

Abstract
This article summarizes efforts to evaluate poly(amido amine) (PAMAM) dendrimers as carriers for oral drug delivery. Specifically, the effect of PAMAM generation, surface charge and surface modification on toxicity, cellular uptake and transepithelial transport is discussed. Studies on Caco-2 monolayers, as models of intestinal epithelial barrier, show that by engineering surface chemistry of PAMAM dendrimers, it is possible to minimize toxicity while maximizing transepithelial transport. It has been demonstrated that PAMAM dendrimers are transported by a combination of paracellular and transcellular routes. Depending on surface chemistry, PAMAM dendrimers can open the tight junctions of epithelial barriers. This tight junction opening is in part mediated by internalization of the dendrimers. Transcellular transport of PAMAM dendrimers is mediated by a variety of endocytic mechanisms. Attachment or complexation of cytotoxic agents to PAMAM dendrimers enhances the transport of such drugs across epithelial barriers. A remaining challenge is the design and development of linker chemistries that are stable in the gastrointestinal tract (GIT) and the blood stream, but amenable to cleavage at the target site of action. Recent efforts have focused on the use of PAMAM dendrimers as penetration enhancers. Detailed in vivo oral bioavailability of PAMAM dendrimer – drug conjugates, as a function of physicochemical properties will further need to be assessed.
Keywords: Poly(amido amine) (PAMAM) dendrimers, oral drug delivery, transepithelial transport, polymers
1. Introduction
1.1 Oral polymeric prodrugs
Conjugation or complexation of therapeutics to polymeric carriers (polymer therapeutics) can result in prolonged circulation half-life, increased concentration at the site of action (such as tumors, or reticuloendothelial system) and decreased non-specific toxicity [1, 2]. Due to their macromolecular nature, polymer therapeutics are generally administered by the parenteral route. It is desirable to develop such systems for oral administration. Oral drug delivery is among the most preferred routes of administration owing to its non-invasive nature, cost-effectiveness and patient compliance [3]. Oral delivery of polymer therapeutics is hindered by several barriers in the gastrointestinal tract (GIT) (Figure 1). These include their large size and hydrophilicity which limit transepithelial transport. Furthermore, variations in pH along with high enzymatic activity in the GIT can adversely affect the stability of polymer therapeutics leading to premature release of the drug or degradation of the constructs [4]. By investigation and optimization of their stability in the GIT, and absorption across the gut, polymer therapeutics can be developed for oral administration.
Figure 1.
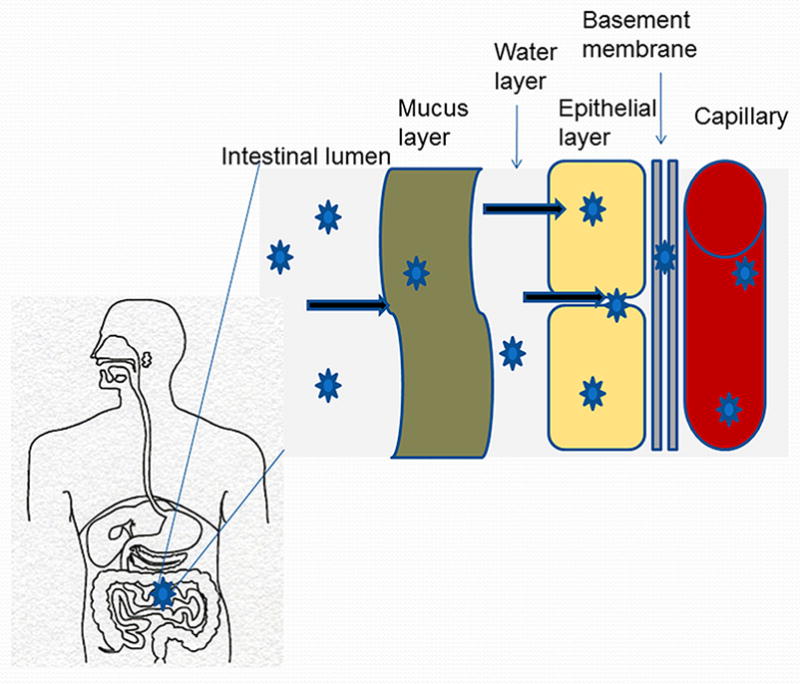
Gastrointestinal barriers to the oral delivery of polymer therapeutics
Owing to their compact structure and high surface charge density, dendritic polymers can be tailor made to permeate across the epithelial barrier of the gut. Polymers with dendritic architecture such as poly(lysine), and poly(amido amine) (PAMAM) have been observed to transport across the epithelial barrier of the intestine, thus showing promise in oral delivery [5–28]. PAMAM dendrimers have been the most widely studied dendritic polymer for oral delivery purposes. They have the advantage of having a very open internal structure and cavities for accommodating guest therapeutic molecules that need to be solubilized or targeted for drug delivery. They have an established divergent synthesis method and are commercially available in different generations and surface groups. The findings summarized in this review emphasizes on the trans-epithelial transport and toxicity of PAMAM dendrimers for oral delivery.
PAMAM dendrimers
Poly(amido amine) or PAMAM dendrimers are a class of hyper-branched polymers originally developed by Tomalia in 1979 [29]. The ethylene diamine core and amido amine branching structure of the PAMAM lead alternatively to amine-terminated full generation or carboxyl-terminated half-generation dendrimers after each addition step in the synthesis (Figure 2A) [30]. For every increase in generation, the number of functional groups doubles, while the dendrimer diameter increases by about 1 nm [31]. Due to their controlled synthesis, these polymers have the unique advantage of having very low polydispersities. A full generation PAMAM dendrimer has primary amine groups on the surface (pKa =6.85) and tertiary amine groups within the core (pKa = 3.86) [31]. The high density of surface functional groups on PAMAM dendrimers presents the opportunity of functionalizing these polymers with various drugs, nucleic acids and imaging system components (Figure 2B) [32–39]. PAMAM dendrimers of certain generations and surface charge can permeate the epithelial barrier of the gut, suggesting their potential as oral drug carriers [6–28]. The purpose of this review is to summarize recent findings on transepithelial transport, cellular uptake and cytotoxicity of PAMAM dendrimers in the context of oral drug delivery and provide insight into the challenges and opportunities that these constructs provide for oral administration of bioactive agents.
Figure 2.

Schematics of PAMAM dendrimers for drug delivery; A. Stepwise addition of ethylene diamine and methacrylic acid to alternatively form a half generation, carboxylic acid PAMAM and a full generation amine-terminated PAMAM generation 2. B. A cartoon representing a dendrimer-based delivery system functionalized with therapeutic agent, biorecognizable moiety, imaging agent, nucleic acid and surface-modifying groups. Ref [58], permission to be obtained.
2. PAMAM toxicity and biodistribution
2.1 Toxicity on Caco-2 cells
Caco-2 cells are human colorectal carcinoma cells that develop a cell polarity when grown in monolayers and allow the study of transepithelial transport [40]. It is known that PAMAM dendrimers demonstrate a generation-, surface charge-, concentration- and incubation time - dependent cytotoxicity profile [8]. Our initial observations reviewed elsewhere [41], suggest that the rank order of cytotoxicity of PAMAM dendrimers is hydroxyl-terminated < carboxyl-terminated < amine-terminated systems. Using the lactate dehydrogenase (LDH) assay on Caco-2 cells, carboxyl-terminated dendrimers of generations 3.5 and 4.5 (G3.5-COOH and G4.5-COOH) have shown to be toxic only at a higher donor concentration of 10.0 mM compared to amine-terminated dendrimers of generations 3.0 and 4.0 which are toxic at 1.0 mM (G3.0-NH2 and G4.0-NH2) [8, 10]. The LDH assay revealed plasma membrane damage of Caco-2 cells by PAMAM dendrimers as a function of generation number, surface charge, incubation time and concentration.
Transmission electron microscopy (TEM) analysis further showed a concentration-, generation- and surface charge-dependent effect of PAMAM dendrimers on Caco-2 microvilli morphology (Figure 3) [18]. Cells treated with a concentration of 0.1 mM or higher G4.0-NH2 showed membrane disruption and loss of Caco-2 microvilli while those treated with G3.5-COOH at the same concentration were unaffected [18]. The extent of disruption and loss of microvilli corresponded with G4.0-NH2 concentration. At lower concentration of 0.01 mM, dendrimers did not influence microvilli morphology as per TEM images. Higher generation cationic dendrimers showed increased intestinal membrane damage compared to lower generation ones [18].
Figure 3.
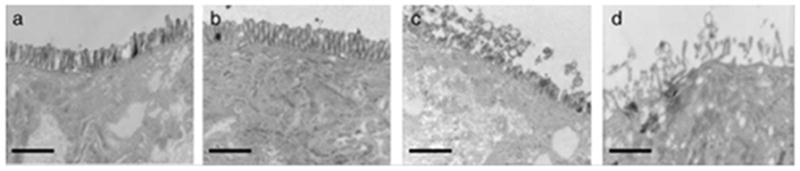
TEM images of Caco-2 cell monolayers after treatment with G4.0-NH2 dendrimers for 2 h: A. control cells; B. 0.01 mM G4.0-NH2; C. 0.1 mM G4.0-NH2; D. 1.0 mM G4NH2 (magnification=12,500x). Scale bars = 1 mm. With permission from Ref [18].
In summary, in vitro toxicity studies on PAMAM dendrimers revealed that cationic systems are non-toxic on Caco-2 cells at lower concentrations of 0.01 mM as per LDH assay and microscopic evaluation. Anionic PAMAM dendrimers are tolerated to a higher extent than cationic dendrimers making it possible to give higher doses of the carboxylic acid-terminated systems. In the range of dendrimers evaluated from generations 0.0 to 4.0 with varying surface functional groups, it was observed that there is a workable non-toxic window for PAMAM dendrimers to be used as carriers for oral drug delivery. These studies set the stage for the in vitro evaluation of transepithelial transport and cellular uptake of PAMAM dendrimers across epithelial barriers.
2.2. Biocompatibility and biodistribution
Preliminary toxicity and immunological evaluation of amine-terminated PAMAM dendrimers G3.0, G5.0 and G7.0 was done in male swiss-webster mice by intravenous administration [42]. Acute toxicity was studied over a period of 7 days, sub-chronic toxicity over 30 days and chronic toxicity over 6 months. For acute and sub-chronic toxicity, intravenous doses of 5 × 10−6 mmol/kg, 5 × 10−5 mmol/kg, and 5 × 10−4 mmol/kg of the dendrimers under study were administered. For chronic toxicity, a dose of 5 × 10−4 mmol/kg was administered intravenously for PAMAM G3.0-NH2 and G5.0-NH2, and 5 × 10−5 mmol/kg for PAMAM G7.0-NH2 [42]. PAMAM G7.0-NH2 was administered at a lower dose for the chronic toxicity study because acute toxicity was observed for PAMAM G7.0-NH2 at the higher dose of 5 × 10−4 mmol/kg [42]. Animals were monitored for routine behavioral abnormalities and changes in body weight. Upon sacrifice, tissues were observed for macroscopic abnormalities. Tissue sections of liver and spleen were fixed and stained with hematoxylin and eosin to observe microscopic abnormalities. Serum samples for the immunogenicity testing were obtained from sets of 2 New Zealand white rabbits to serve as nonimmune controls. The animals were then subcutaneously injected with PAMAM G3.0-NH2, G5-NH2, or G7-NH2 at a dose of 5 × 10−5 mmol. Two subsequent booster shots, at the same dose as the first were given at 3-week intervals. Blood samples were collected 10 days after each injection. Potential biological complications were observed only with PAMAM G7.0-NH2 at the highest dose tested, with 1 out of 5 animal deaths 24 hours post injection [42]. The immunogenicity of the PAMAM dendrimers was studied using two different methods: immunoprecipitation and an Ouchterlony double diffusion assay. Ouchterlony double diffusion assay is an agar immunodiffusion assay for detecting extractable nuclear antigens. No immunological reactions were seen at doses tested. This study was one of the first reports of the in vivo evaluation of PAMAM dendrimers. It was a preliminary evaluation of toxicity of cationic PAMAM dendrimers at a fixed dose and showed that PAMAM toxicity increased with increase in generation and surface charge density of amine groups [42]. PAMAMs of G5.0 or below were well tolerated up to doses 5×10−4 mmol/Kg but higher generation PAMAMs showed biological complications at the same dose. This toxicity study of intravenously administered PAMAM throws light on possible biological complications that may occur upon systemic absorption of orally dosed constructs.
In another study the biocompatibility of cationic PAMAM dendrimers G1.0-G4.0 and anionic PAMAM dendrimers G1.5-G9.5 was systematically investigated to evaluate the effect of dendrimer generation and surface functionality on biological properties in vitro [43]. PAMAM dendrimers were added to fresh rat blood cells in phosphate buffer saline (PBS) and incubated with shaking at 37°C for 1 hour. The hemoglobin released was spectrophotometrically determined. Amine terminated PAMAM dendrimers displayed concentration and generation-dependent hemolysis, and changes in red cell morphology (rounding of cells and clumping) were observed after 1 h even at non-hemolytic concentrations (10 μg/ml) [43]. All cationic PAMAM dendrimers except G1.0 were hemolytic above 1.0 mg/mL. Anionic dendrimers caused no morphological changes to RBCs upto 2.0 mg/mL as per scanning electron microscopy [43]. These results correlated well with in vitro cytotoxicity studies in Caco-2 cells, in that anionic PAMAM dendrimers were more biocompatible than their cationic counterparts and that the cationic dendrimers showed reduced biocompatibility at higher generations.
