

Abstract
Human induced pluripotent stem cells (iPSCs) are a potential source of hepatocytes for liver transplantation to treat end-stage liver disease. In vitro differentiation of human iPSCs into hepatic cells has been achieved using a multistage differentiation protocol, but whether these cells are functional and capable of engrafting and regenerating diseased liver tissue is not clear. We show that human iPSC-derived hepatic cells at various differentiation stages can engraft the liver in a mouse transplantation model. Using the same differentiation and transplantation protocols, we also assessed the ability of human iPSCs derived from each of the three developmental germ layer tissues (that is, ectoderm, mesoderm, and endoderm) to regenerate mouse liver. These iPSC lines, with similar but distinct global DNA methylation patterns, differentiated into multistage hepatic cells with an efficiency similar to that of human embryonic stem cells. Human hepatic cells at various differentiation stages derived from iPSC lines of different origins successfully repopulated the liver tissue of mice with liver cirrhosis. They also secreted human-specific liver proteins into mouse blood at concentrations comparable to that of proteins secreted by human primary hepatocytes. Our results demonstrate the engraftment and liver regenerative capabilities of human iPSC-derived multistage hepatic cells in vivo and suggest that human iPSCs of distinct origins and regardless of their parental epigenetic memory can efficiently differentiate along the hepatic lineage.
Introduction
In the United States, 20,000 patients are actively waiting for a donor liver for transplantation to rescue end-stage liver disease because of, for example, liver cirrhosis or hepatocellular carcinoma. However, the shortage of donor organs means that only 7000 liver transplants are performed annually in the United States (1). Because the need for liver replacement far outstrips current supply, we are forced to critically evaluate alternative approaches to traditional solid organ transplantation. The use of ex vivo adult human hepatocytes is a desirable option for cellular therapies or drug testing. However, these cells have extremely limited proliferation potential and lose function and viability upon isolation. Although there have been great advances in liver stem cell biology (2–4), hepatic stem cells are infrequent within tissue, making their isolation and expansion unfavorable for large-scale applications (5). Attempts to immortalize hepatocytes by introducing telomerase and viral transfection also suffer from the shortcomings of phenotypic changes, poor liver function, and karyotypic abnormalities (6, 7). Recently, there has been a focus on deriving human hepatocytes from other sources, in particular human embryonic stem cells (ESCs) and human induced pluripotent stem cells (iPSCs) (8–12). These pluripotent stem cells have advantages over their adult tissue-specific counterparts because they can be expanded in culture indefinitely while maintaining a normal karyotype and differentiation capacity.
In vitro differentiation of both human ESCs and iPSCs into cells of the hepatic lineage has been achieved (8–12), suggesting a potentially unlimited source of hepatocytes that could be used in drug screening, modeling liver disease, and cell therapy. Multistage hepatic cells resembling definitive endoderm (DE), hepatic progenitors (HPs), and mature hepatocyte (MH)–like cells have been generated from human iPSCs using stepwise differentiation protocols (10, 13). A recent report further demonstrated the feasibility of using in vitro hepatic differentiation of human iPSCs to model several inherited liver diseases (14). Although in vitro culture may recapitulate certain disease features and may be suitable for drug screening purposes, successful regenerative therapy will require hepatic cells that can functionally engraft in the diseased liver. Whether human iPSC-derived hepatic cells retain functionality after engraftment is largely unknown (12, 15), and whether human iPSC-derived hepatic cells at different stages of differentiation have appropriate in vivo functions remains to be determined.
The current differentiation protocols for human iPSCs will continue to benefit from studies of mouse and human ESCs, which serve as the gold standards of pluripotency (8, 13, 16–23). iPSCs resemble ESCs in their pluripotency and offer an alternative to ESC-based therapies that must contend with histocompatibility differences between donor and recipient. However, questions remain whether human iPSCs are as functional and as safe as human ESCs. It has been shown that iPSCs and ESCs can be distinguished by their gene expression signatures (17), even in the absence of transgene expression of the reprogramming factors used to dedifferentiate adult somatic cells into iPSCs (18). From a study of hemangioblastic lineage differentiation from pluripotent stem cell lines, it has been suggested that human iPSC derivatives exhibit limited expansion and early senescence compared to human ESC derivatives (19). There are also questions regarding to what degree the iPSCs of different origins are similar to each other. In the mouse, it has been reported that secondary neurospheres generated from iPSCs derived from different adult tissues varied substantially in their tumor-forming propensity (20). The molecular mechanism underlying this phenomenon is not fully understood, but evidence from studies using mouse iPSCs has suggested that there is epigenetic memory retained in the iPSCs from their adult somatic (parental) cells, and that the memory may influence their differentiation potential (21, 22). On the basis of these mouse iPSC studies, it is reasonable to speculate that the somatic memory of human iPSCs might also affect their differentiation capabilities, which is of importance for disease modeling and cell therapy. Thus, the efficiencies of human iPSCs from multiple origins for generating functional cell types such as hepatocytes need to be determined and compared.
Most of the reported human iPSC lines have been generated from mesoderm-derived adult somatic cells (that is, fibroblasts and blood cells) and a few ectoderm derivatives (keratinocytes and neural progenitors) [reviewed in (16)]. In an effort to generate endoderm-derived human iPSCs for comparative studies as well as for modeling acquired liver diseases, we have recently reported generation of iPSCs from human primary hepatocytes (10). Establishment of a panel of human iPSCs from all three embryonic germ layers provided the opportunity to study molecular and functional similarities and differences across this diverse range of human iPSCs.
Here, we use immunodeficient mice to develop a model of liver injury that allows xenografts of human hepatic cells derived from human iPSCs to be analyzed. This xenograft mouse model enabled the comparative assessment of the in vivo regeneration potential of hepatic cells at various differentiation stages that were derived from human iPSCs. We compared the hepatic differentiation and regeneration capabilities of human iPSC lines of diverse origins that had similar but distinct epigenetic patterns. Our results show that human iPSC-derived hepatic cells at different stages of differentiation can engraft and retain some functionality in vivo regardless of the developmental origins of the iPSCs, and that retained epigenetic memory does not manifest as altered hepatic differentiation capacity.
Results
Human iPSCs derived from diverse developmental origins
As previously described, human iPSC lines derived from human hepatocytes (endoderm), bone marrow mesenchymal stem cells (mesoderm), and liver fibroblasts (mesoderm) were generated using retroviruses expressing genes encoding the transcription factors Oct4, Sox2, Klf4, and c-Myc (10, 23). For this study, we generated iPSCs from human keratinocytes (ectoderm) using the same retroviruses but slightly different culture and reprogramming conditions (fig. S1). The pluripotency of these cell lines has been reported previously (10) and is documented in this study (figs. S1 and S2). All cell lines (15 iPSC lines derived from human tissues originating in each of the three germ layers) proliferated at similar rates, expressed markers of pluripotency, and gave rise to teratomas, demonstrating that they were bona fide human iPSC lines (table S1).
