic reticulum Ca2+ ATPase 2 heterozygote mice keratinocytes. Prog Biophys Mol Biol. 2010;103:81–87. doi: 10.1016/j.pbiomolbio.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Hong JH, Youm JK, Kwon MJ, et al. K6PC-5, a direct activator of sphingosine kinase 1, promotes epidermal differentiation through intracellular Ca2+ signaling. J Invest Dermatol. 2008;128:2166–2178. doi: 10.1038/jid.2008.66. [DOI] [PubMed] [Google Scholar]
- Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–1140. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- Kragballe K. Vitamin D3 analogues. Dermatol Clin. 1995;13:835–839. [PubMed] [Google Scholar]
- Kumar A, Saba JD. Lyase to live by: sphingosine phosphate lyase as a therapeutic target. Expert Opin Ther Targets. 2009;13:1013–1025. doi: 10.1517/14728220903039722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Tucker RW, Hennings H, et al. Chelation of intracellular Ca2+ inhibits murine keratinocyte differentiation in vitro. J Cell Physiol. 1995a;163:105–114. doi: 10.1002/jcp.1041630112. [DOI] [PubMed] [Google Scholar]
- Li L, Tucker RW, Hennings H, et al. Inhibitors of the intracellular Ca(2+)-ATPase in cultured mouse keratinocytes reveal components of terminal differentiation that are regulated by distinct intracellular Ca2+ compartments. Cell Growth Differ. 1995b;6:1171–1184. [PubMed] [Google Scholar]
- Lichte K, Rossi R, Danneberg K, et al. Lysophospholipid receptor-mediated calcium signaling in human keratinocytes. J Invest Dermatol. 2008;128:1487–1498. doi: 10.1038/sj.jid.5701207. [DOI] [PubMed] [Google Scholar]
- Mehta D, Konstantoulaki M, Ahmmed GU, et al. Sphingosine 1-phosphate-induced mobilization of intracellular Ca2+ mediates rac activation and adherens junction assembly in endothelial cells. J Biol Chem. 2005;280:17320–17328. doi: 10.1074/jbc.M411674200. [DOI] [PubMed] [Google Scholar]
- Meyer zu Heringdorf D, Liliom K, Schaefer M, et al. Photolysis of intracellular caged sphingosine-1-phosphate causes Ca2+ mobilization independently of G-protein-coupled receptors. FEBS Lett. 2003;554:443–449. doi: 10.1016/s0014-5793(03)01219-5. [DOI] [PubMed] [Google Scholar]
- Min JK, Yoo HS, Lee EY, et al. Simultaneous quantitative analysis of sphingoid base 1-phosphates in biological samples by o-phthalaldehyde precolumn derivatization after dephosphorylation with alkaline phosphatase. Anal Biochem. 2002;303:167–175. doi: 10.1006/abio.2002.5579. [DOI] [PubMed] [Google Scholar]
- Muller EJ, Caldelari R, Kolly C, et al. Consequences of depleted SERCA2-gated calcium stores in the skin. J Invest Dermatol. 2006;126:721–731. doi: 10.1038/sj.jid.5700091. [DOI] [PubMed] [Google Scholar]
- Palmer AE, Tsien RY. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat Protoc. 2006;1:1057–1065. doi: 10.1038/nprot.2006.172. [DOI] [PubMed] [Google Scholar]
- Reuter J, Braun-Falco M, Termeer C, et al. Erythema annulare centrifugum Darier. Successful therapy with topical calcitriol and 311 nm-ultraviolet B narrow band phototherapy. Hautarzt. 2007;58:146–148. doi: 10.1007/s00105-006-1131-x. [DOI] [PubMed] [Google Scholar]
- Rudolf R, Magalhaes PJ, Pozzan T. Direct in vivo monitoring of sarcoplasmic reticulum Ca2+ and cytosolic cAMP dynamics in mouse skeletal muscle. J Cell Biol. 2006;173:187–193. doi: 10.1083/jcb.200601160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuntabhai A, Burge S, Monk S, et al. Spectrum of novel ATP2A2 mutations in patients with Darier's disease. Hum Mol Genet. 1999a;8:1611–1619. doi: 10.1093/hmg/8.9.1611. [DOI] [PubMed] [Google Scholar]
- Sakuntabhai A, Ruiz-Perez V, Carter S, et al. Mutations in ATP2A2, encoding a Ca2+ pump, cause Darier disease. Nat Genet. 1999b;21:271–277. doi: 10.1038/6784. [DOI] [PubMed] [Google Scholar]
- Stewart LC, Yell J. Vulval Darier's disease treated successfully with ciclosporin. J Obstet Gynaecol. 2008;28:108–109. doi: 10.1080/01443610701844077. [DOI] [PubMed] [Google Scholar]
- Tada J, Hashimoto K. Ultrastructural localization of cell junctional components (desmoglein, plakoglobin, E-cadherin, and beta-catenin) in Hailey-Hailey disease, Darier's disease, and pemphigus vulgaris. J Cutan Pathol. 1998;25:106–115. doi: 10.1111/j.1600-0560.1998.tb01698.x. [DOI] [PubMed] [Google Scholar]
- Uchida Y, Houben E, Park K, et al. Hydrolytic pathway protects against ceramide-induced apoptosis in keratinocytes exposed to UVB. J Invest Dermatol. 2010;130:2472–2480. doi: 10.1038/jid.2010.153. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Yin M, et al. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100:209–219. doi: 10.1016/s0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- White WH. The Pathology of the Central Nervous System in Exophthalmic Goitre. Br Med J. 1889;1:699–700. doi: 10.1136/bmj.1.1474.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Bikle DD. The recruitment of phosphatidylinositol 3-kinase to the E-cadherin-catenin complex at the plasma membrane is required for calcium-induced phospholipase C-gamma1 activation and human keratinocyte differentiation. J Biol Chem. 2007;282:8695–8703. doi: 10.1074/jbc.M609135200. [DOI] [PubMed] [Google Scholar]
- Xie Z, Chang SM, Pennypacker SD, et al. Phosphatidylinositol-4-phosphate 5-kinase 1alpha mediates extracellular calcium-induced keratinocyte differentiation. Mol Biol Cell. 2009;20:1695–1704. doi: 10.1091/mbc.E08-07-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.