Studies on the in vivo biodistribution of 125I-labelled, intravenously administered poly(amido amine) dendrimers in rats have shown that anionic dendrimers circulate longer in the blood than their cationic counterparts [43]. 0.1–1.0 % of dose of PAMAM-NH2 dendrimers was recovered in the blood at 1 hour while 15–40 % of the dose of PAMAM-COOH of various generations was recovered at the same time point. Both types of dendrimers showed high liver accumulation, with the cationic dendrimers showing slightly higher liver concentration (60–90 %) than the anionic dendrimers (25–70 %) [43]. Hydroxyl terminated PAMAM dendrimers below kidney threshold (G5.0-OH) showed about 80% kidney accumulation in mice when intravenously administered at 30 minutes while G6.0-OH accumulated in the liver upto 25 % in the first 30 minutes [44]. PAMAM G7.0-OH, the highest generation tested was long circulating in the plasma with decreased non-specific accumulation in the liver and kidney [44]. Results of biodistribution studies of intravenously administered PAMAM dendrimers can suggest the possible in vivo fate of orally administered PAMAM dendrimers upon systemic absorption into blood circulation.
Recent in vivo studies have focused on establishing the maximum tolerated doses for PAMAM dendrimers of different generations and surface charge, administered orally and intravenously to CD-1 mice [45]. PAMAMs of two different generations 4.0 and 7.0 and three different surface functionalities: amine, carboxylic acid and hydroxyl terminated were tested. Acute toxicity (10 days) was inferred by monitoring routine behavioral changes, body weight changes, and upon animal sacrifice, changes in organ weight, macroscopic tissue abnormalities, blood chemistry and blood picture. It was observed that when intravenously administered, amine terminated dendrimers (both G4.0-NH2 and G7.0-NH2) were tolerated only at doses less than 10 mg/kg [45]. This finding is in agreement with a previous study by Roberts et al. summarized above, where biological complications were observed for G7.0-NH2 dendrimers, dosed at 5.0 × 10−4 mmol/Kg which translates to about 58.3 mg/kg of the dendrimer [42]. In the same study, lower generation dendrimers: G3.0-NH2 and G5.0-NH2, dosed at 5×104 mmol/Kg (about 3.4 and 14.4 mg/Kg respectively) were non-toxic [42]. In contrast carboxyl- (G3.5-COOH and G6.5-COOH) and hydroxyl- (G4.0-OH and G7.0-OH) terminated dendrimers were safely administered intravenously at 50-fold or higher doses (Figure 4). A similar trend was observed for orally administered doses of PAMAM, but they were tolerated at higher doses than when administered intravenously (unpublished data). Orally administered PAMAM G4.0-NH2 was tolerated upto 100 mg/Kg, which was the highest dose tested while anionic and hydroxyl terminated PAMAMs of the corresponding generation were tolerated at 300 mg/Kg. PAMAM G7.0-NH2 was found to be toxic at 50 mg/Kg while the anionic and hydroxyl-terminated PAMAMs of corresponding generations were tolerated at 300 mg/Kg. Blood analysis showed decreased levels of fibrinogen, platelets and high levels of fibrin degradation products (FDP) in groups treated with amine-terminated dendrimers as compared to control. This suggested the occurrence of both pro- and anti-coagulation processes similar to disseminated intravascular coagulation (DIC). DIC is a complex systemic thrombo-hemorrhagic disorder involving generation of intravascular fibrin and consumption of pro-coagulants and platelets. The resultant clinical condition is characterized by intravascular coagulation and hemorrhage, observed in the animals treated with amine terminated dendrimers [45]. Overall these studies revealed that PAMAM dendrimers showed similar toxicity trends when administered orally and intravenously, with the higher generation cationic dendrimers being more toxic than their lower generation counterparts and the anionic dendrimers being less toxic than the cationic ones. PAMAM dendrimers were tolerated at higher doses when administered orally as compared to intravenous administration. This could be due to a rate limiting absorption process that reduces exposure of the dendrimers to blood. As a consequence, it seems that the biologically compatible working range of cationic dendrimers increases almost 10-fold when administered orally, making it possible to administer higher doses of drugs being delivered by PAMAM-NH2. However, detailed histological evaluation of the gastrointestinal epithelium needs to be carried out to understand possible tissue toxicity due to PAMAM exposure.
Figure 4.
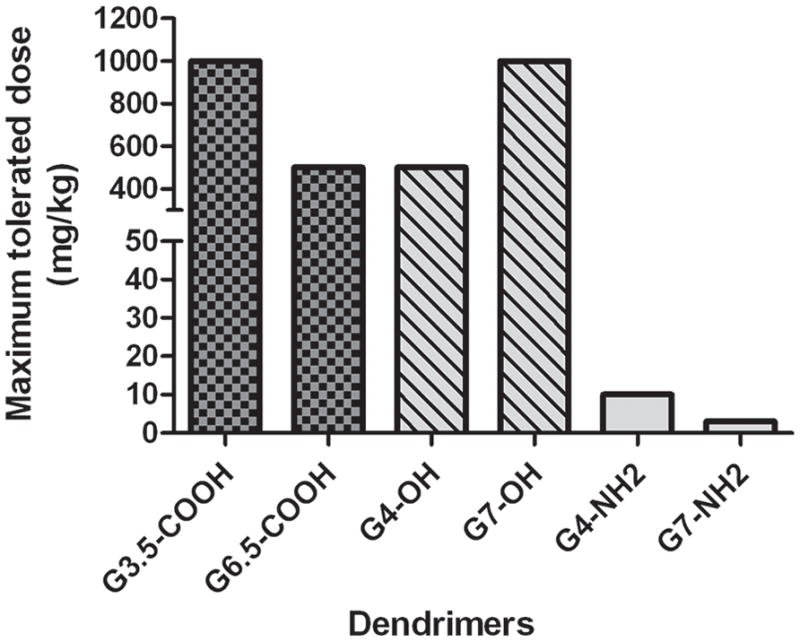
Maximum Tolerated Doses (MTD) in mg/Kg of intravenously administered PAMAM dendrimers to CD-1 mice (n=5). Ref [45], permission to be obtained.
An attempt to understand histological damage to intestinal epithelial barrier was carried out recently. Lin et al., evaluated the intestinal membrane damage in SD rats of amine-terminated PAMAMs generations 0.0–3.0 (0.05 to 0.5% w/v), when evaluating in situ absorption of hydrophilic molecules in the presence of PAMAM dendrimers [26]. The activities of LDH and the amounts of protein released from the small intestinal membranes were measured by an in situ loop method 4 hours after administration. G2.0-NH2 at 0.5% (w/v) significantly increased the activities of lactate dehydrogenase (LDH) and the amounts of protein released from the intestinal membranes, while no significant increase in toxic markers was observed at concentrations of 0.05% (w/v) and 0.1% (w/v) [26]. However, the activities and amounts of these toxic markers were less than those in the presence of 3% (v/v) Triton X-100 used as a positive control. Intestinal absorption of 5(6)-Carboxyfluorescein (CF), a water-soluble dye was not enhanced by pretreatment with G2.0-NH2 (0.5%, w/v), although the co-administration with G2.0-NH2 significantly increased the intestinal absorption of CF, suggesting that the absorption-enhancing effect of G2.0-NH2 is reversible and might not cause the continuous and irreversible membrane toxicity in the rat small intestine. This data correlates with cytotoxicity and in vivo data of PAMAM-NH2 dendrimers, with the toxicity increasing as a function of generation and concentration. At the highest dose employed of 0.5% w/v of PAMAM G2.0, signs of histological toxicity observed were found to be reversible.
3. Transepithelial transport and intracellular fate
An important barrier to oral absorption of PAMAM dendrimers is limited transepithelial transport. Several methods are available to study transport of compounds across the epithelial barrier of the gut [40]. These include, but are not limited to, isolated intestinal tissue techniques, cell culture monolayer systems such as Caco-2 cells, and in situ perfusion models. The effect of PAMAM generation, surface group, concentration and incubation time with cells on transport across epithelial barriers has been extensively studied on epithelial monolayers and isolated intestinal tissue in vitro [6–12, 14–20, 22]. In an initial study, the isolated intestinal tissue model using the everted sac technique was employed to assess the transepithelial transport of PAMAM dendrimers [6]. This technique, along with the Ussing chamber technique involving isolated intestinal tissue, provides mechanistic insights and additionally allows the comparison of differences in the segmental transport throughout different regions of the GIT [40]. The everted sac setup involves everting an intestinal segment, 2–4 cm long over a glass rod, 3 mm in diameter, and submerging the setup in culture medium containing the compound to be tested, that then contacts the mucosal side of the intestine, on the outside of the sac [46]. The model is a simple, quick, reproducible and inexpensive technique. However, the volume inside the sac is small, because of which sink conditions cannot be established. The Ussing chamber setup involves mounting of a small segment of the intestine between two compartments: the serosal and the mucosal side [46]. It is also possible to connect electrodes in the two compartments to measure transepthelial electrical resistance (TEER) and therefore tight junction modulation. A small amount of sample is needed to evaluate absorption. However, the preparation of the intestinal epithelial layer can be complicated. Incomplete removal of the serosal muscle layer can result in false measurements of transport [46]. These ex vivo techniques may be useful to determine the effect of penetration enhancers on the barrier properties and viability of different segments in the intestine [40]. Maintaining tissue viability is critical and difficult in employing isolated tissues to evaluate transport.
125I-labelled PAMAM dendrimers were first evaluated for their uptake and transport in vitro using the everted intestinal sac system in rat [6]. Amine-terminated cationic PAMAM dendrimers showed greater tissue uptake than their serosal transfer rates across everted rat intestinal sacs at each time point. In contrast anionic G2.5 and G3.5 showed greater serosal transfer rates (3.4–4.4 AL/mg protein/h) than their tissue uptake (0.6–0.7 AL/mg protein/h) in the same model. Anionic G5.5 had greater tissue uptake (30–35%) than that of G2.5 and G3.5 (15–20%), and the serosal transfer of G5.5 (60–70% recovered in serosal fluid) was less than that of G2.5 and G3.5 (80–85% recovered in serosal fluid) [6]. This study demonstrated that PAMAM generation and surface charge influence their transepithelial transport and by selecting a suitable range of size and charge, PAMAM dendrimers can potentially be used as carriers for oral drug delivery.
A variety of pre- and post-epithelial factors influence the transport of compounds across the isolated intestinal tissues including everted sacs. To avoid the influence of pre- and post-epithelial factors, and gain a detailed understanding of the influence of physicochemical properties of dendrimers on the extent and mechanism of transepithelial transport, a series of studies were conducted to examine the effect of the physicochemical and structural properties of PAMAMs of various generations and surface charges on their transport across epithelial cell culture monolayers [7–12, 14–20, 22]. Amongst cultured cells, Madin-Darby canine kidney (MDCK) cells, Caco-2 monolayers, and IPEC-J2 monolayers have been used to assess the transepithelial transport of PAMAM dendrimers. When cultured as monolayers, the cells undergo differentiation, maintain a cell polarity and develop a transepithelial resistance [40]. The polarity in the cell line makes it possible to study the directionality of uptake. The possibility to measure transepithelial resistance allows the study of tight junction regulation in cultured monolayer cells. Both cellular uptake and transepithelial transport can be studied. The transport of compounds from the apical to basolateral side can also be measured in the presence of inhibitors of certain active transport pathways, thereby providing mechanistic insight into the intestinal transport of compounds under study. Cell monolayers can also remain viable for a longer period of time compared to tissue isolates, although more limited set of enzymes and active proteins are present in cultured monolayers compared to the isolated tissue systems. Cultured intestinal cell monolayers are useful in rank ordering permeabilities of compounds of the same class. A study attempting to correlate in vitro results in Caco-2 cell monolayers to in vivo data has demonstrated that compounds with apparent permeability coefficients (Papp) above 1 × 10−6 cm/sec are likely to be well absorbed [47]. However, cultured monolayers such as the Caco-2 cell monolayers widely used to assess PAMAM intestinal transport lack mucous secretion and therefore do not present a mucosal barrier to transport [40]. They also lack the cellular heterogeneity found in the intestinal mucosa and more closely represent the colonic than the small intestinal epithelium. Similar to isolated intestinal models, the cultured intestinal cells do not account for other clearance and instability mechanisms that may contribute to low oral bioavailability like hepatic first pass clearance and enzymatic degradation in the GIT [40].