Human iPSCs from tissues of different developmental origins exhibit similar but distinct epigenetic patterns
To gain insight about the epigenetic patterns of human iPSCs derived from tissues of different origins, we performed a genome-wide methylation analysis on all three germ layer–derived human iPSC lines, their parental cells, and human ESCs. The overall methylation patterns of all human iPSCs including endoderm tissue origin iPSCs (iH10, iH11, and iH14), mesoderm iPSCs (iM2, iM3, iM7, iLC1, and iLC2), and ectoderm iPSCs (iK1, iK2, and iK3) are highly similar to those of human ESCs (H1 and H9) and are distinct from those of the parental cells (Fig. 1A and fig. S3). In hierarchical clustering and heat maps of these human iPSC lines based on the top 5% most variable features (Fig. 1B), we were able to detect one cluster of CpG loci that are hypermethylated only in human ESCs but not in human iPSCs or parental cells (Fig. 1B, asterisk), indicating a certain level of common epigenetic memory in iPSCs retained from the adult somatic cells from which they were derived. We also found methylation patterns that are common to the same germ layer origin iPSCs and their parental cells but distinct from those of all other cells (that is, different origin iPSCs and their parental cells, and ESCs), indicating a certain level of tissue-specific epigenetic memory preserved in all of these distinct iPSCs (tables S2 to S4). Nonetheless, the overall methylation patterns of human iPSCs were highly similar to one another and to human ESCs rather than to their parental cells even in the hierarchical clustering and heat map that were generated based on the top 5% most variable features (Fig. 1B).
Fig. 1.
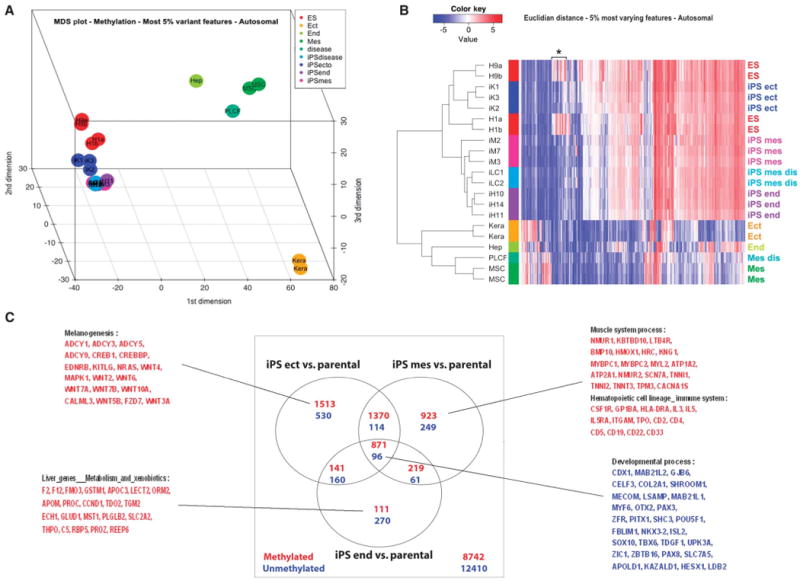
DNA methylation analysis of human iPSCs, their parental cells, and ESCs. (A) Multidimensional scaling plot showing the relationships among all individual human iPSC lines analyzed in our study. Euclidian distance and the top 5% most variable autosomal loci across all samples analyzed were used. Color code for each group of cell lines analyzed is shown in the figure. iK1, iK2, and iK3 are iPSC lines derived from adult human keratinocytes (Kera). iM2, iM3, and iM7 are iPSC lines derived from human bone marrow mesenchymal stem cells (MSCs). iLC1 and iLC2 are iPSC lines derived from adult human fibroblasts (PLCF). iH11, iH14, and iH10 are from iPSC lines derived from primary human hepatocytes (Hep). Both H1 and H9 are human ESC lines. Mes, mesoderm; Ect, ectoderm; End, endoderm. (B) Hierarchical clustering using Euclidian distance and top 5% most variable autosomal loci across all analyzed samples. Color bars on the side identify the different sample groups. The color code expresses the log2 methylation ratio. (C) Venn diagrams showing the number of autosomal loci differentially methylated between each iPSC group and the corresponding parental cells, as obtained from our linear model analysis (adjusted P < 0.001). Some of the representative genes associated with the differentially methylated features, based on functional gene set enrichment analysis, are listed for both common and distinct changes occurring during reprogramming among different source-derived iPSCs (hypermethylated, red; hypomethylated, blue).
We further analyzed the list of loci differentially methylated between each of the iPSC lines and their parental cells and compared the lists among the distinct developmental groups (Fig. 1C; differentially methylated features between iPSCs and parental cells, P < 0.001). In addition to common sets of loci that either gained or lost DNA methylation during reprogramming, tissue-specific methylation changes were also evident in the reprogramming processes of somatic cells from each germ layer (Fig. 1C). This was further confirmed by functional gene set enrichment analyses. Genes involved in tissue-specific biological pathways (for example, genes involved in liver metabolism in hepatocytes) were among those that gained methylation during reprogramming (Fig. 1C). These results suggest that human adult somatic cells undergo both common and distinct changes associated with their tissue of origin during reprogramming to iPSCs.
Together, these results revealed that human iPSCs from various sources are similar to one another and to human ESCs, regardless of retained epigenetic memory and distinct epigenetic changes that occurred during the reprogramming process (Fig. 1, fig. S3, and tables S2 to S4).
Human iPSCs derived from distinct sources show a similar propensity for hepatic differentiation in vitro
To determine whether the observed molecular differences among human iPSC lines from distinct sources could influence their ability to differentiate along the hepatic lineage in vitro, we evaluated the directed hepatic differentiation capabilities for 15 iPSC lines and 2 ESC lines (table S1) using our multistage differentiation protocol (Fig. 2A) (1). All 15 human iPSC lines, regardless of their origin, generated each stage of liver differentiation (DE, HP, and MH cells) with comparable efficiencies (Fig. 2, B to D). At the end of stage 1, all iPSCs gave rise to DE cells with ∼90% efficiency as measured by negative staining for SSEA3 (a human pluripotent stem cell marker) and positive staining for CXCR4 (a DE stage-specific cell marker) (Fig. 2B). Ninety percent to 100% of the DE cells differentiated into HP cells expressing α-fetoprotein (AFP) at day 10 (Fig. 2C); the HP cells then differentiated into MH cells expressing albumin (ALB) and α-1-antitrypsin (AAT) at day 20 (Fig. 2D) after the induction of hepatic differentiation. These results were also comparable to the hepatic differentiation potential and efficiency of human ESCs (Fig. 2, B to F). In addition, mature hepatic cells derived from human iPSCs generated from cells from distinct origins or from human ESCs exhibit comparable in vitro functional capabilities as measured by secretion of albumin and the activities of five major cytochrome P450 enzymes in the liver (CYP3A4, CYP1A2, CYP2C9, CYP2C19, and CYP2D6; Fig. 2, E and F, and fig. S4).
Fig. 2.
