

Abstract
Cancer stem cells (Tumor-initiating stem-like cells: TISCs) are resistant to chemotherapy and are associated with metastatic hepatocellular carcinoma (HCC), which is commonly observed in hepatitis C virus (HCV)-infected patients with obesity or alcohol abuse. However, it is unknown whether the TLR4-NANOG pathway serves as a universal oncogenic signaling in the genesis of TISCs and HCC. We aimed to determine whether Tlr4 is a putative proto-oncogene for TISCs in liver oncogenesis due to different etiologies and how Tlr4 is regulated at the transcriptional and epigenetic levels. CD133+/CD49f+ TISCs were isolated using FACS from HCC developed in HCV Core Tg mice fed alcohol, diethylnitrosamine-treated mice, and alcoholic patients with or without HCV infection. CD133+/CD49f+ cells isolated from the animal models and patients are tumorigenic both in vitro and in a xenograft model, and Tlr4 or Nanog silencing with shRNA attenuates their tumor initiating property. Functional oncogene screening of a cDNA library identified the organ size control pathway targets Yap1 and AKT activator Igf2bp3 as NANOG-dependent genes that inhibit TGF-β signaling in TISCs. Tlr4 expression is higher in TISCs compared to CD133−/CD49f+ cells, and DNA hypomethylation, histone acetylation and de-methylation drive Tlr4 induction through NOTCH signaling or Hif-1α. Taken together, Tlr4 is induced by hypoxia and NOTCH signaling and is a universal proto-oncogene responsible for the genesis of TLR4-NANOG dependent TISCs, and this pathway serves as a novel therapeutic target for HCC.
Keywords: Alcohol, HCV, HCC, Tumor-initiating stem-like cells, Obesity
HCV, diabetes, alcohol, and HCC
Chronic liver damage caused by viral infection, alcohol, or metabolic syndrome can result in increased risk for HCC which is the third most common cancer in the world 1. Clearly, understanding the molecular mechanisms of HCV-induced hepatocarcinogenesis is required for the eventual development of improved therapeutic modalities for this disease 2. In particular, chronic infection with HBV or HCV represents a major risk factor for HCC 1. HCV affects more than 170 million people worldwide 1,3,4.
Ample epidemiological evidence suggests that there is a strong connection between HCV and obesity/alcoholic liver diseases (ALD). First, the prevalence of HCV is significantly higher among obesity/alcoholics than in the general population; for example, while the HCV positive rate in the general population of the U.S. is roughly 1%, it is 16% for alcoholics and nearly 30% for alcoholics with liver diseases 5. Second, the presence of HCV infection correlates with the severity of the disease in alcoholic subjects, i.e, HCV-infected patients with obesity and ALD develop liver cirrhosis and HCC at a significantly younger age than uninfected ALD patients, suggesting that alcohol and HCV work synergistically to cause liver damage 6. Many studies also support synergistic interactions between HCV and alcoholism in hepatocarcinogenesis 7–11. Heavy alcohol consumption/obesity and viral hepatitis synergistically increase the risk for HCC among blacks and whites in the U.S. 10. HCC odds ratio increases to 48.3-fold or 47.8 from 8.1 or 8.6 by having concomitant alcohol abuse or obesity in HBV/HCV infected patients, respectively 9,10. Indeed, our recent result demonstrates that the incidence of spontaneous HCC induction in the HCV core transgenic mice is increased 2-fold by chronic alcohol intake.
Recent studies with mice expressing HCV proteins have shed pivotal insights into the mechanisms underlying this synergism. The HCV core protein causes overproduction of reactive oxygen species which appears to be responsible for mitochondrial DNA damage 3,12,13. Thus, these core-induced perturbations such as oxidant stress and insulin resistance, which are also known risk factors for ALD, may underlie the synergism reproduced in alcohol-fed core transgenic mice 14. TLR2 and TLR4 are markedly upregulated in hepatocytes, Kupffer cells, and peripheral monocytes of patients with chronic hepatitis C. TLR-mediated signals result in liver disease associated with hepatitis B, hepatitis C, alcoholic non-alcoholic steatohepatitis, and hepatic fibrosis 15. The most devastating consequence of the synergism between viral hepatitis and alcohol is HCC 7–11.