The influence of size, charge, incubation time, and dendrimer concentration of amine-terminated PAMAM dendrimers G0.0 to G4.0 across Caco-2 cell monolayers was first studied by El-Sayed et al [8]. In this study it was observed that the basolateral to apical (BA) permeability of each dendrimer was generally higher than the corresponding apical to basolateral (AB) permeability. This could probably be attributed to the difference in tight junction characteristics at the apical and basolateral sides. The effect of amine-terminated PAMAM dendrimers (PAMAM-NH2) on paracellular pathway was investigated through TEER measurements and the permeability of a known paracellular permeability marker, 14C-mannitol. TEER data correlated with the permeability profiles of the PAMAM-NH2 dendrimers, in which prolonged interaction of the dendrimers with the epithelial surface, and increased surface charge density due to increased generation number and/or concentration, can result in modulation of the tight junctions, which would lead to the decline in TEER values [8]. The influence of surface charge of PAMAM dendrimers on transepithelial transport of 14C-mannitol across Caco-2 cells and their cytotoxicity was further evaluated [10]. Neutral PAMAM dendrimers with hydroxyl surface terminal groups (PAMAM–OH) did not significantly influence TEER or 14C-mannitol permeability across Caco-2 monolayers. Anionic, carboxylic acid-terminated PAMAMs (PAMAM–COOH) had a generation-dependent effect on TEER and 14C-mannitol permeability. G-0.5, G0.5 and G1.5 caused no decline in TEER values and no significant increase in 14C-mannitol permeability, whereas G2.5 and G3.5 caused a significant decline in TEER compared to control values and a 6-fold increase in 14C-mannitol permeability [10]. PAMAM G4.5 also caused a decline in TEER values; however, this was due to cytotoxicity as evidenced by LDH assay. The decrease in TEER values and the increase in 14C-mannitol permeability suggested that the amine-terminated dendrimers enhance net paracellular transport by modulating tight junctions of Caco-2 cell monolayers.
In a subsequent study it was demonstrated that in addition to enhancing paracellular transport, PAMAM dendrimers are also translocated across the epithelial barrier of Caco-2 cells by endocytosis mechanisms [9]. The AB permeability of G2.0–NH2 across Caco-2 cell monolayers was investigated at 37°C and 4°C to evaluate the contribution of an energy-dependent endocytotic process for transport. The permeability of G2.0 NH2 was significantly lower at 4°C than at 37°C, suggesting endocytotic mechansims of transport. Since the BA permeability of G2.0–NH2 was observed to be higher than the corresponding AB permeability, the contribution of an efflux system, namely P-glycoprotein (P-gp), to PAMAM transport was evaluated. The AB and BA permeabilities of 14C-paclitaxel, a known P-gp substrate, were measured in the absence and presence of G2.0-NH2. The BA permeability of 14C-paclitaxel was greater than the AB permeability, indicating a functioning P-gp efflux system in the utilized Caco-2 cell monolayers [9]. There was no significant difference in AB and BA permeability of 14C-paclitaxel in the presence of G2.0-NH2. The AB and BA permeability of G2.0–NH2 did not change in the presence of paclitaxel, which suggests G2.0-NH2 is not a P-gp substrate and is therefore not subject to the P-gp efflux system [9].
Since these initial studies demonstrated that PAMAM dendrimers are transported by both para- and transcellular routes, more detailed studies on the mechanism of transport of PAMAM dendrimers have been carried out which are summarized below:
The transepithelial permeabilities of amine-terminated (G2.0-NH2 and G4.0-NH2), hydroxyl-terminated (G2.0-OH), and carboxyl-terminated (G1.5-COOH and G3.5-COOH) PAMAM dendrimers, conjugated to fluorescein isothiocyanate (FITC) across Caco-2 cells were evaluated, with 14C-Mannitol as a marker of paracellular permeability [15]. 14C-Mannitol permeability significantly increased in the presence of charged PAMAM dendrimers indicating the opening of tight junctions (Figure 5A) [15]. Decline in TEER values correlated with the increase in 14C-mannitol permeability with native PAMAM G2.0-NH2 dendrimers causing the greatest decline in TEER (Figure 5B). FITC labeled PAMAM G4.0-NH2 dendrimers caused a decline in TEER up to 32% during the experiment, and only G4.0-NH2-FITC (1:8) had a reversible effect in Caco-2 cells where the TEER value of cells returned to 92.1% of the original reading after 24 h (Figure 5B). Immunofluorescence microscopy was used to further study changes in the barrier function of tight junctions. Tight junctions are protein complexes that include occludin amongst other proteins. Occludin is responsible for the fusion of adjacent plasma membranes to form the junction, which functions to maintain cellular integrity. A clear band of occludin protein was observed for untreated Caco-2 cells, whereas exposure of charged PAMAM dendrimers resulted in increased accumulation of occludin protein as evidenced by the disruptive staining pattern encircling Caco-2 cells (Figure 5C) [15]. This pattern indicated the opening of the tight junctions upon incubation with cationic and anionic PAMAM dendrimers under study. Actin filaments are responsible for binding adjacent cells, thereby forming a single unit. Incubation of Caco-2 cell monolayers with rhodamine phalloidin, a specific probe for filamentous actin, revealed a clear, continuous staining pattern. Upon incubation with cationic and anionic PAMAM dendrimers, however, the cells show a clear disruption of actin staining [15]. These data correspond with the observations that cationic and anionic dendrimers cause a decline in TEER and enhance 14C-mannitol permeability. These studies further confirmed that PAMAM dendrimers modulated tight junction proteins occludin and actin and that increased permeability of dendrimers is partly due to opening of tight junctions, which can be reversible depending on the concentration, generation and surface charge of the dendrimers.
Figure 5.

Transepithelial permeability of 14C-mannitol and tight junction modulation in presence of PAMAM dendrimers; (a). The permeability of 14C-mannitol (3.3 μM) in the presence of fluorescently labeled and unlabeled PAMAM dendrimers of donor concentration 1.0 mM across Caco-2 cell monolayers at incubation times of (□) 60 min and (■) 120 min. Permeability values are not reported (**) for dendrimers that cause toxicity. Results are reported as mean +/− standard error of the mean (n = 9). (*) Denotes a significant increase in permeability compared to control (P < 0.001). (b). Transepithelial electrical resistance of Caco-2 cells in the presence of fluorescently labeled PAMAM dendrimers as a function of time: (◆) HBSS transport medium, (■) G2.0-NH2, (▲) G2.0-OH, (×) G1.5-COOH, (+) G2.5-COOH, (●) G3.5-COOH, (◇) G4NH2-FITC (1:8). Results are reported as mean +/− standard error of the mean (n = 9). (c). Staining of the tight junction protein occludin. (A) Caco-2 cells with no polymer treatment. Caco-2 cells incubated for 120 min with 1.0 mM: (B) G2.0-NH2; (C) G2.0-OH, (D) G1.5-COOH; (E) G2.5-COOH; (F) G3.5-COOH. Main panels illustrate the xy plane; horizontal bars illustrate the xz plane; vertical bars illustrate the yz plane. Scale bars equal 100.00 μm. With permission from Ref [15].
The endocytotic pathway is an important route for intracellular uptake of macromolecules. The internalization and subcellular trafficking of FITC-labeled cationic and anionic PAMAM dendrimers was investigated on cell suspensions of Caco-2 cells [18]. Confocoal microscopy revealed that both cationic and anionic PAMAM dendrimers were internalized within 20 min, and differentially colocalized with endocytosis markers clathrin (endocytosis marker that aids in the formation of a coated pit upon invagination), EEA-1 (membrane-bound protein component specific to early endosomes), and LAMP-1 (a lysosomal protein marker) [18]. Significant colocalization coefficients with clathrin suggested a clathrin-dependent receptor-mediated endocytosis process of cellular uptake for the PAMAMs. The clathrin colocalization was independent of incubation time showing a constant presence of dendrimers in early endosomes. Colocalization of dendrimers with LAMP-1 increased proportionally with time and suggested that lysosomal trafficking of dendrimers is a function of incubation time and surface charge.
To provide further insight into the endocytosis mechanisms of PAMAM transport across Caco-2 cell monolayers, the effect of endocytosis inhibitors on G4.0-NH2 uptake and transport across Caco-2 cells was examined [22]. The endocytosis inhibitors used were brefeldin A, colchicine, filipin and sucrose. Brefeldin A induces tubulation of secondary endosomes and lysosomes, which compromises microtubule trafficking of transport vesicles within the cell. Colchicine interferes with microtubule trafficking through binding to tubulin subunits. Filipin is known to inhibit caveolae-mediated endocytosis by binding to cholesterol, a major component of glycolipid microdomains and caveolae. Hyperosmolarity conditions generated by increased sucrose concentration primarily blocks membrane internalization and clathrin recycling via the coated-pit pathway. A significant reduction in G4.0-NH2 uptake and permeability was observed in the presence of endocytosis inhibitors. Brefeldin A reduced G4.0-NH2 uptake 2-fold to 22.1 pmol/mg protein/min at 8 μM. Likewise, G4.0-NH2 absorption was reduced almost 3-fold to 18.5 pmol/mg protein/min in the presence of colchicine [22]. The co-incubation with filipin resulted in reduced G4.0-NH2 uptake 3-fold to 16.5 pmol/mg protein/min, and sucrose reduced the rate of G4.0-NH2 uptake almost 3-fold to 20.1 pmol/mg protein/min [22]. The apparent permeability of G4.0-NH2 was 11.6 × 10−5 cm/s, which reduced to 5.7 × 10−6 cm/s in the presence of brefeldin A, 3.4 × 10−6 cm/s with colchicine, 7.3 × 10−6 cm/s with flipin and 9.7 × 10−6 cm/s with sucrose (Figure 6). Riboflavin was used as a positive control ligand in the endocytosis study. These findings support previous results that in addition to paracellular transport, cationic dendrimers are also endocytosed. However it was shown that PAMAM G4.0-NH2 is nonspecifically internalized in coated vesicles at the plasma membrane.
Figure 6.
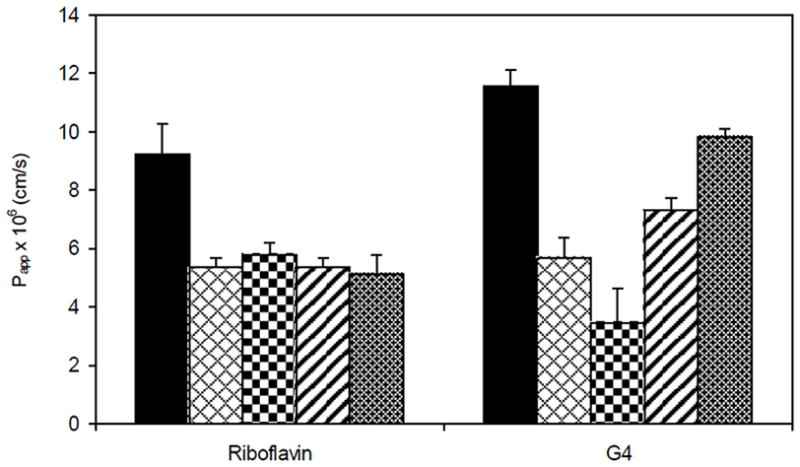
Reduced apparent permeability (Papp × 106 cm/s) of Riboflavin (500 nM) and G4.0-NH2 (1 μM) across Caco-2 cell monolayers in the presence of endocytosis inhibitors: Bars from left to right indicate: 1–5 μM brefeldin A; 10 μM colchicine; 1 μg/ml filipin; 200 mM sucrose. Results are reported as mean ± SD (n = 3). **, p < 0.01: ***, p < 0.001. With permission from Ref [22]
While the above studies were conducted on amine terminated systems, given that surface charge may influence the pathway of cellular uptake, the mechanisms of PAMAM G3.5-COOH dendrimer cellular uptake, intracellular trafficking, transepithelial transport and tight junction modulation in Caco-2 cell monlayers was evaluated [27]. Chemical inhibitors blocking clathrin-, caveolin- and dynamin dependent endocytosis pathways were used to investigate the mechanisms of dendrimer cellular uptake and transport across Caco-2 cells using flow cytometry and confocal microscopy, where PAMAM G3.5 was labeled with oregon green to facilitate detection. The Inhibitors used included chemicals known to prevent clathrin-mediated endocytosis: phenylarsine oxide and monodansyl cadaverine. Monodansyl cadaverine is known to stabilize clathrin-coated pits on the cell membrane, thereby preventing internalization while the mechanism of phenylarsine oxide is unknown. To inhibit caveolin-mediated endocytosis, filipin and genistein were used. Filipin acts by binding cholesterol while genistein inhibits protein tyrosine kinases to block internalization by caveolae. Dynamin-dependent endocytosis was inhibited by dynasore. Dynasore is known to block vesicular endocytosis by selectively inhibiting dynamin 1 and dynamin 2 GTPases, which are responsible for vesicle scission during both clathrin- and caveolin-mediated endocytosis. G3.5 PAMAM dendrimer showed reduction in cellular uptake in the presence of all endocytosis inhibitors tested, suggesting the involvement of both clathrin- and caveolin-mediated endocytosis pathways in cellular uptake [27]. The greatest reduction in uptake of PAMAM G3.5 was shown in the presence of dynasore. Dendrimers showed decreased uptake in the presence of both clathrin inhibitors, with a greater decrease seen in the presence of monodansyl cadaverine [27]. PAMAM G3.5 dendrimers also showed reduced uptake in the presence of both caveolin inhibitors. Once taken up by cells, PAMAM G3.5 colocalized with early endosomes (EEA-1) and lysosomes (LAMP-1) [27]. Over time, the trafficking pathway was saturated, causing dendrimer retention in early endosomes once lysosomes were occupied. PAMAM G3.5 significantly increased occludin staining relative to untreated cells, indicating tight junction opening. In cells treated with dynasore, no difference could be detected in occludin staining between cells treated with dendrimers or dynasore alone. A significant finding was that PAMAM dendrimers were unable to open the tight junctions in cells where dynamin-dependent endocytosis was inhibited, suggesting that dendrimer internalization is in part responsible for modulating cellular tight junctions (Figure 7). It was found that G3.5 dendrimer internalization by dynamin-dependent mechanisms promotes tight junction opening, suggesting that this dendrimer acts on intracellular cytoskeletal proteins to modulate tight junctions, thus catalyzing its own transport via the paracellular route [27]. More studies are needed to evaluate whether such phenomenon is indeed responsible for tight junction opening of other dendrimers with a different surface charge or generation, and to further delineate the contributions of extracellular vs intracellular factors in opening of the tight junctions in the presence of dendrimers.