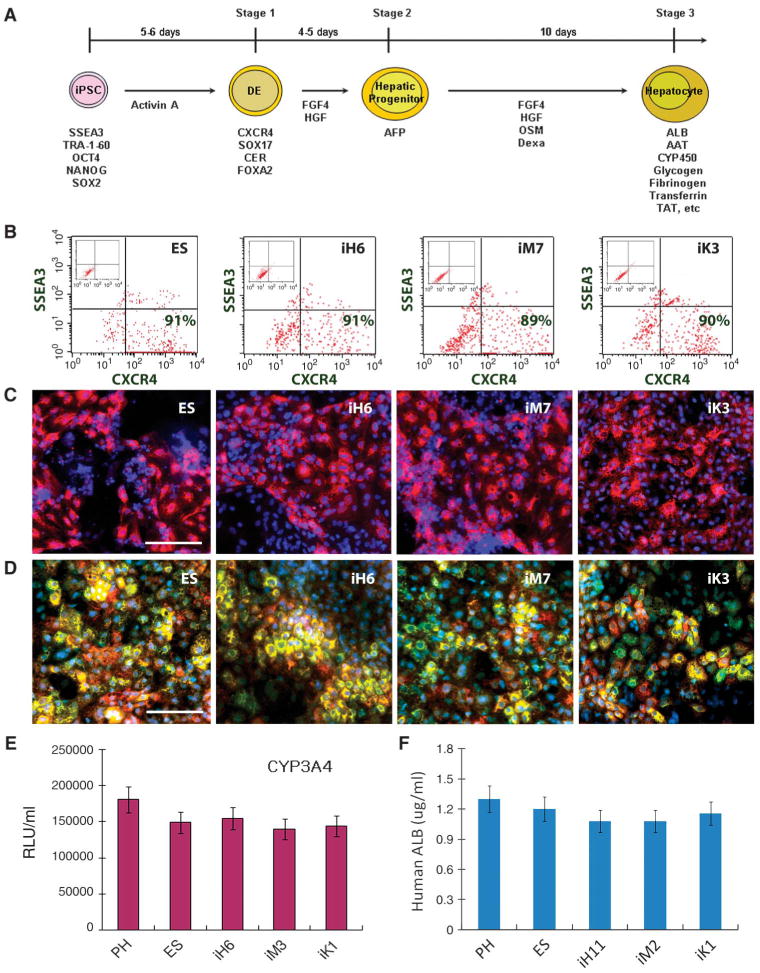
Comparison of directed hepatic differentiation in vitro among iPSC lines from different developmental origins. (A) Steps along the differentiation pathway from human iPSCs to hepatocyte-like cells. The key factors that are responsible for directing each stage of differentiation and the time course of commitment to the hepatic lineage are shown. Stage-specific markers including genes and surface phenotypes are indicated under the cells at each differentiation stage. (B to D) Using this protocol, all human ESCs and human iPSCs of different origins were able to differentiate into (B) DE (definitive endoderm, SSEA3- CXCR4+), (C) HP (hepatic progenitors, AFP+, red), and (D) MH (mature hepatocyte-like cells, ALB+ green, and AAT+ red) with a comparable efficiency to one another. Representative results are shown with the H1 human ESC line and human iPSC lines derived from each germ layer tissue. Magnification, ×100. Scale bar, 100 μm. (E and F) All of these human ESC- and iPSC-derived MH cells display cytochrome P450 (CYP450) metabolism (E) and albumin (ALB) secretion (F) (n = 3 experiments, means ± SEM). Representative results are shown for the H1 human ESC line and one of each germ layer tissue–derived human iPSC lines. Day 20 MH cells were incubated with CYP3A4 pGlo substrates; at 3 hours after treatment, 50 μl of culture medium was removed and read on a luminometer. CYP3A4 activity is expressed as relative luminescence units (RLU)/ml of culture medium (n = 6 experiments). All iPSC lines exhibited similar amounts of CYP450 enzyme activities when compared to primary hepatocytes (PHs) and human ESCs. Human ALB secretion in hepatic differentiation culture (F). The cultured medium of human ESC- or iPSC-derived day 20 MH cells (∼1 million/ml for 2 days) was analyzed using human ALB ELISA assays.
Multistage hepatic cells differentiated from diverse origin human iPSCs engraft mouse liver after transplantation
Recent reports have shown in vitro hepatic differentiation by human iPSCs (10–12), and that human iPSC-derived hepatocyte-like cells could integrate into mouse liver parenchyma and express albumin (12). However, the in vivo functionality of the multistage hepatic cells derived from human iPSCs has yet to be documented. To investigate engraftment by human iPSCs from multiple tissue origins and their potential to contribute to liver regeneration, we developed a liver injury transplantation model taking advantage of NOD/Lt-SCID/IL-2R γ g-/-(NSG) mice, which are currently one of the best rodent models for human cell engraftment analysis (24, 25). Chronic liver injury was induced in the NSG mice by treating them for 4 weeks with dimethylnitrosamine (DMN), which results in liver tissue damage that mimics liver cirrhosis (histology and liver function are shown in fig. S5) (26–28). Human ESC- and iPSC-derived hepatic cells (0.1 × 106 to 2 × 106 per mouse) were intravenously injected into these mice to assess their ability to engraft and regenerate the injured liver (Fig. 3A). About 89% of the NSG mice with liver injury transplanted with human hepatic cells survived until tissue harvesting, whereas ∼60% of the mice that did not receive human cells died within the first 3 weeks (Fig. 3B). The survival rates are comparable among mice with liver injury that received hepatic cells at different stages of differentiation (Fig. 3B). Eight weeks after human cell transplantation, mouse liver and blood were analyzed to measure liver engraftment and in vivo functions of the human ESCs and iPSCs and their multistage hepatic derivatives (Figs. 3 and 4 and Table 1). Human ESC- and iPSC-derived DE cells differentiated into mature and functional human hepatocyte-like cells as evidenced by the production of human albumin and CYP2E1 cytochrome P450 proteins (Fig. 3D). We further evaluated HP and MH cells derived from human ESCs and iPSCs for their ability to repopulate mouse liver (Fig. 3, E and F). Human hepatic cells at each differentiation stage showed engraftment ranging from 2 to 17% as evidenced by positive staining for human albumin and CYP2E1 in mouse liver.
Fig. 3.
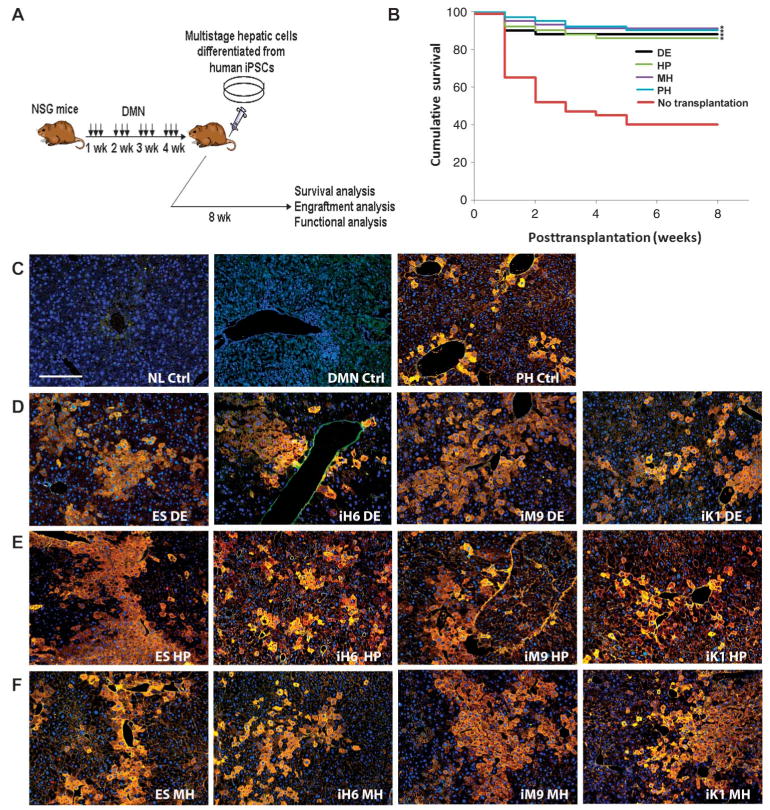
In vivo engraftment and regeneration potential of hepatic cells derived from human iPSCs and ESCs. (A) Diagram depicts liver injury model, transplantation protocol, and analyses used for the in vivo study. (B) Survival rates for NSG mice treated with DMN for 4 weeks that were given human iPSC-derived hepatic cells compared to control mice that did not receive donor cells. The day of transplantation is day 0. Transplanted mice (n = 16 to 20 per group) showed higher survival rates when compared with control mice (n = 17). *P < 0.05, compared with control mice by a log-rank test. (C) Healthy normal (NL) mice, 4-week DMN-treated mice that did not receive human cells, and mice that received human PH cells were used as negative and positive controls, respectively. The intraperitoneal injection of DMN caused hepatic fibrosis, which was reflected by the disruption of tissue architecture and the marked increase in fibrosis in DMN-injected mouse liver. Neutrophils and mononuclear cells infiltrated the livers of the DMN-treated mice (DMN control). Magnification, ×100; scale bar,100 μm. (D to F) CYP2E1 (red) and ALB (green) costaining (yellow) images of DMN-treated NSG mouse liver 8 weeks after intravenous transplantation with human ESC- or iPSC-derived (D) day 5 DE cells (2 × 106), (E) day 10 HP cells (2 × 106), and (F) day 20 MH cells (2 × 106). Representative images of hepatic cells derived from ESCs (H1 and H9) and iPSCs originating from each germ layer are shown.