TLRs signaling in HCC
The TLR signaling pathway is upregulated in chronic liver diseases. Many different cell types in the liver express TLRs 15. Chronic alcohol consumption activates other TLRs, such as TLR1, 2 and 6–9, which further increases the TNF-α response to LPS in mice15. Human monocytes exposed to ethanol for a week develop hypersensitivity to LPS through decreased IRAK-M expression, which activates mitogen-activated protein kinase (MAPK) and NF-κB through TLR4 signaling, leading to activation of NF-κB, AP-1, and ERK 16. The direct mechanistic evidence has recently been attained by our research using mice expressing the HCV non-structural protein NS5A in a hepatocyte-specific manner. These mice when fed alcohol for 12 months, develop liver tumors in a manner dependent on TLR4 induced by NS5A 17. This NS5A-induced TLR4 is activated by endotoxemia associated with alcohol intake, leading to accentuated TLR4 signaling which in turn upregulates the stem cell marker Nanog required for TLR4-dependent liver oncogenesis. This finding on the NS5A-TLR4-Nanog axis in synergistic oncogenesis, is beginning to shed a novel insight into molecular mechanisms for HCC in alcoholic HCV patients.
Tumor-initiating stem-like cells and HCC
Stem cells have three major characteristics, self renewal, asymmetric and multiple cell division (clonality), and plasticity. The liver has a high regenerative potential, and hepatic small oval progenitor cells around the peripheral branches of the bile ducts, the canals of Hering, can differentiate into biliary epithelial cells and hepatocytes 18. These oval liver progenitor cells share molecular markers with adult hepatocytes (albumin, cytokeratin 7 [CK7], CK19, oval cell markers [OV-6, A6, and OV-1], chromogranin-A, NCAM [neural cell adhesion molecule]) and fetal hepatocytes (α-fetoprotein) 18,19. They are also positive for more common stem cell markers such as CD34+, Thy-1+, c-Kit+, and Flt-3+ (FMS-like tyrosine kinase 3) 20. Thus, it currently remains unclear whether these stem cells are derived from the bone marrow and just migrate to this periportal niche or whether they represent true resident liver stem/progenitor cells. Binding of stroma-derived factor-1α (SDF-1α) to its surface receptor CXCR4 activates oval hepatic cells 21. Forty percent of HCC have clonality, and thus are considered to originate from progenitor/stem cells 19,22–24. Recent studies of HCC have centered on TISCs, including detection of TISCs in cancer, identification of TISCs markers, and isolation of TISCs from human HCC cell lines. TISCs were identified as a CD117+/CD133+ hepatic precursors in regenerating liver tissue 25 and a CD45−/CD90+ subpopulation of tumor cells in HCC 26. The CD90+ cells are not present in the normal liver and, when injected into immunodeficient mice, create tumors repeatedly. In human HCC and HCC cell lines, specifically CD133+ cells, not CD133− cells, had the ability to self-renew, create differentiated progenies, and form tumors 27. One potential reason for this chemoresistance may lie in the plasticity of cancer stem cells with dysregulated signaling and gene expression.
Nanog is one of the core transcription factors found in pluripotent embryonic stem cells (ESCs) 28. It is essential for maintaining self-renewal and pluripotency of both human and mouse embryonic stem cells 29–32. Overexpression of Nanog induces and maintains the pluripotency and self-renewing characteristics of ESCs under what normally would be differentiation-inducing culture conditions 33. Recently, Nanog expression has been reported in human neoplasms, including germ cell tumors 34–37,breast carcinomas 37, osteosarcoma 38, and HCC 39. Ectopic expression of Nanog induces an oncogenic potential in NIH3T3 40.
Nanog-positive cancer stem cells induced by HCV and alcohol
Alcohol synergistically enhances the progression of liver disease and the risk for liver cancer caused by HCV. TLR4 is induced by hepatocyte-specific transgenic (Tg) expression of the HCV nonstructural protein NS5A, and this induction mediates synergistic liver damage and tumor development by alcohol-induced endotoxemia 17. The stem/progenitor cell marker, Nanog, is up-regulated as a novel downstream gene by TLR4 activation and the presence of CD133/Nanog-positive cells in liver tumors of alcohol-fed NS5A Tg mice 17. Transplantation of p53-deficient hepatic progenitor cells transduced with TLR4 results in liver tumor development in mice following repetitive lipopolysaccharide (LPS) injection, but concomitant transduction of Nanog short-hairpin RNA abrogates this outcome 17. Despite the common understanding that TLR4 is one of the pattern recognition receptors expressed predominantly by innate immune cells such as macrophages and lymphocytes, our study demonstrates that hepatocytes can be the primary cellular site of both TLR4 upregulation and its pathologic consequences in the context of HCV infection. Therefore, the TLR4-dependent mechanism synergizes liver disease by HCV and alcohol and is partly dependent on Nanog, a TLR4 downstream gene.
Nanog transduction alone is not as effective as TLR4 activation in liver tumorigenesis, as shown by our cell transplantation experiment 17. We believe that TLR4 activation induces other tumor-driver genes which cooperatively work with Nanog to cause liver oncogenesis. Thus, Nanog is still essential for TLR4-dependent oncogenesis, but it alone is poorly oncogenic. In our previous work using a cell line, we demonstrated that TLR4 promoter up-regulation by NS5A is mediated by PU.1, Oct-1, and AP-1 elements 41. The similar transcriptional mechanism may underlie TLR4 induction in primary hepatocytes.