Figure 7.

Transpeithelial transport and cellular uptake mechanism of PAMAM G3.5-COOH; A–D. Occludin staining in the presence and absence of Oregon green labeled G3.5-COOH dendrimers in Caco-2 cells treated with HBSS or Dynasore. A. G3.5-COOH/HBSS, B. HBSS only C. G3.5-COOH/Dynasore and D. Dynasore only. Main panels illustrate the xy plane; horizontal bars illustrate the xz plane; vertical bars illustrate the yz plane. Scale bars equal 21 μm. E. Quantification of Occludin staining. Results are reported as mean +/− standard deviation with n=4. (***) indicates p<0.001. With permission from Ref [27].
More recent reports have focused on modeling the porosity of epithelial layers as a function of dendrimer generation, surface charge, concentration and incubation time [25]. Mathematical calculations show that the increase in concentration, incubation time and generation number of cationic G0.0-NH2 to G2.0-NH2 and anionic G2.5-COOH to G3.5-COOH result in increasing porosity of epithelial cell monolayers. These findings postulate the hypothesis that PAMAM permeability through Caco-2 monolayers is potentially a result of disorganization of tight junctions and associated increase in membrane porosity as well [25].
Together, these studies show that PAMAM dendrimers are transported across Caco-2 cell monolayers by a combination of the paracellular pathway and an energy-dependent process, such as endocytosis (Figure 8). These studies have set the stage for evaluation of PAMAM dendrimers as drug carriers across epithelial barriers and to see how their surface modification influences toxicity and transport.
Figure 8.
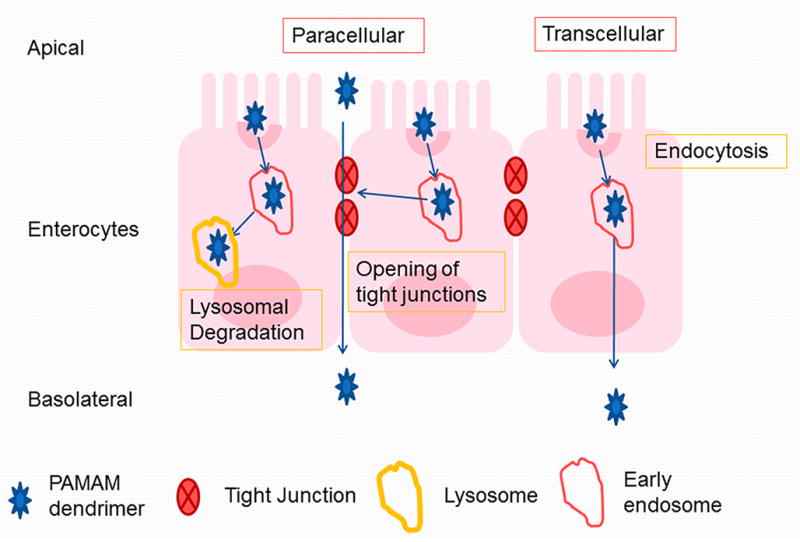
Transepithelial transport mechanisms of PAMAM dendrimers (adapted from Ref [27]).
4. Surface modifications to reduce toxicity and increase transepithelial transport
While relative to their size, PAMAM dendrimers have shown appreciable permeability across epithelial barriers, it would be desirable to increase the transepithelial transport of PAMAM dendrimers and reduce their toxicity. One way to achieve this is by surface modification. Lauric acid is a biocompatible penetration enhancer and is known to enhance membrane permeability, uptake and transport through the gut [48]. Jevprasesphant et al conjugated lauroyl chains to cationic PAMAM dendrimers, generation 2.0 to 4.0 and assessed its cytotoxicity and permeability across Caco-2 cell monolayers [11, 12]. The fatty acid conjugation to PAMAM generation 2.0, 3.0 and 4.0 dendrimers, with 6 and 9 lauroyl chains per molecule of PAMAM reduced toxicity and improved permeability. The reduction in cytotoxicity can be explained by shielding of the positively charged amine groups to interact with negatively charged cell surface proteins [11]. PAMAM permeability generally increased with an increase in generation, increase in the number of lipid chains, and concentration [12]. The apparent permeability values for G2L6 (PAMAM G2.0 modified with 6 lauroyl chains) and G3L6 (PAMAM G3.0 modified with 6 lauroyl chains) were approximately twice those of the corresponding unmodified PAMAM dendrimers at the same concentrations. A significant increase in Papp was observed for G3L9 (PAMAM G3.0 modified with 9 lauroyl chains) but not with G2L9 (PAMAM G2.0 modified with 9 lauroyl chains). This inconsistency could be attributed to aggregation observed for G2L9, as per the hydrodynamic size measurements. However, lauroyl-modified dendrimers did not reduce TEER values as much as unmodified dendrimers [12]. The decrease in TEER when cells were exposed to lauroyl-dendrimer conjugates was less (50% for G2L6) than the decrease observed with unmodified dendrimers (80% for PAMAM G2.0) suggesting that the lauroyl-modified dendrimers used a combination of paracellular and transcellular pathways. Of the dendrimers tested, PAMAM G4.0-NH2 conjugated to 9 lauroyl chloride chains showed maximum permeability (8.03 × 10−6 cm/sec) compared to lower generation PAMAMs G2.0 and G3.0 and PAMAMs G2.0-G4.0 modified with 6 lauric acid chains on the surface [12].
The mechanism of transport of PAMAM G3.0-NH2 and lauroyl-modified G3.0-NH2 across Caco-2 cell monolayers has been investigated [13]. Cellular internalization of dendrimers was demonstrated using flow cytometry, confocal microscopy and transmission electron microscopy (TEM). The higher cell fluorescence intensity observed with the FITC-labeled lauroyl–dendrimer conjugates suggests that lauroyl moieties enhance dendrimer penetration across Caco-2 cells [13]. Optical sectioning of cells incubated with FITC-conjugated G3.0-NH2 and lauroyl-modified FITC-conjugated G3.0-NH2 using confocal laser scanning microscopy revealed colocalisation of the dendrimers with a marker for cell nuclei (4V,6-diamidino-2-phenylindole, DAPI), further suggesting cellular internalization of the dendrimers [13]. TEM images of Caco-2 cell monolayers revealed localization of the dendrimers intracellularly in multivesicular bodies. Addition of a gold label to dendrimers did not change this pattern. Early endosomes containing dendrimer-gold composites were found below the apical surface. In addition, endosomes appeared to aggregate to produce multivesicular bodies found throughout the cell, indicative of intracellular vesicular transport [13]. This study helped in the understanding of how surface modification by lauroyl groups may enhance cellular uptake of nanoparticulate dendrimer-drug complexes in HT-29 cells and potentially increase drug delivery across physiological cell barriers like the colonic epithelium. In addition, surface modification with lauroyl moieties appears to reduce the extent of lysosomal accumulation of the dendrimer-gold nanoparticles, suggesting potentially less exposure of the dendrimer conjugate to the highly acidic lysosomal environment and hydrolytic enzymes. This is especially important when delivering drug conjugates with linkers labile to acidic conditions and lysosomal enzymatic cleavage. Thus, dendrimer modification with lauroyl chains may enhance its cellular uptake into human intestinal epithelial cells and may have the potential for improving drug delivery via the oral route.
In addition to surface modification with lauroyl chains, as discussed above, PAMAM dendrimers can be modified with small molecular weight compounds such as fluorescein labels for the purpose of studying transport mechanisms [15]. This labeling can potentially influence physicochemical properties of the carrier and influence transport. Cationic PAMAM-NH2 (G2.0 and G4.0), neutral PAMAM-OH (G2.0-OH), and anionic PAMAM-COOH (G1.5 and G3.5) dendrimers were conjugated to fluorescein isothiocyanate (FITC) in varying stoichiometric ratios and their permeability was measured in the apical-to-basolateral direction [15]. 14C-Mannitol permeability was measured in the presence of unlabeled and FITC labeled PAMAM dendrimers. At feed molar ratios of 1:1, 1:4, and 1:8 (G4.0-NH2:FITC) the toxicity of G4.0-NH2 dendrimers reduced with an increase in FITC label as per the WST-1 assay. WST-1 (4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) assay is a colorimetric assay that determines the number of viable cells in proliferation. G4.0-NH2-FITC (1:8) displayed the highest relative permeability with an increase in mannitol flux, possibly due to a higher degree of tight junctional modulation [15]. This observation suggested that if cationic dendrimers are modified with hydrophobic groups resulting in partial masking of amine groups, they reduce cytotoxicity and can influence permeability.
To examine this phenomenon in more detail, a subsequent study was carried out where generations 2 and 4 PAMAM-NH2 dendrimers were fully or partially acetylated and their transport across and uptake by Caco-2 cells were evaluated and compared with native dendrimers [16]. It was shown that surface-acetylated PAMAM G4.0-NH2 retained permeability across Caco-2 monolayers comparable to that of native PAMAM dendrimers while reducing toxicity. The decrease in toxicity enabled the dendrimers to be tolerated at higher concentrations by the cells. The native PAMAM G4.0-NH2 was toxic at high concentrations of 0.1mM using the WST-1 assay. However, partial modification was sufficient to reduce cytotoxicity (Figure 9A) [16]. The cytotoxicity data further suggested a linear relationship between the number of surface amine groups and cellular toxicity for a particular dendrimer generation (Figure 9B), with the cytotoxicity of PAMAM G4.0-NH2 being higher than PAMAM G2.0-NH2. Interestingly, a linear correlation was observed for cell viability with surface density of amine groups, irrespective of dendrimer generation (Figure 9C). Due to their branched covalent architecture, PAMAM dendrimers can keep surface moieties in a relatively fixed (but flexible) three dimensional space compared to linear random coil polymers where there is limited control over surface density of charge. The observation that percent cell viability correlated linearly with charge density irrespective of PAMAM generation suggests that PAMAM architecture also plays a role in interaction with cell surface. PAMAM G4.0-NH2 acetylated up to a 50% surface coverage, showed 2 fold increase in permeability at higher concentrations (0.1mM) as compared to the native PAMAM G4.0-NH2 at lower concentrations (0.01mM) (Figure 9D) [16]. Surface modification of PAMAM dendrimers also enhanced 14C-mannitol permeability as a function of concentration and had a less drastic effect on TEER reduction compared to unmodified PAMAM dendrimers. The permeability enhancement of acetylated dendrimers could possibly be attributed to a reduction of repulsive forces between surface amine groups, which would render the dendrimer structure more compact and result in increased permeability [16]. The key finding of this study was that the cytotoxicity of the cationic PAMAM G4.0 can be reduced by surface acetylation while maintaining its permeability. This creates a window of opportunity to use cationic PAMAM dendrimers for oral delivery of bioactive agents conjugated to the surface that will mask the surface amine groups.
Figure 9.

Cytotoxicity and transepithelial transport of acetylated PAMAM G4.0-NH2; (A) In vitro cell viability of Caco-2 cells after incubation with PAMAM dendrimers (1 mM) for 3 hours. Results are reported as mean +/−SEM (n = 9) where G4NH2, G4A32, G4A60: PAMAM G4.0 with 0, 32 and 60 amine groups acetylated respectively; (B) Relationship between cell viability and number of surface amine groups for (■) G4.0-NH2 (R2 = 0.99) and (▲) G2.0-NH2 analogues (R2 = 1); (C). Relationship between cell viability and surface density (R2 = 0.93); (D) Permeability of G4.0-NH2, G4A32, and G4A60 across Caco-2 cell monolayers after 120 min. Results are reported as mean +/− SEM (n=6). (*) (p < 0.05) and (**) (p < 0.01) denote a significant difference in permeability when compared to permeability of unmodified PAMAM dendrimer at 0.01 mM. (×) Permeability is not evaluated due to cytotoxicity. With permission from Ref [16].