Fig. 4.
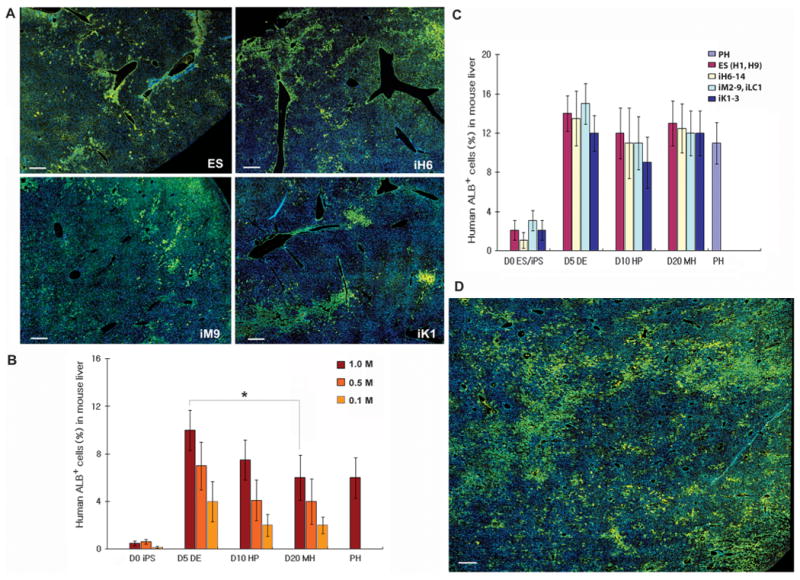
In vivo engraftment efficiency of human iPSC-derived multistage hepatic cells. (A) Large area scan images of human albumin (ALB, green) staining in mouse liver transplanted with 2 × 106 DE cells derived from human iPSCs of different origins and from human ESCs. Liver images were taken using the motorized Nikon Ti-E microscope with an Encoded Motorized XY stage and a function in NIS-Elements to generate these montage images. Magnification, ×200; scale bar, 100 μm. (B) The engraftment percentages in mouse liver for multistage hepatic cells derived from human iPSCs (iH6-14) were calculated on the basis of human-specific ALB-positive staining and the large area scan images of multiple different liver lobes and lobules obtained from the recipient NSG mice 8 weeks after transplantation of 0.1 to 1 × 106 cells (n = 3 to 10 mice, means ± SEM). *P < 0.05 by Mann-Whitney test. Similar results were observed with human ESCs and human iPSCs from different tissue origins. (C) The engraftment percentages in mouse liver for multistage hepatic cells derived from diverse origin human iPSCs were calculated on the basis of human ALB-positive staining and the large-area scan images of multiple different liver lobes and lobules obtained from the recipient NSG mice 8 weeks after transplantation with 2 × 106 cells (n = 3 to 12 mice, means ± SEM). (D) An increased percentage of human ALB-positive cells was detected in mouse liver when a higher dose (7 × 106) of human iPSC-derived DE cells was transplanted. Magnification, ×200; scale bar, 100 μm.
Table 1.
Human liver protein secretion from transplanted human iPSC-derived hepatic cells. Human liver protein secretion was measured using ELISA for each protein in serum or plasma obtained from mice that had been transplanted with hepatic cells (2 × 106 cells per mouse) at different differentiation stages derived from human ESCs or iPSCs, or transplanted with primary human hepatocytes (PHs). Both undifferentiated (day 0) iPSCs and PH cells were used as controls. There were no marked differences in albumin secretion for hepatic cells derived from human iPSCs of different origins (A), although the concentrations of albumin secreted by these hepatic cells were higher than for undifferentiated iPSCs. Similar results were observed for the secretion of other liver proteins (transferrin, AAT, and fibrinogen) (B) (results of human endoderm–derived iPSCs are shown). DE, definitive endoderm; HP, hepatic progenitors; MH, mature hepatocyte-like cells derived from human iPSCs; PSCs, pluripotent stem cells; iPSend, endoderm-derived human iPSCs; iPSmes, mesoderm-derived human iPSCs; iPSect, ectoderm-derived iPSCs; ND, not detected.
| Day 0 (iPSCs) | Day 5 (DE) | Day 10 (HP) | Day 20 (MH) | PHs | |
|---|---|---|---|---|---|
| A. Human albumin secretion from distinct origin human iPSC- and ESC-derived hepatic cells in host serum (n = 3 to 10, means ± SEM) | |||||
| Human PSCs (ng/ml) | |||||
| H1, H9 (ESCs) | 1.9 ± 0.5 | 50 ± 12 | 35 ± 10 | 38 ± 11 | |
| iH6-14 (iPSend) | 1.5 ± 0.5 | 46 ± 13 | 37 ± 11 | 40 ± 9 | |
| iM2-9, iLC1-2 (iPSmes) | 2.1 ± 0.8 | 48 ± 14 | 34 ± 9 | 38 ± 12 | 40 ± 8 |
| iK1-3 (iPSect) | 1.8 ± 0.5 | 45 ± 11 | 32 ± 12 | 39 ± 10 | |
| B. Human liver protein secretion from human endoderm iPSC-derived hepatic cells in host serum/plasma (n = 4 to 10, means ± SEM) | |||||
|
| |||||
| Transferrin (ng/ml) | 9 ± 0.8 | 101 ± 18 | 62 ± 13 | 86 ± 15 | 58 ± 11 |
| AAT (ng/ml) | 1.1 ± 0.5 | 8.1 ± 1.4 | 7.8 ± 1.1 | 7.9 ± 1.2 | 8.2 ± 1.3 |
| Fibrinogen (μg/ml) | ND | 1.1 ± 0.2 | 0.9 ± 0.1 | 0.9 ± 0.1 | 0.9 ± 0.1 |
To more accurately determine the hepatic engraftment efficiency and to overcome the limitations of conventional imaging (that is, taking photographs of only a small area of a tissue section), we took advantage of the automated “Scan Large Image” method, which scans the entire area of a tissue section (Fig. 4). With this approach, we were able to calculate more precise engraftment percentages for human hepatic cells expressing albumin (ALB+) by scanning multiple different lobules and lobes reflecting the entire mouse liver (Fig. 4A; representative scan images for human iPSCs of each tissue origin are shown). Cells from all three differentiation stages showed liver engraftment, with DE cells displaying a higher engraftment efficiency (4 to 10%) than HP and MH cells (2 to 6%) (0.1 to 1 million transplanted cells per mouse, Fig. 4B). The overall engraftment potential (9 to 15%) of differentiated cells was comparable to that of human primary hepatocytes (PHs, ∼11%) and was higher than that of undifferentiated human ESCs or human iPSCs (1 to 3%) (2 million cells transplanted, P < 0.01, Fig. 4C). In addition, we compared the human iPSCs from multiple origins regarding differences in their potential for liver engraftment. Cells from each differentiation stage (DE, HP, and MH) were used for the comparative study. Regardless of their tissue of origin, human iPSC-derived hepatic cells displayed equivalent liver engraftment when compared with ESC-derived hepatic cells as well as PH cells (2 million cells transplanted per mouse, Fig. 4C). The higher engraftment efficiency of DE cells that we observed with lower cell doses (Fig. 4B; 0.1 to 1 million transplanted cells) was not apparent when 2 million cells were used for transplantation (Fig. 4C). Distribution of human cells in mouse liver tissue sections was easily analyzed using the scan method; many of the human cells were detected as focal patterns associated with blood vessels (Fig. 4A, 2 million DE cells were transplanted), and some were evenly distributed in the liver parenchyma especially in a mouse receiving a higher cell dose (Fig. 4D, 7 million DE cells were transplanted). We observed increased engraftment with a higher cell dose (∼35% with 7 million cells transplanted, Fig. 4D) compared with a lower cell dose (∼13% with 2 million transplanted, Fig. 4A). Liver engraftment by human cells was sustained for long periods (>7 months) without signs of tumor formation.