In other studies, cationic PAMAM G4.0 was conjugated with positively-charged amino acids (ornithine and arginine) and their permeability across Caco-2 and IPEC-J2 cell monolayers investigated [19, 20]. Polyamines are transported into the cell by a carrier-mediated transport mechanism, i.e., the polyamine transporter protein (PAT) system. It was hypothesized that conjugation of the polyamines to PAMAM dendrimers may increase their permeability due to increased dendrimer uptake by a combination of paracellular pathway as well as by active transport of the PAT system [49]. Cytotoxicity in both the Caco-2 and IPEC-J2 cells was evaluated by the (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) or MTT assay. This assay is a colorimetric assay that measures the reduction of MTT by mitochondrial succinate dehydrogenase, a mitochondrial enzyme that can be correlated with cell viability. No statistically significant differences in toxicity were found between the amino acid-modified and unmodified constructs, possibly because the amino acids that mask the surface amine groups of the dendrimer contribute one (arginine) and two (ornithine) primary amine groups to the constructs [19, 20]. The ornithine modified conjugates showed a slightly higher toxicity (statistically insignificant) than the arginine and unmodified conjugates, probably due to increased number of amine groups contributed by ornithine. The AB and BA Papp for the PAMAM dendrimers were measured across Caco-2 and IPEC-J2 cell monolayers grown in transwell inserts. A concentration and incubation time-dependent enhancement in permeability was observed from the apical to basolateral side for the polyamine modified PAMAMs [19, 20]. Decrease in TEER with increase in permeability for the polyamine-conjugated dendrimers suggests that paracellular transport is one of the mechanisms of transepithelial transport. The authors suggested that the polyamine transporter (PAT) system is a metabolism-dependent transport system for polyamines and may be involved in the enhanced transport of the amino-acid modified PAMAM dendrimers. However further investigations are needed to understand the involvement of polyamine transporter protein (PAT) in the transport mechanisms of polyamine-conjugated dendrimers. While Caco-2 cells are human colorectal carcinoma cells and lack mucous secretion and a mucous barrier to transport, IPEC-J2 is a primary cell line, which produces glycocalyx-bound mucin and is derived from jejunal epithelia isolated from neonatal, unsuckled piglet. Results from the two different studies help us to understand the differences between these two epithelial permeability models. Specifically, the results help evaluate the potential of the polyamine-conjugated dendrimers to cross the mucosal barrier. It is also possible that the acidic molecules such as acidic mucopolysaccharides on the mucous membrane contribute to transport of polyamine conjugated PAMAMs and further mechanistic studies with IPEC-J2 are needed to study this phenomenon. It appears that IPEC-J2 cells form tighter junctions as compared to Caco-2 cell monolayers, and as a result, the decrease in TEER with the modified and native dendrimers and the apparent permeability values from apical to basolateral at an incubation time of 90 minutes were significantly lower with IPEC-J2 (0.23 ± 0.04 × 10–6 cm/sec for unmodified PAMAM G4.0) as compared to Caco-2 cell monolayers (0.34 ± 0.04 × 10–6 for unmodified PAMAM G4.0) [19, 20].
PEGylation of drug carriers, including PAMAM dendrimers, is known to improve biocompatibility and pharmacokinetics in vivo [50, 51]. Surface PEGylating PAMAM dendrimers for oral delivery may affect the mechanism and extent of transport. PEG chains can also serve as linkers for a dendrimer-drug conjugate to control drug release. The effect of surface PEGylation on PAMAM cytotoxicity and transepithelial transport across Caco-2 cell monolayers was investigated [24]. PEGylation of anionic dendrimers G3.5-COOH and G4.5-COOH with poly(ethylene glycol) (PEG) of molecular weight 750 kDa did not significantly alter cytotoxicity up to a concentration of 0.1 mM [24]. Confocoal microscopy experiment revealed that dendrimer PEGylation reduced the opening of tight junctions. The decrease in tight junction modulation was associated with a decrease in transepithelial transport of the PEGylated dendrimers. PEGylation of G3.5-COOH dendrimers significantly decreased cellular uptake and transepithelial transport while PEGylation of G4.5 dendrimers led to a significant increase in uptake, but also a significant decrease in transport, yielding apparent permeabilities 60–70% lower than native dendrimers (Figure 10) [24]. A PEG chain of 750 kDa, due to its flexible random coil conformation can wrap around the rigid, spherical dendrimer and shield some of the negative charge on the dendrimer surface. It was hypothesized that for PAMAM G3.5, one stealth-like PEG chain sufficiently reduced the interactions with the cells, leading to decreased cellular uptake for all PEGylation ratios [24]. However, for PAMAM G4.5, addition of 1 PEG chain increased uptake as compared to unmodified G4.5. This was explained by a theoretically calculated surface-charge density of the dendrimers. The higher surface-negative charge density of PAMAM G4.5 (1.6 charges/nm2) compared to PAMAM G3.5 (1.3 charges/nm2), may have caused repulsion with the negatively charged cell membrane [24]. Addition of 1 PEG chain may have reduced the surface charge, and therefore repulsion, increasing uptake compared to unmodified PAMAM G4.5. However, addition of more PEG chains to PAMAM G4.5 may have shielded more of the negative charge, again, creating a stealth system and decreasing cellular uptake, as in PAMAM G3.5. The cellular uptake data was correlated with the zeta potential of the constructs [24]. A zeta potential around −30 mV promoted cellular uptake. PAMAM G4.5 with zeta potentials more negative than −30 mV had reduced cellular uptake (possibly due to repulsion of like charges) while pegylated PAMAM G3.5 with zeta potentials greater than −30 mV also showed lower uptake (possibly due to stealth effects of PEG chains) [24]. A balance between charge based cell membrane interaction of the anionic PAMAMs and charge-shielding due to PAMAM might affect cellular uptake and also transepithelial transport.
Figure 10.

Cellular uptake and transepithelial transport of PAMAM G3.4 and G4.5; Uptake of G3.5 (A) and G4.5 (B) native and differentially PEGylated dendrimers at 0.02 mM for 30 and 60 minutes in Caco-2 cells. n=3, Mean ± standard deviation. (*), (**) and (***) denote significant differences from unmodified dendrimers with p<0.05, p<0.01 and p<0.001 respectively. Apparent permeability of G3.5 (C) and G4.5 (D) native and differentially PEGylated dendrimers at 0.1 mM after a 2 hour incubation time with Caco-2 cell monolayers. n=3, Mean ± standard deviation. (*) denotes a significant difference from unmodified dendrimers with p<0.001. With permission from Ref [24].
Together the above studies demonstrate that by engineering the surface groups of PAMAM dendrimers, it is possible to alter cytotoxicity, permeability and cellular uptake. It was shown that surface modification by uncharged groups (PEG, lauroyl, acetyl) reduced toxicity by charge masking of the primary amine groups. Hydrophobic surface modifiers such as the acetyl groups and lauric acid increased permeability while hydrophilic polymers such as PEG reduced permeability. Thus, surface modification provides a tool to reduce toxicity and influence permeability of PAMAM dendrimers across epithelial barriers. Figure 11 summarizes the effect of surface modification on cytotoxicity, cellular uptake and permeability across epithelial barrier of the gut.
Figure 11.

Effect of surface modification on PAMAM cytotoxicity, transepithelial permeability and cellular uptake.
5. Transepithelial transport of PAMAM – drug complexes and conjugates
Drugs can be attached to PAMAM dendrimers by covalent conjugation, complexed by ion-ion and van der waals interactions, or incorporated in the empty spaces within branches. Although, there has been extensive research on dendrimer-based drug carriers for a variety of routes of delivery, very few studies have demonstrated transepithelial transport of such conjugates or complexes.
Earlier studies by D’Emanuele and coworkers used PAMAM G3.0-NH2 or lauroyl-modified PAMAM G3.0-NH2 conjugated to propranolol in varying stoichiometric ratios of propranolol and lauric acid [14]. Propranolol was chosen as an example of a poorly soluble drug known to be a substrate of the P-glycoprotein (P-gp) efflux transporter. P-glycoprotein reduces the absorption of orally administered drugs and decreases bioavailability. Co-administration of P-gp inhibitors such as cyclosporine A can increase bioavailability by blocking secretion. Propranolol was conjugated to PAMAM via a chloroacetyl spacer. PAMAM G3.0 was chosen because it has been shown to effectively permeate Caco-2 cell monolayers and was not a P-gp substrate [9]. Lauroyl modification of PAMAM G3.0-NH2 is known to increase its permeability across Caco-2 monolayers. Cytotoxicity of the constructs was determined by the MTT assay and decreased with an increase in the amount of propranolol and lauric acid conjugated, attributed to the shielding of positive surface charge of primary amine groups on PAMAM G3.0-NH2. The BA apparent Papp of propranolol, a P-gp substrate, was greater than AB Papp, indicating that P-gp efflux system is functionally active in the Caco-2 cell monolayers used in this study [14]. The AB Papp of free propranolol was increased in the presence of the P-gp inhibitor cyclosporin A but not in the presence of PAMAM G3.0-NH2, which was therefore not functioning as a P-gp inhibitor. The AB transport increased and BA transport decreased when propranolol was conjugated to PAMAM G3.0-NH2 by covalent attachment. The extent of enhancement of AB transport was not dependent on the number of attached propranolol molecules over the range under study (2–6 moles of propranolol per mole of G3.0-NH2). There was a 3.5 fold enhancement of AB transport of propranolol when 6 lauroyl chains were attached to PAMAM G3.0-NH2 containing 2 propranolol moieties [14]. There was no difference in the permeability of propranolol conjugates in the presence and absence of cyclosporin A and the permeability decreased at 4°C as compared to 37°C [14]. Moreover, the decrease in TEER when cells were exposed to propranolol conjugates was less than that observed with unmodified dendrimers. This suggests that the transport of the conjugates across Caco-2 cell monolayers involves endocytosis-mediated transepithelial transport via a transcellular route. Overall this study showed that conjugation of propranolol with dendrimers increased solubilization and enabled circumvention of the P-gp efflux pump and increased apical to basolateral transport of the dendrimers across Caco-2 monolayers [14]. However, detailed evaluation of the stability of the conjugated system in presence of Caco-2 cell culture medium warranted further investigation.
Naproxen, a poorly water-soluble drug, was conjugated to PAMAM G0.0 either directly by an amide bond or by ester bonds using either l-lactic acid or diethylene glycol as a linker [52]. The type of linkage between the dendrimer and drug affected the release characteristics of the drug from the delivery system. While the direct amide linkage was stable under the conditions of plasma and liver homogenate tested, the ester linkage could be tailored to be stable in plasma and release the drug at the desired site of action in the liver [17, 52]. Hydrolysis of the conjugates was measured at 37 °C in a range of pH from 1.2 to 8.5. The amide conjugate and both ester conjugates were chemically stable at all pH values over 48 h of incubation. Naproxen was enzymatically released from both ester conjugates in plasma; the lactic ester conjugate (G0-lact-Nap) hydrolyzed slowly with only 25% of naproxen released after 24 h, and the diethylene glycol ester conjugate (G0-deg-Nap) cleaved rapidly following pseudo first order kinetics (t1/2 = 51 min) [52]. The lactic ester conjugate, G0-lact-NAP, hydrolyzed slowly in 80% human plasma and in 50% rat liver homogenate (t1/2 = 180 min) while G0-deg-NAP was hydrolyzed more rapidly in 80% human plasma (t1/2 = 51 min) and was rapidly cleaved in 50% liver homogenate (t1/2 = 4.7 min) [17]. The length of the spacer affected enzymatic release of the drug. The longer spacer (diethylene glycol) is likely to cause less steric hindrance for enzymatic cleavage and hence may cause faster cleavage of drug in the plasma and liver homogenate. The amount of naproxen transported across Caco-2 cell monolayers was higher when conjugated to G0.0 [17]. To further increase permeability, PAMAM G0.0 was surface modified with one lauroyl chain per molecule of PAMAM. Permeability studies across Caco-2 cell monolayers showed a significant enhancement in naproxen transport when conjugated with lauroyl-modified PAMAM G0.0 through a diethyleneglycol spacer as compared to non-modified conjugate [17]. The study shows that PAMAM based drug conjugates with appropriate linkers have potential as carriers for the oral delivery of low solubility drugs such as naproxen.
In another study Kolhatkar et al complexed SN-38, a potent camptothecin analogue to PAMAM G4.0-NH2 (Figure 12A) and assessed the transport of the dendrimer-drug complex across Caco-2 monolayers [21]. 7-Ethyl-10-hydroxy-camptothecin (SN-38) is a potent topoisomerase I poison and a biologically active metabolite of irinotecan hydrochloride (CPT-11). SN-38 has potent antitumor activity, approximately 1,000-fold more active than CPT-11, but suffers from poor aqueous solubility and dose-limiting toxicity. It was hypothesized that complexing SN-38 to G4.0-NH2 will result in increased solubility and permeability of the drug and will help reduce dose related toxicity of SN-38. When complexed with PAMAM G4.0-NH2, SN-38 showed up to 10 fold higher permeability and 100 fold higher uptake than free SN-38 (Figure 12C) [21]. However, the complex was not stable under acidic conditions. Only 10% of the drug remained complexed with the PAMAM after 30 minutes of exposure to pH 5.5 buffer (Figure 12B) [21]. These studies indicate that while complexation is a simple and viable approach, it has distinct drawbacks of instability and premature release.
Figure 12.

Gastrointestinal stability and transpeithelial transport of PAMAM-SN38 complex. A. Schematic representation of G4S5 complex. B. Stability of polymer-SN-38 complexes G4S5 (open squares, solid line) and G4S11 (filled squares, solid line) at pH 7.4, and G4S5 (open circles, dotted line) and G4S11 (filled circles, dotted line) at pH 5. G4S5, G4S11: 5 and 11 moles of SN38 complexed to PAMAM G4.0 respectively. C. Permeability of G4S5, G4S11 and SN-38, across Caco-2 cell monolayers after 120 min With permission from Ref [21].
Covalent conjugates of PAMAM G3.5-COOH and SN-38 with glycine and β-alanine spacers were synthesized, characterized (Figure 13) and evaluated for cytotoxicity, gastrointestinal stability and enzymatic release in the liver environment, as well as transepithelial transport in vitro [38, 53]. Under similar conditions, carboxyl-terminated anionic dendrimers have shown less toxicity than cationic PAMAM dendrimers, and yet maintained comparable transepithelial transport. Hence PAMAM G3.5-COOH was used in this study. Glycine has been used to attach SN-38 and other camptothecin analogues to polymers such as poly(ethylene glycol) (PEG), poly(glutamic acid), and pegylated poly(L-lysine) dendrimer showing increased aqueous solubility [54–56]. Alanine differs from glycine in the presence of an additional methylene group and could thus act as a relatively extended spacer. The two spacers were chosen in order to compare the possible effect of linker length on the extent of drug loading, rate of drug release, and activity of the conjugates. The spacers connected the drug and the dendrimer via an ester linkage that is likely to be cleaved in the presence of carboxylesterase and also likely to be sterically hindered to prevent acidic or basic hydrolysis. SN-38 is a topoisomerase I inhibitor and is known to exhibit characteristic G2/M phase cell cycle arrest. The progression of HCT-116 cells through the cell cycle after 24 h treatment with 2 × IC50 concentration of either conjugate or free drug was evaluated using flow cytometry. As expected, SN-38 caused cell cycle arrest in the G2/M phase (73%) with considerable reduction in population of cells in the G0/G1 (4%) phase relative to untreated cells (61%) [53]. Upon exposure to SN38 conjugated with G3.5 via a glycine spacer (G3.5-gly-SN38), a significant accumulation of cells in the G2/M phase (50%) was observed with accompanying substantial decrease in G0/G1 (7.0%). Similarly, in the case of SN38 conjugated to G3.5 via a β-alanine spacer (G3.5-βala-SN38), the accumulation of 52% cell population in the G2 phase was observed. These results suggest that the dendrimer-drug conjugates have a similar mechanism of action as SN-38 and the conjugates affect cells due to release of the free drug. Fluorescence microscopy was used to examine and compare the effect of conjugates and free drug on cell and nuclear morphology (Figure 14). HCT-116 cells were treated for 36 h with either free SN-38 or conjugates at 1/3 IC50, stained with the nuclear dye, DAPI, and observed under the confocal fluorescence microscope. Untreated cells appeared mostly uniform with regularly shaped nuclei. In contrast, cells treated with either SN-38 or the dendrimer-drug conjugates appeared heterogeneous [53]. Nuclear fragments were distinctly visible, along with swollen and sparsely condensed nuclei. Mitotic cells were also occasionally seen in the treated cells. Although these observations are not conclusive evidence of apoptosis or necrosis, they do suggest that the free drug and conjugates have the same mode of action indicating that the activity of the conjugates is due to the release of the free drug.
Figure 13.

Synthetic Schemes of G3.5-Gly-SN38 (top) and G3.5-βAla-SN38 (bottom) conjugates. With permission from Ref [28, 53]; Table: Characteristics of G3.5 PAMAM-SN38 conjugates. With permission from Ref [53]
Figure 14.
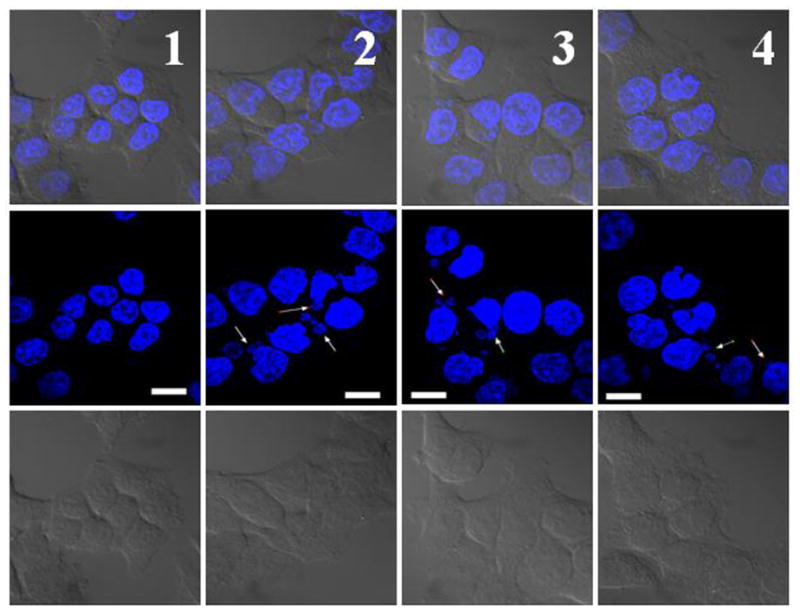
Nuclear fragmentation in HCT-116 cells treated with drug/conjugates. Untreated cells (column 1); 5 nM SN38 (column 2); 40 nM G3.5-gly-SN38 (column 3); 120 nM G3.5-βala-SN38 (column 4). Scale bar is 10 μm. Arrows indicate nuclear fragments. From bottom: 1st row, differential interference contrast image; 2nd row, fluorescence image; 3rd row, overlay of differential interference contrast and fluorescence images. With permission from Ref [53].
The conjugates were further evaluated for stability in the GIT. They were shown to be relatively stable in the gastric and intestinal environments, minimizing the chance of premature drug release upon oral administration and reducing non-specific toxicity of the drug [28]. Of the conjugates evaluated, G3.5-gly-SN38 showed a more favorable profile of gastrointestinal stability, transepithelial transport and cytotoxicity against colorectal carcinoma cells and effective release of free drug in the presence of liver carboxylesterases. G3.5-βala-SN38 was mostly stable while G3.5-gly-SN38 showed 10%, 20%, and 56% SN-38 release in simulated gastric, intestinal and liver environments for up to 6, 24 and 48 hours, respectively (Figure 15A–C) [28]. The β-alanine spacer rendered the conjugate more stable to hydrolytic and enzymatic degradation, which prevented release of the drug in desired enzymatic milieu. The impact of linker chemistry on SN-38 release was directly correlated with the IC-50 values of the conjugates in colorectal cancer cells. G3.5-gly-SN38 conjugates had six-fold greater efficacy than G3.5-βala-SN38 conjugates due to higher SN-38 release from the G3.5-gly-SN38 conjugate [28]. There were distinct differences in the transepithelial transport of the conjugates across Caco-2 cell monolayers. The SN-38 flux was concentration-dependent for G3.5-gly-SN38 while it was unchanged for G3.5-βala-SN38 between treatment with 10 and 100 μM concentrations (Figure 15D) [28]. This suggests that G3.5-gly-SN38 may be transported primarily by a concentration gradient-driven process, such as paracellular diffusion, whereas a saturable process, such as transcellular transport, potentially governs G3.5-βala-SN38 transport.
Figure 15.

Stability and transepithelial transport of G3.5-Gly-SN38 and G3.5-βAla-SN38 conjugates. Release of SN38 was monitored in simulated conditions of the stomach for 2 hours (A), intestine for 24 hours (B) and liver for 48 hours (C). Mean ± standard deviation (n=2). G3.5-Gly-SN38 is represented by red squares and G3.5-βAla-SN38 is represented by purple circles. Buffers without enzymes are depicted as dashed lines with open symbols and buffers with enzymes are depicted as solid lines with filled symbols. (D) Equivalent SN38 flux across differentiated Caco-2 monolayers treated with G3.5-SN38 conjugates and SN38. Equivalent SN38 flux was calculated by multiplying the measured molar flux of the conjugates with the number of SN38 molecules per dendrimer. Mean ± standard deviation (n=4). (***) indicates a significant difference with p<0.001. With permission from Ref [28].
The choice of linker is critical to the stability of the dendrimer-drug conjugate in the harsh conditions of the gastrointestinal tract and to the efficient release of the drug at the site of action. In the context of solid state malignancies, both the GIT and tumor physiology can have a common range of enzymes and pH. It is a chemical paradox to design a linker perfectly stable in one condition and completely hydrolyzed in the other. It is therefore important to strike a balance between stability in the GIT and site-specific release. Identifying tumor-specific elevated enzymes and designing linkers to be specifically cleaved by them can potentially overcome this problem.
6. In vivo oral bioavailability of PAMAM – drug complexes
The work summarized in the above sections includes the in vitro evaluation of the transepithelial transport of dendrimer-drug conjugates or complexes. In vitro models lack the variables of gastric emptying, intestinal tract motility and enzymatic environment of the gastrointestinal tract present in in vivo models. However, they are useful to provide mechanistic insight into transepithelial transport and absorption and help come up with strategies to enhance the same. The choice of technique will depend on the research question and a combination of different models will ultimately provide an answer to the absorption characteristics of PAMAM dendrimers. Two recent studies have evaluated the in vivo bioavailability of drugs when orally delivered by PAMAM dendrimers.
The extent of complexation of silybin, a potent hydrophobic alkaloid by PAMAM dendrimers G1.5, G2.0, G2.5, and G3.0 with different concentrations was determined and compared in different pH conditions [57]. G1.5 and G2.5 incorporated 4 and 6 moles of silybin per mole of PAMAM while G2.0 and G3.0 incorporated 20 and 32 moles of silybin per mole of PAMAM. Compared to the carboxylic acid-terminated half-generation dendrimers, the amine-terminated full generation dendrimers have an additional electrostatic attachment between their surface amine groups and the oppositely-charged phenolic hydroxyl groups of silybin. This could result in a higher incorporation of silybin in full generation dendrimers. The in vitro release experiments in simulated gastric (SGF) and intestinal (SIF) fluids demonstrated that the G2.0-silybin and G3.0-silybin complexes released up to 20% silybin in SGF in 2 hours and up to 90% silybin release was observed in SIF at 10 hours [57]. The complexes were not highly stable in SIF indicating that majority of the drug is going to be released from the PAMAM in the small intestine effectively resulting in co-delivery of the PAMAM with the drug. The oral absorption kinetics of PAMAM G2.0-silybin complex was investigated in rats. The relative oral bioavailability of a potent alkaloid silybin was enhanced 2-fold by complexing with amine terminated PAMAM G2.0-NH2 when administered by oral gavage to male rats [57]. Part of the drug might have remained complexed to the dendrimer explaining an extended Tmax (15 minutes) of absorption as compared to silybin alone (Tmax = 10 minutes) [57].
The cellular uptake in Caco-2 cells, transport across rat intestinal segments and oral absorption pharmacokinetics of doxorubicin complexed with amine terminated PAMAM G3.0-NH2 in rats by oral gavage has also been investigated [23]. Doxorubicin, which is a P-gp substrate with poor bioavailability after oral administration, was selected as a model drug. The release of doxorubicin from doxorubicin–PAMAM (1:2 molar ratio) complex appears to be slower and less (74.5% during 24 hour) as compared to that from the free doxorubicin solution (more than 95% during 3 hour) in the presence of N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES) [23]. TES is a buffer in the pH range of 6.0–8.0. However, in the GIT the complex can be subjected to lower pH in the acidic range and also to enzymatic degradation. Release in simulated GIT conditions was not evaluated in this study. A higher accumulation of doxorubicin in Caco-2 cells was observed in the presence of 10 mg/mL of Cyclosporin A (CsA), as compared to that treated with free doxorubicin, due to the inhibition of P-gp efflux by CsA [23]. Cyclosporin A (CsA) is known to be an inhibitor of the P-gp system. However, the doxorubicin uptake was higher when complexed with PAMAM compared to free doxorubicin alone or free doxorubicin with P-gp inhibitor. The addition of CsA did not cause significant increase of uptake of doxorubicin. There was also significantly higher transport of doxorubicin by using a doxorubicin–PAMAM complex compared to free doxorubicin in everted intestinal rat segments of duodenum, ileum and jejunum. The transport of doxorubicin in the ileum treated with the doxorubicin–PAMAM complex was slightly higher than that in the duodenum and the jejunum. At 90 min, the transport efficiency of the doxorubicin PAMAM complex from the mucosal side to the serosal side was 4–7 times higher than that of the free doxorubicin solution [23]. There was also a slight increase in permeability of mannitol across small intestinal mucosa of all segments upon incubation with doxorubicin–PAMAM complex, indicative of an increased paracellular transport in the presence of the complex. A 300 fold increase in bioavailability of the doxorubicin was observed when delivered with the PAMAM as compared to the free drug following a single oral dose in rats [23]. The absorption kinetics were evaluated using a simple non-compartmental model. The vast differences in area under the curve values of doxorubicin in blood in presence and absence of the dendrimer could be due to the solubilization effect of the PAMAM on doxorubicin, a highly hydrophobic drug. The GIT stability profile of the two dendrimer-drug complexes of doxorubicin and silybin has not been investigated in detail. Thus, there might be partial or complete release of the free drug complexed to the dendrimer in the GIT before absorption. The increased amount of drug detected in the blood stream could therefore also be due to increased solubilization by the dendrimer or intestinal penetration enhancement of the free drug by the dendrimer.