Because albumin is a secreted protein, it could potentially be taken up by surrounding mouse liver cells, giving a false-positive result. Using polymerase chain reaction (PCR) of genomic DNA isolated from the liver tissue of the transplanted mice, we therefore confirmed that the cells detected as albumin-positive were indeed of human origin (fig. S6). From these results, we conclude that the human iPSC-derived hepatic cells have the inherent capacity to integrate into the mouse hepatic parenchyma in vivo. To rule out the possibility that the engrafted human hepatic cells may have fused with mouse liver cells, we showed by PCR (fig. S7) that no mouse genes were detected in the human ALB+ hepatic cells isolated from transplanted mice.
To further determine whether the engrafted human iPSC-derived hepatic cells are producing liver proteins, we measured in vivo secretion of human liver-specific proteins in serum or plasma obtained from mice that had been transplanted with hepatic cells at each stage of differentiation derived from human iPSCs of diverse origins (Table 1). We were able to detect measurable concentrations of multiple human liver-specific proteins including albumin, transferrin, AAT, and fibrinogen [protein (∼46, 101, 8.1, and 1100 ng/ml) was detected for mice transplanted with 2 million human hepatic DE cells; Table 1]. Although the measured concentrations of albumin were lower than expected given the engraftment efficiencies (Fig. 4), the concentrations of these liver proteins secreted from the engrafted human ESC- and iPSC-derived multistage hepatic cells were comparable to those obtained from mice that had been transplanted with human primary hepatocytes (Table 1). The protein concentrations detected correlated with the liver engraftment efficiencies (Fig. 4C). In the absence of directed differentiation, all undifferentiated iPSCs showed uniformly lower engraftment efficiencies (Fig. 4, B and C, and Table 1) and liver protein secretion, suggesting only low levels of spontaneous in vivo differentiation of transplanted human iPSCs. This propensity for spontaneous hepatic differentiation appears to be comparable among human iPSC lines from multiple origins. Together, these data demonstrate that human iPSC-derived multistage hepatic cells, regardless of their origins, can engraft damaged mouse liver and contribute to regeneration, functionally producing and secreting human liver proteins albeit at low concentrations (Figs. 3 and 4 and Table 1).
Discussion
Our study demonstrates the liver regenerative potential of several differentiation stages of hepatic cells derived from human iPSCs. The iPSC-derived, in vitro–differentiated cells resembling DE, HP, or MH cells (Fig. 2 and fig. S4) can engraft within host liver parenchyma while retaining the ability to proliferate and further differentiate into functional hepatocytes (Figs. 3 and 4 and Table 1). Together with a previous report showing that human ESC-derived DE cells can mature into hepatocyte-like cells in vivo (8), our results suggest that earlystage hepatic cells such as DE and HP cells can differentiate into mature and functional hepatocyte-like cells in the adult mouse liver environment. The engraftment and functionality of human iPSC-derived hepatic cells were comparable to those of human primary hepatocyte control cells (Figs. 3 and 4 and Table 1). Detection of human liver proteins in the serum or plasma of mice receiving human hepatic cells suggests a good correlation between human protein secretion and engraftment efficiency. We also observed a cell dose effect; a higher cell dose showed more engraftment (Fig. 4D, ∼35% repopulation with 7 million DE cells). The liver engraftment potential of hepatic cell derivatives from human iPSCs will be useful for creating humanized mice using iPSCs from patients with liver disease. Such mice will be valuable for disease modeling in vivo and to further assess functionality of iPSCs and their hepatic derivatives for potential cell replacement therapy.
On the basis of our in vivo human cell engraftment data (Figs. 3 and 4 and figs. S6 and S7) and increased human rather than mouse liver protein secretion (Table 1 and fig. S5), we suggest that the cirrhosisrelated animal deaths in nontransplanted mice may be due to the decreased function (that is, decreased synthesis of major mouse liver proteins) of the injured mouse liver, and that transplanted human cells contributed to the regeneration of the DMN-injured mouse liver. This liver cirrhosis model (that is, chronic DMN-treated NSG mice), like most biological models, may not recapitulate the human condition precisely. The concentrations of human liver proteins secreted into mouse blood (Figs. 3 and 4, fig. S5, and Table 1) are lower than expected when compared with other immunodeficient transgenic mouse models of liver injury, for example, Fah-/-/Rag2-/-/Il2rg-/- or alb-uPA severe combined immunodeficient (SCID) mice (29, 30). The limitations of our xenograft model of liver cirrhosis (created by injection of DMN into immunodeficient NSG mice) include (i) a high surgical mortality preventing intraportal or intrasplenic delivery of donor cells and (ii) extensive liver fibrosis, which may have hampered secretion of human liver proteins from engrafted human cells and prevented the high levels of human cell engraftment (∼90%) seen in other mouse models (29, 30). However, this functional mouse model of liver cirrhosis and repair by human hepatic cells does support the possibility that, in the future, iPSC-derived hepatic cells could be used for cellular therapy in patients with chronic liver disease.
At a higher cell dosage (2 million or more cells transplanted per mouse), we did not observe significant differences in engraftment efficiency among DE, HP, and MH cells derived from human iPSCs (Fig. 4C). However, the DE cells displayed slightly higher engraftment efficiencies at lower cell doses (Fig. 4B). This can be attributed to a higher proliferation capability of the DE cells (which are at an earlier developmental stage) after engraftment.
Tumor formation has been a concern associated with human ESC-or iPSC-based cellular therapy. We did not observe tumor formation in any organ of the transplanted mice for the duration of this study (up to 7 months after transplantation into the NSG mice, which is equivalent to >30 years of human life), regardless of the sources or stages of the donor cells. The possible explanation for the lack of tumor formation includes (i) the high efficiency of the directed hepatic differentiation, (ii) the relatively low dose of human cells transplanted, and (iii) the transplantation route (intravenous) used in this study. Whether transplantation of a higher number of cells by a more localized transplantation route such as intrahepatic or intraportal infusion will result in tumor formation needs to be addressed in a future study.
The influence of iPSC origin on their ability to differentiate into blood cells has been documented in mice (21, 22). Here, we investigated the potential differences among the human iPSCs of multiple origins regarding their hepatic differentiation potential. We have previously generated iPSCs from human hepatocytes (10) to model acquired liver diseases that carry somatic mutations only in liver cells. It is, however, much more convenient and less invasive to obtain other tissues (for example, fibroblasts, blood cells, and keratinocytes) compared to a liver biopsy. Therefore, it is of paramount importance to compare hepatic and nonhepatic origin iPSCs for their hepatic differentiation potential and in vivo function. Through global DNA methylation analyses, we found that all of the human iPSC lines used in this study (including both hepatic and nonhepatic origin iPSCs) exhibit highly similar methylation patterns, but distinct epigenetic signatures of iPSCs from each germ layer origin can be identified (Fig. 1, fig. S3, and tables S2 to S4). From the in vitro differentiation studies, we showed that these cell lines display similar efficiencies at each stage of hepatic cell differentiation (Fig. 2). To further elucidate any possible functional differences by a more stringent in vivo test, we transplanted the human iPSC-derived differentiated hepatic cells and assessed their engraftment efficiencies (Figs. 3 and 4). Our results showed that the differentiated cells from various origin human iPSCs have similar capabilities to engraft host liver tissue and to produce donor (that is, human) liver-specific proteins.
A recent report showed a tendency of early-passage mouse iPSCs to differentiate preferentially into the cells of origin (that is, bloodderived iPSCs differentiating into blood cells) and increased expression of Oct4 during passage of mouse iPSCs (22). We also initially hypothesized that human hepatocyte-derived iPSCs might preferentially differentiate into hepatic cells. However, such passage or cell origin effects were not observed in our human iPSC-based hepatic differentiation study. We believe that this is mainly due to a high differentiation efficiency of human iPSCs into hepatic lineage cells (∼90%), making small differences not easily distinguishable (10). Given that passage-related Oct4 expression by iPSCs is inversely correlated with the efficiency of differentiation into the cells of origin (22), we performed real-time PCR for OCT4 among early and late passages of human iPSCs (fig. S8). The expression levels of OCT4 of all three germ layer–derived human iPSC lines were similar to one another and to human ESCs regardless of passage number, which correlates with our hepatic differentiation results across all iPSCs of different origins and passages.
This study, however, does not exclude the possibility that for certain human cell types (for example, blood cells), epigenetic memory may have a more marked impact on the efficiency of directed differentiation as observed in mouse studies (21, 22). Future work should elucidate the role of epigenetic memory in cell fate determination among human iPSCs from distinct sources using improved DNA methylation arrays or whole-genome sequencing. Epigenetic memory may be advantageous for directed differentiation of certain tissue types where efficient differentiation remains a bigger challenge.
In summary, these studies demonstrate the in vivo liver regenerative potential of human iPSC-derived hepatic cells at different stages of differentiation and suggest that epigenetic memory retained in human iPSCs does not influence directed hepatic differentiation. Further studies are needed to determine whether the hepatic derivatives of patient-specific iPSCs can also functionally engraft in mouse liver and recapitulate disease features for the purpose of creating in vivo disease models. Safety studies and detailed studies of the function of engrafted iPSC-derived hepatic cells will be required to establish the clinical potential of iPSC-based cell transplantation for treating liver disease.
Materials and Methods
Hepatic differentiation of human iPSCs
ESCs and iPSCs were cultured in standard ESC maintenance media (10). Human ESC lines WA09 (H9) and WA01 (H1) (WiCell) were cultured on irradiated mouse embryo fibroblast (MEF) feeder layers in the ESC medium. Alternatively, cells were cultured on Matrigel using mTeSR (StemCell Technologies). This study was done in accordance with Johns Hopkins Institutional Stem Cell Research Oversight regulations and following a protocol approved by the Johns Hopkins Institutional Review Board.
Confluent cultures (50 to 60%) were placed in RPMI medium [supplemented with GlutaMAX and 0.5% defined fetal bovine serum (FBS) and activin A (100 ng/ml) (R&D Systems)] for 5 to 6 days to induce DE stage cells (10). DE cells were passaged with 0.05% trypsin– 0.53 mM EDTA and plated on collagen I–coated dishes in minimal MDBK-MM medium (Sigma) supplemented with GlutaMAX and bovine serum albumin (BSA) (0.5 mg/ml), fibroblast growth factor 4 (FGF4) (10 ng/ml), and hepatocyte growth factor (HGF) (10 ng/ml). Day 10 HP cells were used for experiments. HP cells were switched to complete hepatocyte culture medium containing 5% defined FBS, FGF4 (10 ng/ml), HGF (10 ng/ml), oncostatin M (10 ng/ml) (PeproTech), and 10-7 M dexamethasone (Sigma). Differentiation was continued for another 10 days to generate MH cells (10).
Transplantation of human iPSC-derived hepatic cells
NSG (Jackson) mice were used as recipients of human cells because the NSG mouse is a new generation of severely immunodeficient mouse, which accepts human cells much more easily compared with any other immunodeficient rodent models (24). Six- to 9-week-old NSG mice received DMN (5 mg/kg, Sigma, 1.0% dissolved in saline, intraperitoneally) 3 consecutive days per week for 4 weeks to induce chronic liver injury (26–28). One day after the final DMN injection, 0.1 × 106 to 2 × 106 human iPSC/ESC-derived hepatic cells were intravenously transplanted to the DMN-treated mice (25). Because of a high mortality rate associated with surgery for intrasplenic injection in the highly immunodeficient NSG mice especially after hepatotoxin treatment, tail vein injections were used for all the experiments. Recipient mouse liver and blood were harvested at 8 weeks after human cell transplantation. Additionally, some of these mice were killed at 4 weeks, 4 months, and 7 months after transplantation. Human hepatocytes that are producing the albumin protein were identified in mouse liver by an antibody specifically recognizing human but not mouse albumin. Liver images were taken using the Nikon Ti-E microscope with CoolSNAP HQ2 camera. Serum and plasma were separated from mouse blood and stored at −80°C for liver function tests. All animal experiments were conducted following experimental protocols previously approved by Johns Hopkins Institutional Animal Care and Use Committee.
Fluorescence-activated cell sorting analysis and immunofluorescence
Antibodies used were antibody against SSEA3 488 (eBioscience, 1:100), CXCR4 (BioLegend, 1:100), ALB (DAKO, 1:200), AAT (Thermo, 1:200), and CYP2E1 (Thermo, 1:100). Secondary antibodies used were Alexa Fluor Series (Invitrogen). For immunofluorescence, cells were fixed with 4% paraformaldehyde and permeabilized/blocked with 0.1% Triton X-100 and 0.3% BSA in phosphate-buffered saline (PBS).
Cytochrome P450 assay
CYP activity was assessed using the pGlo kit (Promega, CYP1A2-V8771, CYP3A4-V8901, CYP2D6-V8891, CYP2C19-V8881, and CYP2C9-V8791) according to the manufacturer's instruction for nonlytic CYP450 activity estimation. iPSC-derived MH cells were incubated with medium supplemented with pGlo substrates. At 3 hours after exposure, 50 μl of medium was removed and read on a luminometer (GLOMAX, model 9101-002). CYP activities are expressed as relative light units per milliliter of media. For normalization, the CYP-measured, iPSC-derived hepatic cells were dissociated from the culture dish with 0.05% trypsin/0.53 mM EDTA, fixed with 4% paraformaldehyde for 20 min, stained with fluorescein isothiocyanate (FITC)–conjugated anti-human ALB antibody in a permeabilizing solution (0.1% Triton X-100 and 0.3% BSA in PBS) for 1 hour, and analyzed using flow cytometry for ALB. Then, the measured CYP luminescence values were normalized by considering the percentage of ALB-expressing hepatocytes (10).
Enzyme-linked immunosorbent assay
Serum and plasma obtained from mice that had been transplanted with each stage of hepatic cells differentiated from iPSCs/ESCs were used for detecting human-specific liver protein secretion. Human ALB and transferrin (Alpha Diagnostic) and AAT and fibrinogen (GenWay) were measured using enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer's instruction. Serum and plasma obtained from mice that had not been transplanted with human cells or transplanted with human PHs were used as a negative and a positive control, respectively.