7. PAMAM dendrimers as intestinal penetration enhancers
Conventional penetration enhancers that have been developed to improve the intestinal permeability of poorly absorbable drugs include surfactants, fatty acid based enhancers, and bile salts which are known to effectively increase the absorption of poorly bioavailable drugs across the large intestine [48]. With its large surface area, the small intestine offers the opportunity for increased absorption. Cationic PAMAM dendrimers have been recently explored as a class of penetration enhancers that act on increasing the permeability of hydrophobic and hydrophilic small molecules as well as hydrophilic macromolecules through the small intestine [26, 57]. Co-delivery of poorly-absorbable drugs with PAMAM dendrimers that can act as penetration enhancers will help solubilize the drug and improve its permeability leading to increased oral bioavailability.
In situ intestinal models have been used to evaluate PAMAM dendrimers as penetration enhancers of hydrophilic small molecules and macromolecules [26]. An in situ method allows the gut to remain in the anesthetized animal so that the normal function of the intestine remains intact where it is possible to access both luminal and serosal sides [40]. This is achieved by cannulation of the lumen and vascular bed. An in situ model preserves many of the in vivo features of the gut morphology and physiology and allows the assessment of absorption without the interference of gastric emptying and motility. Such perfusion studies may be useful to overcome problems of low and variable absorption encountered in an in vivo model setting.
The penetration enhancement was achieved at non-toxic doses and was reversible [26]. Effects of amine-terminated PAMAM generations 0–3 were examined on the absorption of 5(6)-carboxyfluorescein, a hydrophilic small-molecular weight dye. An in situ closed loop method in SD rats was used to evaluate absorption enhancement. The absorption enhancing effects of PAMAM on the small intestinal segment were concentration and generation dependent. Of the dendrimers tested, PAMAM G2.0-NH2 showed maximum absorption enhancement, up to 11.1 fold, at 0.5 % w/v. At this concentration, PAMAM G2.0-NH2 caused membrane damage as evaluated by the LDH assay [26]. However, the damage was found to be reversible. The absorption-enhancing effects of G2.0-NH2 were tested in the small intestinal segment at 0.5% w/v for hydrophillic macromolecular compounds like fluorescein isothiocyanate-dextrans (FDs) of various molecular weights, calcitonin and insulin. PAMAM G2.0-NH2 was effective in increasing the absorption of hydrophilic, macromolecular FDs upto 4000 dalton through the small intestine [26]. However, PAMAM G2.0-NH2 did not significantly increase the permeability of hydrophilic macromolecules of a molecular weight above 4,000 dalton, such as insulin and calcitonin [26]. Absorption enhancement across the small intestinal segment was found to be a function of molecular weight with the absorption enhancement ratio being highest (about 11.0) for 5(6)-carboxyfluorescein, a small molecular weight hydrophilic compound [26]. It has been speculated that amine-terminated PAMAM dendrimers modulated the tight junctions leading to an increase in absorption of the smaller sized hydrophilic molecules tested. However, the enlargement of the pore radius at the tight junction may not have been enough to allow larger sized macromolecules to pass through [26]. Unlike conventional absorption enhancers, the absorption enhancing effects for PAMAM G2.0-NH2 observed in the small intestine was much greater than that seen in the large intestine in this study. These differences could be due to the physiological variation such as the thickness of mucous layer and the number and tightness of tight junctions. Further research is needed in order to elucidate the mechanism of penetration enhancement and to understand the intestinal segmental differences in the enhancement capacity of PAMAM dendrimers.
The study summarized above investigated cationic PAMAM dendrimers as penetration enhancers. Generations 0.0 to 3.0 were utilized and showed reversible plasma membrane damage at high concentrations (0.5% w/v) as evaluated by the LDH assay. Previous research has shown in Caco-2 cell monolayers and isolated intestinal tissue that along with cationic PAMAM dendrimers, anionic, carboxyl-terminated PAMAM dendrimers can also translocate across the intestinal barrier effectively while being less toxic to the epithelium as compared to the cationic dendrimers [6, 27]. Anionic PAMAM dendrimers also need to be evaluated for absorption enhancing effects in vivo. Several surface modifications for PAMAMs such as acetyl groups, fatty acids and amino acids have shown increased permeability in Caco-2 cell monolayers or isolated intestinal tissue [11, 16]. Studying the effect of these surface modifications on the penetration enhancing effect of PAMAM on hydrophobic and hydrophilic drugs in vivo could lead to the establishment of a structure-activity relationship between surface modification and absorption-enhancement effect.
8. Challenge to in vivo translation
PAMAM dendrimers have been systematically evaluated in vitro for transepithelial toxicity and transport. However, there have been few in vivo reports of their oral bioavailability and biocompatibility. To the best of our knowledge, to date, there has been no published record of in vivo demonstration of the intact intestinal translocation of a dendrimer-drug conjugate. Ongoing work in our lab is focused on understanding the in vivo oral toxicity and bioavailability profile of PAMAM dendrimers and to orally deliver intact PAMAM-drug conjugates. The primary challenges are in the choice of linker chemistry and in vitro to in vivo extrapolation of the data. It is critical to achieve linker stability sufficient to prevent gastrointestinal toxicity and a release rate efficient and specific enough for effective therapy. Identifying site-specific elevated enzymes and designing linkers to be specifically cleaved by them is necessary to overcome this problem.
In vitro to in vivo correlation of toxicity and bioavailability data also remains a challenge. There are no established correlations to extrapolate non-toxic concentrations obtained from in vitro data to dosing for in vivo. Hence, in vivo oral toxicity profiles need to be established for the different generations and surface modifications of PAMAM dendrimers.
In vitro models employed to assess transepithelial transport have been useful to rank the translocation of PAMAM dendrimers of different generations and surface functionalities. However, they lack the variables of mucous membrane barrier, gastrointestinal transit time and/or enzymatic milieu pertinent to the gastrointestinal tract. Systematic modeling and correlation of in vitro transport to in vivo absorption can facilitate high throughput screening of PAMAM-based conjugates and complexes.
The hydrophilic and macromolecular nature of the PAMAM dendrimers is likely to cause low oral bioavailability and the charged PAMAM dendrimers are also likely to be toxic. However, some of these problems can be overcome by optimizing the dosage, surface modifications to reduce toxicity and increase permeability and delivering highly potent drugs. It is possible that a combination of penetration enhancement and transepithelial translocation will be beneficial for oral drug delivery using PAMAM dendrimers. There is, therefore, a window of opportunity for the PAMAM dendrimers to be used as carriers for oral delivery of drugs.
Acknowledgments
This work was supported by National Institute of Health (R01-EB007470 and R01DE019050) and the Utah Science Technology and Research (USTAR) Initiative.
Abbreviations
- AB
Apical to basolateral
- BA
Basolateral to apical
- CF
5(6)-carboxyfluorescein
- CPT-11
Irinotecan hydrochloride
- CsA
Cyclosporin A
- DAPI
4V,6-diamidino-2-phenylindole
- DIC
Disseminated intravascular coagulation
- EEA-1
Early endosome antigen-1
- EPR
Enhanced permeability and retention
- FD
Fluorescein isothiocyanate-labeled dextrans
- FDP
Fibrin degradation product
- FITC
Fluorescein isothiocyanate
- GIT
Gastrointestinal tract
- GX.0-NH2
Amine-terminated PAMAM dendrimer generation X.0
- GX.0-OH
Hydroxyl-terminated PAMAM dendrimer generation X.0
- GX.5-COOH
Carboxyl-terminated PAMAM dendrimer generation X.5
- G3.5-gly-SN38
SN38 conjugated with G3.5-COOH via a glycine spacer
- G3.5-βala-SN38
SN38 conjugated with G3.5-COOH via a β-alanine spacer
- GXLY
Amine-terminated PAMAM dendrimer of generation X.0 modified with Y moles of lauric acid per mole of PAMAM
- G0-lact-NAP
Naproxen conjugated to G0.0 via a lactic acid spacer
- G0-deg-NAP
Naproxen conjugated to G0.0 via a diethyleneglycol spacer
- GX.0-NH2-FITC
Fluorescein isothiocyanate-labeled amine terminated PAMAM dendrimer of generation X.0
- LAMP-1
Lysosome-associated membrane protein 1
- LDH
Lactate dehydrogenase
- MDCK
Madin-Darby canine kidney
- MTT
(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)
- PAMAM-NH2
Amine-terminated PAMAM dendrimer
- PAMAM-COOH
Carboxylic-terminated PAMAM dendrimer
- PAMAM-OH
Hydroxyl-terminated PAMAM dendrimer
- Papp
Apparent permeability coefficient
- P-gp
P-glycoprotein
- PAMAM
Poly(amido amine)
- PAT
Polyamine transport
- PBS
Phosphate Buffer Saline
- PEG
Poly(ethylene glycol)
- SGF
Simulated gastric fluid
- SIF
Simulated intestinal fluid
- SN-38
7-ethyl-10-hydroxy-camptothecin
- TEER
Transepithelial electrical resistance
- TEM
Transmission electron microscopy
- TES
N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid
- WST-1
(4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 2.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 3.Hillery A, Lloyd AW, Swarbrick J. Drug delivery and targeting. CRC Press; 2003. [Google Scholar]
- 4.Ponchel G, Irache JM. Specific and non-specific bioadhesive particulate systems for oral delivery to the gastrointestinal tract. Adv Drug Delivery Rev. 1998;34:191–219. doi: 10.1016/s0169-409x(98)00040-4. [DOI] [PubMed] [Google Scholar]
- 5.Florence AT, Sakthivel T, Toth I. Oral uptake and translocation of a polylysine dendrimer with a lipid surface. J Controlled Release. 2000;65:253–259. doi: 10.1016/s0168-3659(99)00237-0. [DOI] [PubMed] [Google Scholar]
- 6.Wiwattanapatapee R, Carreño-Gómez B, Malik N, Duncan R. Anionic PAMAM dendrimers rapidly cross adult rat intestine in vitro: a potential oral delivery system? Pharm Res. 2000;17:991–998. doi: 10.1023/a:1007587523543. [DOI] [PubMed] [Google Scholar]
- 7.Tajarobi F, El-Sayed M, Rege B, Polli J, Ghandehari H. Transport of poly amidoamine dendrimers across Madin-Darby canine kidney cells. Int J Pharm. 2001;215:263–267. doi: 10.1016/s0378-5173(00)00679-7. [DOI] [PubMed] [Google Scholar]
- 8.El-Sayed M, Ginski M, Rhodes C, Ghandehari H. Transepithelial transport of poly (amidoamine) dendrimers across Caco-2 cell monolayers. J Controlled Release. 2002;81:355–365. doi: 10.1016/s0168-3659(02)00087-1. [DOI] [PubMed] [Google Scholar]
- 9.El-Sayed M, Rhodes CA, Ginski M, Ghandehari H. Transport mechanism (s) of poly (amidoamine) dendrimers across Caco-2 cell monolayers. Int J Pharm. 2003;265:151–157. doi: 10.1016/s0378-5173(03)00391-0. [DOI] [PubMed] [Google Scholar]
- 10.El-Sayed M, Ginski M, Rhodes CA, Ghandehari H. Influence of surface chemistry of poly (amidoamine) dendrimers on Caco-2 cell monolayers. J Bioact Compat Polym. 2003;18:7–22. [Google Scholar]
- 11.Jevprasesphant R, Penny J, Jalal R, Attwood D, McKeown NB, D’Emanuele A. The influence of surface modification on the cytotoxicity of PAMAM dendrimers. Int J Pharm. 2003;252:263–266. doi: 10.1016/s0378-5173(02)00623-3. [DOI] [PubMed] [Google Scholar]