Statistical analysis
Hepatic differentiation and engraftment results are expressed as means ± SEM.
For pairwise comparisons, an unpaired Student's t test or Mann-Whitney test (95% confidence interval) was used. Survival data were analyzed with the Kaplan-Meier test. The statistical significance of the survival rates was determined by a log-rank test. For all tests, P < 0.05 was considered significant.
DNA methylation measurement
Methylation status across the genome was measured using the Illumina Infinium comprehensive DNA methylation assays, which enable investigation of a large number of genomic loci (27,578 CpG measurements spanning 14,495 genes). Methylation estimates, obtained from the Illumina BeadStudio software, were processed and analyzed using functions and methods available through the R/Bioconductor biostatistical computing environment (31, 32). At each genomic locus, the log2 ratio between probe intensities for methylated and unmethylated probes was obtained and further used in all subsequent analyses. For our generalized linear model and gene set enrichment analyses, all genomic features located within 1500 base pairs (bp) from a transcription starting site of a known gene, as obtained from the National Center for Biotechnology Information (NCBI) ENTREZ gene database, were averaged. For clustering analyses, both original and averaged methylation estimates were used, with comparable results. All analyses were performed with all measured autosomal genomic loci, excluding the nonautosomal ones, to avoid confounding with gender. All results described were obtained using the log2 ratio between unmethylated and methylated probes; however, similar results were also obtained using the β statistics [methylated/(unmethylated + methylated)].
Multidimensional scaling plot and hierarchical clustering
We used classical multidimensional scaling (principal coordinates or components analysis) (33) to display in a multidimensional space the distance between the individual samples analyzed, based on their methylation status, using both averaged and unaveraged methylation estimates. To this end, we computed the Euclidian distance between all analyzed samples using methylation status estimates at all measured autosomal loci. For this analysis, the variance across all samples analyzed was used to filter genomic loci, showing no variation across samples. Similarly, hierarchical clustering was performed to further show the relationships among the analyzed cell lines using Euclidian distance and average linkage clustering method. All analyses were also performed with one-squared correlation as the distance, using both averaged and unaveraged methylation estimates with similar results.
Differential methylation analysis
Differential methylation across the analyzed sample groups was investigated using functions and methods implemented in the R/Bioconductor (31, 32) package limma (34, 35). At each genomic locus, the averaged log2 ratio between probe intensities for methylated and unmethylated probes was used, and nonautosomal loci were excluded from the analysis to avoid confounding with gender. A fixed-effects linear model was fit for each individual locus to estimate methylation differences between the compared groups of samples. An empirical Bayes approach was applied to moderate SEs of normalized logarithmic fold change (M values). Finally, for each feature, moderated t statistics, log odds ratios of differential methylation (B statistics), and raw and adjusted P values [false discovery rate (FDR) control by the Benjamini and Hochberg method] (36) were obtained.
Analysis of functional annotation
Gene set enrichment analyses were performed as previously described (37, 38) using statistical packages from the R/Bioconductor project (31, 32). Briefly, to capture biological processes and signaling pathways associated with differential methylation, we performed Analysis of Functional Annotation (AFA), which is conceptually similar to Gene Set Enrichment Analysis (GSEA) (39, 40). The gene lists used were obtained from the Gene Ontology database (41, 42), the KEGG pathway database (43), and the Molecular Signature Database (MsigDb) (40). We first evaluated functional theme enrichment for each group comparison performed by one-sided Wilcoxon test using the moderated t statistics obtained from our linear model analyses to rank the features/loci. Similarly, we also tested for gene sets over- and underrepresented in overlapping and nonoverlapping lists of differentially methylated features across contrasts between iPSCs and their parental counterpart using cross-tabulation and the χ2 test. Multiple testing correction was performed with the Benjamini and Hochberg method (36).
Supplementary Material
Acknowledgments
Funding: Supported by NIH grant DKO70971 and by Maryland Stem Cell Research Fund grants 103655, 103974, 108592, and 108563.
Footnotes
Author contributions: Y.-Y.J. conceived and designed the experiments. H.L., Y.K., and Y.-Y.J. performed the experiments. Y.-Y.J., H.L., and L.M. analyzed the experiments. Y.-Y.J. wrote the manuscript. S.S. provided critical discussion and contributed to preparation of the manuscript.
Competing interests: Y.-Y.J. and S.S. have filed a patent for the development of iPSCs from human hepatocytes (patent no. JHU3390-1WO). The other authors declare no competing interests.
Supplementary Material: www.sciencetranslationalmedicine.org/cgi/content/full/3/82/82ra39/DC1
Methods: Fig. S1. Human keratinocyte-derived iPSC (iKera) colony formation and characterization.
Fig. S2. iKera cells can differentiate into all three primary germ layers in vitro and in vivo.
Fig. S3. Multidimensional scaling plot based on genome-scale DNA methylation analysis of human iPSCs and their parental cells, and hESCs.
Fig. S4. Multiple CYP450 enzyme activity analyses for human iPSC-derived hepatocytes.
Fig. S5. DMN mouse liver histology and function before and after the transplantation of human iPSC-derived hepatic cells.
Fig. S6. Detection of human DNA within transplanted mouse livers.
Fig. S7. Mouse gene analysis in engrafted human albumin-positive cells.
Fig. S8. Effect of passaging on pluripotency gene OCT4 expression.
Table S1. Human iPSCs and ESCs used for this study and their pluripotency.
Table S2. Genes differentially methylated between endoderm cells (iPSCend and their parental cells) versus all other cell lines (all other iPSCs, other parental cells, and ESCs).
Table S3. Genes differentially methylated between mesoderm cells (all iPSCmes and their parental cells) versus all other cell lines (all other iPSCs, other parental cells, and ESCs).
Table S4. Genes differentially methylated between ectoderm cells (iPSCect and their parental cells) versus all other cell lines (all other iPSCs, other parental cells, and ESCs).