- 12.Jevprasesphant R, Penny J, Attwood D, McKeown NB, D’Emanuele A. Engineering of dendrimer surfaces to enhance transepithelial transport and reduce cytotoxicity. Pharm Res. 2003;20:1543–1550. doi: 10.1023/a:1026166729873. [DOI] [PubMed] [Google Scholar]
- 13.Jevprasesphant R, Penny J, Attwood D, D’Emanuele A. Transport of dendrimer nanocarriers through epithelial cells via the transcellular route. J Controlled Release. 2004;97:259–267. doi: 10.1016/j.jconrel.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 14.D’Emanuele A, Jevprasesphant R, Penny J, Attwood D. The use of a dendrimer-propranolol prodrug to bypass efflux transporters and enhance oral bioavailability. J Controlled Release. 2004;95:447–453. doi: 10.1016/j.jconrel.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Kitchens KM, Kolhatkar RB, Swaan PW, Eddington ND, Ghandehari H. Transport of poly (amidoamine) dendrimers across Caco-2 cell monolayers: influence of size, charge and fluorescent labeling. Pharm Res. 2006;23:2818–2826. doi: 10.1007/s11095-006-9122-2. [DOI] [PubMed] [Google Scholar]
- 16.Kolhatkar RB, Kitchens KM, Swaan PW, Ghandehari H. Surface acetylation of polyamidoamine (PAMAM) dendrimers decreases cytotoxicity while maintaining membrane permeability. Bioconjugate Chem. 2007;18:2054–2060. doi: 10.1021/bc0603889. [DOI] [PubMed] [Google Scholar]
- 17.Najlah M, Freeman S, Attwood D, D’Emanuele A. In vitro evaluation of dendrimer prodrugs for oral drug delivery. Int J Pharm. 2007;336:183–190. doi: 10.1016/j.ijpharm.2006.11.047. [DOI] [PubMed] [Google Scholar]
- 18.Kitchens KM, Foraker AB, Kolhatkar RB, Swaan PW, Ghandehari H. Endocytosis and interaction of poly (amidoamine) dendrimers with Caco-2 cells. Pharm Res. 2007;24:2138–2145. doi: 10.1007/s11095-007-9415-0. [DOI] [PubMed] [Google Scholar]
- 19.Pisal DS, Yellepeddi VK, Kumar A, Palakurthi S. Transport of surface engineered polyamidoamine (PAMAM) dendrimers across IPEC-J2 cell monolayers. Drug Delivery. 2008;15:515–522. doi: 10.1080/10717540802321826. [DOI] [PubMed] [Google Scholar]
- 20.Pisal DS, Yellepeddi VK, Kumar A, Kaushik RS, Hildreth MB, Guan X, Palakurthi S. Permeability of surface-modified polyamidoamine (PAMAM) dendrimers across Caco-2 cell monolayers. Int J Pharm. 2008;350:113–121. doi: 10.1016/j.ijpharm.2007.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kolhatkar RB, Swaan P, Ghandehari H. Potential oral delivery of 7-Ethyl-10-Hydroxy-camptothecin (SN-38) using poly (amidoamine) dendrimers. Pharm Res. 2008;25:1723–1729. doi: 10.1007/s11095-008-9572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitchens KM, Kolhatkar RB, Swaan PW, Ghandehari H. Endocytosis inhibitors prevent poly (amidoamine) dendrimer internalization and permeability across Caco-2 cells. Mol Pharmaceutics. 2008;5:364–369. doi: 10.1021/mp700089s. [DOI] [PubMed] [Google Scholar]
- 23.Ke W, Zhao Y, Huang R, Jiang C, Pei Y. Enhanced oral bioavailability of doxorubicin in a dendrimer drug delivery system. J Pharm Sci. 2008;97:2208–2216. doi: 10.1002/jps.21155. [DOI] [PubMed] [Google Scholar]
- 24.Sweet DM, Kolhatkar RB, Ray A, Swaan P, Ghandehari H. Transepithelial transport of PEGylated anionic poly (amidoamine) dendrimers: Implications for oral drug delivery. J Controlled Release. 2009;138:78–85. doi: 10.1016/j.jconrel.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin Y, Khanafer K, El-Sayed MEH. Quantitative evaluation of the effect of poly (amidoamine) dendrimers on the porosity of epithelial monolayers. Nanoscale. 2010;2:755–762. doi: 10.1039/b9nr00407f. [DOI] [PubMed] [Google Scholar]
- 26.Lin Y, Fujimori T, Kawaguchi N, Tsujimoto Y, Nishimi M, Dong Z, Katsumi H, Sakane T, Yamamoto A. Polyamidoamine dendrimers as novel potential absorption enhancers for improving the small intestinal absorption of poorly absorbable drugs in rats. J Controlled Release. 2011;149:21–28. doi: 10.1016/j.jconrel.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 27.Goldberg D, Ghandehari H, Swaan P. Cellular entry of G3. 5 poly (amido amine) dendrimers by clathrin-and dynamin-dependent endocytosis promotes tight junctional opening in intestinal epithelia. Pharm Res. 2010;27:1547–1557. doi: 10.1007/s11095-010-0153-3. [DOI] [PubMed] [Google Scholar]
- 28.Goldberg D, Vijayalakshmi N, Swaan P, Ghandehari H. G3. 5 PAMAM dendrimers enhance transepithelial transport of SN38 while minimizing gastrointestinal toxicity. J Controlled Release. 2011;150:318–325. doi: 10.1016/j.jconrel.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomalia DA, Baker H, Dewald J, Hall M, Kallos G, Martin S, Roeck J, Ryder J, Smith P. A new class of polymers: starburst-dendritic macromolecules. Polymer. 1985;17:117–132. [Google Scholar]
- 30.Kolhatkar R, Sweet D, Ghandehari H. Multifunctional Pharmaceutical Nanocarriers. Vol. 4. Springer Science; 2008. Functionalized Dendrimers as Nanoscale Drug Carriers; pp. 201–232. [Google Scholar]
- 31.Tomalia DA, Naylor AM, Goddard WA. Starburst dendrimers: molecular-level control of size, shape, surface chemistry, topology, and flexibility from atoms to macroscopic matter. Angew Chem, Int Ed. 1990;29:138–175. [Google Scholar]
- 32.Gu S, Zhao X, Zhang L, Li L, Wang Z, Meng M, An G. Anti-angiogenesis effect of generation 4 polyamidoamine/vascular endothelial growth factor antisense oligodeoxynucleotide on breast cancer in vitro. J Zhejiang Univ Sci B. 2009;10:159–167. doi: 10.1631/jzus.B0820175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bhadra D, Bhadra S, Jain S, Jain N. A PEGylated dendritic nanoparticulate carrier of fluorouracil. Int J Pharm. 2003;257:111–124. doi: 10.1016/s0378-5173(03)00132-7. [DOI] [PubMed] [Google Scholar]
- 34.Kukowska-Latallo JF, Bielinska AU, Johnson J, Spindler R, Tomalia DA, Baker JR. Efficient transfer of genetic material into mammalian cells using Starburst polyamidoamine dendrimers. Proc Natl Acad Sci U S A. 1996;93:4897–4902. doi: 10.1073/pnas.93.10.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thiagarajan G, Ray A, Malugin A, Ghandehari H. PAMAM-camptothecin conjugate inhibits proliferation and induces nuclear fragmentation in colorectal carcinoma cells. Pharm Res. 2010;27:2307–2316. doi: 10.1007/s11095-010-0179-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Asthana A, Chauhan AS, Diwan PV, Jain NK. Poly(amidoamine) (PAMAM) dendritic nanostructures for controlled site specific delivery of acidic anti-inflammatory active ingredient. AAPS PharmSciTech. 2005;6:536–542. doi: 10.1208/pt060367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yiyun C, Na M, Tongwen X, Rongqiang F, Xueyuan W, Xiaomin W, Longping W. Transdermal delivery of nonsteroidal anti inflammatory drugs mediated by polyamidoamine (PAMAM) dendrimers. J Pharm Sci. 2007;96:595–602. doi: 10.1002/jps.20745. [DOI] [PubMed] [Google Scholar]
- 38.Cheng Y, Qu H, Ma M, Xu Z, Xu P, Fang Y, Xu T. Polyamidoamine (PAMAM) dendrimers as biocompatible carriers of quinolone antimicrobials: an in vitro study. Eur J Med Chem. 2007;42:1032–1038. doi: 10.1016/j.ejmech.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 39.Ma M, Cheng Y, Xu Z, Xu P, Qu H, Fang Y, Xu T, Wen L. Evaluation of polyamidoamine (PAMAM) dendrimers as drug carriers of anti-bacterial drugs using sulfamethoxazole (SMZ) as a model drug. Eur J Med Chem. 2007;42:93–98. doi: 10.1016/j.ejmech.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 40.Borchardt SPR, Wilson G. Pharmaceutical Biotechnology. Plenum Press; New York and London: 1996. Models for assessing drug absorption and metabolism. [Google Scholar]
- 41.Kitchens KM, El-Sayed MEH, Ghandehari H. Transepithelial and endothelial transport of poly (amidoamine) dendrimers. Adv Drug Delivery Rev. 2005;57:2163–2176. doi: 10.1016/j.addr.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 42.Roberts JC, Bhalgat MK, Zera RT. Preliminary biological evaluation of polyamidoamine (PAMAM) Starburst [TM] dendrimers. J Biomed Mater Res. 1996;30:53–65. doi: 10.1002/(SICI)1097-4636(199601)30:1<53::AID-JBM8>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 43.Malik N, Wiwattanapatapee R, Klopsch R, Lorenz K, Frey H, Weener JW, Meijer EW, Paulus W, Duncan R. Dendrimers: relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J Controlled Release. 2000;65:133–148. doi: 10.1016/s0168-3659(99)00246-1. [DOI] [PubMed] [Google Scholar]
- 44.Sadekar S, Ray A, Janat-Amsbury M, Peterson C, Ghandehari H. Comparative biodistribution of PAMAM dendrimers and HPMA copolymers in ovarian-tumor-bearing mice. Biomacromolecules. 2011;12:88–96. doi: 10.1021/bm101046d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greish K, Thiagarajan G, Herd H, Price R, Bauer H, Hubbard D, Burckle A, Sadekar S, Yu T, Anwar A, Ray A, Ghandehari H. Size and surface charge significantly influence toxicity of silica and dendritic nanoparticles. Nanotoxiciology. doi: 10.3109/17435390.2011.604442. accepted. [DOI] [PubMed] [Google Scholar]
- 46.Qiu Y, Chen Y, Liu L, Zhang GGZ. Developing solid oral dosage forms: pharmaceutical theory and practice. Academic Press; 2008. [Google Scholar]
- 47.Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun. 1991;175:880–885. doi: 10.1016/0006-291x(91)91647-u. [DOI] [PubMed] [Google Scholar]
- 48.Aungst BJ. Intestinal permeation enhancers. J Pharm Sci. 2000;89:429–442. doi: 10.1002/(SICI)1520-6017(200004)89:4<429::AID-JPS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 49.Cullis PM, Green RE, Merson-Davies L, Travis N. Probing the mechanism of transport and compartmentalisation of polyamines in mammalian cells. Chem Biol. 1999;6:717–729. doi: 10.1016/s1074-5521(00)80019-8. [DOI] [PubMed] [Google Scholar]
- 50.Parveen S, Sahoo SK. Nanomedicine: clinical applications of polyethylene glycol conjugated proteins and drugs. Clin Pharmacokinet. 2006;45:965–988. doi: 10.2165/00003088-200645100-00002. [DOI] [PubMed] [Google Scholar]
- 51.Okuda T, Kawakami S, Maeie T, Niidome T, Yamashita F, Hashida M. Biodistribution characteristics of amino acid dendrimers and their PEGylated derivatives after intravenous administration. J Controlled Release. 2006;114:69–77. doi: 10.1016/j.jconrel.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 52.Najlah M, Freeman S, Attwood D, D’Emanuele A. Synthesis, characterization and stability of dendrimer prodrugs. Int J Pharm. 2006;308:175–182. doi: 10.1016/j.ijpharm.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 53.Vijayalakshmi N, Ray A, Malugin A, Ghandehari H. Carboxyl-terminated PAMAM-SN38 conjugates: synthesis, characterization, and in vitro evaluation. Bioconjugate Chem. 2010;21:1804–1810. doi: 10.1021/bc100094z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao H, Rubio B, Sapra P, Wu D, Reddy P, Sai P, Martinez A, Gao Y, Lozanguiez Y, Longley C. Novel prodrugs of SN38 using multiarm poly (ethylene glycol) linkers. Bioconjugate Chem. 2008;19:849–859. doi: 10.1021/bc700333s. [DOI] [PubMed] [Google Scholar]
- 55.Singer JW, Vries P, Bhatt R, Tulinsky J, Klein P, Li C, Milas L, Lewis RA, Wallace S. Conjugation of camptothecins to poly (l Glutamic Acid) Ann N Y Acad Sci. 2000;922:136–150. doi: 10.1111/j.1749-6632.2000.tb07032.x. [DOI] [PubMed] [Google Scholar]
- 56.Fox ME, Guillaudeu S, Frechet JMJ, Jerger K, Macaraeg N, Szoka FC. Synthesis and in vivo antitumor efficacy of PEGylated poly (l-lysine) dendrimer-camptothecin conjugates. Mol Pharmaceutics. 2009;6:1562–1572. doi: 10.1021/mp9001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang X, Wu Z, Gao W, Chen Q, Yu B. Polyamidoamine dendrimers as potential drug carriers for enhanced aqueous solubility and oral bioavailability of silybin. Drug Dev Ind Pharm. 2011;37:419–427. doi: 10.3109/03639045.2010.518150. [DOI] [PubMed] [Google Scholar]
- 58.Thiagarajan G, Ghandehari H. The biomedical engineering handbook. 4. CRC Press; Dendrimers for drug delivery. In press. [Google Scholar]