References and Notes
- 1.Locke JE, Sun Z, Warren DS, Sheets TP, Holzer H, Shamblott MJ, Montgomery RA, Cameron AM. Generation of humanized animal livers using embryoid body-derived stem cell transplant. Ann Surg. 2008;248:487–493. doi: 10.1097/SLA.0b013e318185e821. [DOI] [PubMed] [Google Scholar]
- 2.Herrera MB, Bruno S, Buttiglieri S, Tetta C, Gatti S, Deregibus MC, Bussolati B, Camussi G. Isolation and characterization of a stem cell population from adult human liver. Stem Cells. 2006;24:2840–2850. doi: 10.1634/stemcells.2006-0114. [DOI] [PubMed] [Google Scholar]
- 3.Lázaro CA, Rhim JA, Yamada Y, Fausto N. Generation of hepatocytes from oval cell precursors in culture. Cancer Res. 1998;58:5514–5522. [PubMed] [Google Scholar]
- 4.Rogler LE. Selective bipotential differentiation of mouse embryonic hepatoblasts in vitro. Am J Pathol. 1997;150:591–602. [PMC free article] [PubMed] [Google Scholar]
- 5.Czyz J, Wiese C, Rolletschek A, Blyszczuk P, Cross M, Wobus AM. Potential of embryonic and adult stem cells in vitro. Biol Chem. 2003;384:1391–1409. doi: 10.1515/BC.2003.155. [DOI] [PubMed] [Google Scholar]
- 6.Dalgetty DM, Medine CN, Iredale JP, Hay DC. Progress and future challenges in stem cell–derived liver technologies. Am J Physiol Gastrointest Liver Physiol. 2009;297:G241–G248. doi: 10.1152/ajpgi.00138.2009. [DOI] [PubMed] [Google Scholar]
- 7.Delgado JP, Parouchev A, Allain JE, Pennarun G, Gauthier LR, Dutrillaux AM, Dutrillaux B, Di Santo J, Capron F, Boussin FD, Weber A. Long-term controlled immortalization of a primate hepatic progenitor cell line after Simian virus 40 T-antigen gene transfer. Oncogene. 2005;24:541–551. doi: 10.1038/sj.onc.1208089. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal S, Holton KL, Lanza R. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells. 2008;26:1117–1127. doi: 10.1634/stemcells.2007-1102. [DOI] [PubMed] [Google Scholar]
- 9.Song Z, Cai J, Liu Y, Zhao D, Yong J, Duo S, Song X, Guo Y, Zhao Y, Qin H, Yin X, Wu C, Che J, Lu S, Ding M, Deng H. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res. 2009;19:1233–1242. doi: 10.1038/cr.2009.107. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Ye Z, Kim Y, Sharkis S, Jang YY. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology. 2010;51:1810–1819. doi: 10.1002/hep.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sullivan GJ, Hay DC, Park IH, Fletcher J, Hannoun Z, Payne CM, Dalgetty D, Black JR, Ross JA, Samuel K, Wang G, Daley GQ, Lee JH, Church GM, Forbes SJ, Iredale JP, Wilmut I. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010;51:329–335. doi: 10.1002/hep.23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Touboul T, Hannan NR, Corbineau S, Martinez A, Martinet C, Branchereau S, Mainot S, Strick-Marchand H, Pedersen R, Di Santo J, Weber A, Vallier L. Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology. 2010;51:1754–1765. doi: 10.1002/hep.23506. [DOI] [PubMed] [Google Scholar]
- 14.Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-Doran I, Griffin J, Ahrlund-Richter L, Skepper J, Semple R, Weber A, Lomas DA, Vallier L. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest. 2010;120:3127–3136. doi: 10.1172/JCI43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu T, Wang Y, Tai G, Zhang S. Could co-transplantation of iPS cells derived hepatocytes and MSCs cure end-stage liver disease? Cell Biol Int. 2009;33:1180–1183. doi: 10.1016/j.cellbi.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Stadtfeld M, Hochedlinger K. Induced pluripotency: History, mechanisms, and applications. Genes Dev. 2010;24:2239–2263. doi: 10.1101/gad.1963910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chin MH, Mason MJ, Xie W, Volinia S, Singer M, Peterson C, Ambartsumyan G, Aimiuwu O, Richter L, Zhang J, Khvorostov I, Ott V, Grunstein M, Lavon N, Benvenisty N, Croce CM, Clark AT, Baxter T, Pyle AD, Teitell MA, Pelegrini M, Plath K, Lowry WE. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111–123. doi: 10.1016/j.stem.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marchetto MC, Yeo GW, Kainohana O, Marsala M, Gage FH, Muotri AR. Transcriptional signature and memory retention of human-induced pluripotent stem cells. PLoS One. 2009;4:e7076. doi: 10.1371/journal.pone.0007076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng Q, Lu SJ, Klimanskaya I, Gomes I, Kim D, Chung Y, Honig GR, Kim KS, Lanza R. Hemangioblastic derivatives from human induced pluripotent stem cells exhibit limited expansion and early senescence. Stem Cells. 2010;28:704–712. doi: 10.1002/stem.321. [DOI] [PubMed] [Google Scholar]
- 20.Miura K, Okada Y, Aoi T, Okada A, Takahashi K, Okita K, Nakagawa M, Koyanagi M, Tanabe K, Ohnuki M, Ogawa D, Ikeda E, Okano H, Yamanaka S. Variation in the safety of induced pluripotent stem cell lines. Nat Biotechnol. 2009;27:743–745. doi: 10.1038/nbt.1554. [DOI] [PubMed] [Google Scholar]
- 21.Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, Yabuuchi A, Takeuchi A, Cunniff KC, Hongguang H, McKinney-Freeman S, Naveiras O, Yoon TJ, Irizarry RA, Jung N, Seita J, Hanna J, Murakami P, Jaenisch R, Weissleder R, Orkin SH, Weissman IL, Feinberg AP, Daley GQ. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, Natesan S, Wagers AJ, Melnick A, Evans T, Hochedlinger K. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol. 2010;28:848–855. doi: 10.1038/nbt.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye Z, Zhan H, Mali P, Dowey S, Williams DM, Jang YY, Dang CV, Spivak JL, Moliterno AR, Cheng L. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–5480. doi: 10.1182/blood-2009-04-217406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDermott SP, Eppert K, Lechman ER, Doedens M, Dick JE. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood. 2010;116:193–200. doi: 10.1182/blood-2010-02-271841. [DOI] [PubMed] [Google Scholar]
- 25.Jang YY, Collector MI, Baylin SB, Diehl AM, Sharkis SJ. Hematopoietic stem cells convert into liver cells within days without fusion. Nat Cell Biol. 2004;6:532–539. doi: 10.1038/ncb1132. [DOI] [PubMed] [Google Scholar]
- 26.Ogiso T, Nagaki M, Takai S, Tsukada Y, Mukai T, Kimura K, Moriwaki H. Granulocyte colony-stimulating factor impairs liver regeneration in mice through the up-regulation of interleukin-1β. J Hepatol. 2007;47:816–825. doi: 10.1016/j.jhep.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 27.Nishibe Y, Kaneko H, Suzuki H, Abe T, Matsuura Y, Takaku H. Baculovirus-mediated interferon alleviates dimethylnitrosamine-induced liver cirrhosis symptoms in a murine model. Gene Ther. 2008;15:990–997. doi: 10.1038/gt.2008.29. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida T, Ogata H, Kamio M, Joo A, Shiraishi H, Tokunaga Y, Sata M, Nagai H, Yoshimura A. SOCS1 is a suppressor of liver fibrosis and hepatitis-induced carcinogenesis. J Exp Med. 2004;199:1701–1707. doi: 10.1084/jem.20031675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, Strom S, Kay MA, Finegold M, Grompe M. Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/-mice. Nat Biotechnol. 2007;25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tateno C, Yoshizane Y, Saito N, Kataoka M, Utoh R, Yamasaki C, Tachibana A, Soeno Y, Asahina K, Hino H, Asahara T, Yokoi T, Furukawa T, Yoshizato K. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am J Pathol. 2004;165:901–912. doi: 10.1016/S0002-9440(10)63352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ihaka R, Gentleman R. R: A language for data analysis and graphics. J Comput Graphic Stat. 1996;5:299–314. [Google Scholar]
- 32.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Tierney L, Yang JY, Zhang J. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gower JC. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika. 1966;53:325–338. [Google Scholar]
- 34.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 35.Smyth GK, Michaud J, Scott HS. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics. 2005;21:2067–2075. doi: 10.1093/bioinformatics/bti270. [DOI] [PubMed] [Google Scholar]
- 36.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B. 1995;57:289–300. [Google Scholar]
- 37.Daniel VC, Marchionni L, Hierman JS, Rhodes JT, Devereux WL, Rudin CM, Yung R, Parmigiani G, Dorsch M, Peacock CD, Watkins DN. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009;69:3364–3373. doi: 10.1158/0008-5472.CAN-08-4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaeffer EM, Marchionni L, Huang Z, Simons B, Blackman A, Yu W, Parmigiani G, Berman DM. Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer. Oncogene. 2008;27:7180–7191. doi: 10.1038/onc.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 40.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledgebased approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la Cruz N, Tonellato P, Jaiswal P, Seigfried T, White R Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.