Epigenetic regulation of tumor-initiating stem-like cells
Cellular memory mechanisms enable cells to remember their chosen fate over many cell divisions. The dynamic interactions between epigenetic regulators and DNA components orchestrate transcriptional memory system for several hundred genes. DNA methylation is a major epigenetic modification of the genome particularly as it relates to pluripotency and differentiation of stem cells 42–44. Genomic methylation patterns in somatic differentiated cells are generally stable and heritable. In contrast, aberrant genome-wide DNA hypomethylation links to tumorigenesis 45,46. In TISCs, DNA methylation patterns and histone modifications are suspected be reprogrammed genome-wide, leading to generation of cells with altered gene expression profile for stem cell-like characters 47. However, epigenetic mechanism and implications in HCC remain to be elucidated, and the role of epigenetic reprogramming in generation of cancer stem cells is yet to be investigated.
In summary, alcohol, obesity, and HCV synergistically induce liver tumor development via induction and activation of TLR4 in mice. Pharmacologic inhibition of TLR4 signaling may become a novel therapeutic strategy for HCV-associated liver tumors.
Fig. 1.
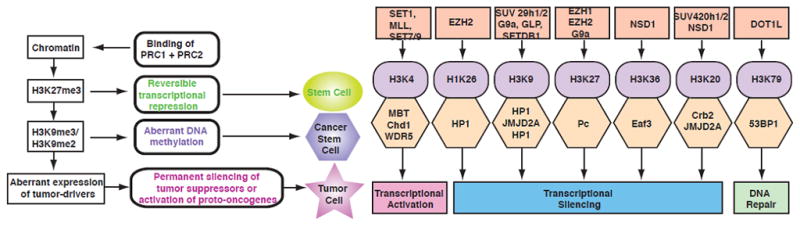
These diagrams depict histone lysine methyltransferases, target lysine methylation sites in histones, methyl-lysine binding protein modular domains and the consequent epigenetic regulation (right), as well as the hypothetical epigenetic mechanisms of cancer stem cell generation (left). Modified from original figures shown in references 47,48.
Acknowledgments
We thank Akiko Ueno and Raul Lazaro for performing mouse experiments, Naomi Anderson (USC) for technical support and critical reading, Ratna Ray (Saint Louis Univ.) for providing HCV Ns5a Tg mice, Michael Karin (UCSD) for suggestions and reagents, Steve Weinman (Univ. Kansas) for critiques and discussions. This project was supported by NIH grants 1R01AA018857-01, P50AA11999 (Animal Core, Morphology Core, and Pilot Project Program), R24AA012885 (Non-Parenchymal Liver Cell Core), Zumberge Foundation, AI83025U19, RC2AA019392-01, CA123328, and CA108302. Microscopy was performed by the Cell and Tissue Imaging Core of the USC Research Center for Liver Diseases (P30 DK048522). Statistical analysis was performed by Dr. Susan Groshen and Ms. Lingyun Ji in Norris Comprehensive Cancer Center Biostatistics Core supported by NIH/NCI P30 CA 014089. Animal imaging was performed by the USC Molecular Imaging Center supported by NIH/NVRR S10. Tissue pathological slide preparation was performed by Ms. Moli Chen Translational Pathology Core of Norris Comprehensive Cancer Center.
Footnotes
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Okuda K. Hepatocellular carcinoma. J Hepatol. 2000;32:225–37. doi: 10.1016/s0168-8278(00)80428-6. [DOI] [PubMed] [Google Scholar]
- 2.Crippin JS, McCashland T, Terrault N, Sheiner P, Charlton MR. A pilot study of the tolerability and efficacy of antiviral therapy in hepatitis C virus-infected patients awaiting liver transplantation. Liver Transpl. 2002;8:350–5. doi: 10.1053/jlts.2002.31748. [DOI] [PubMed] [Google Scholar]
- 3.Okuda M, et al. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–75. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 4.Yao F, Terrault N. Hepatitis C and hepatocellular carcinoma. Curr Treat Options Oncol. 2001;2:473–83. doi: 10.1007/s11864-001-0069-6. [DOI] [PubMed] [Google Scholar]
- 5.Heintges T, Wands JR. Hepatitis C virus: epidemiology and transmission. Hepatology. 1997;26:521–6. doi: 10.1002/hep.510260338. [DOI] [PubMed] [Google Scholar]
- 6.Brechot C, Nalpas B, Feitelson MA. Interactions between alcohol and hepatitis viruses in the liver. Clin Lab Med. 1996;16:273–87. [PubMed] [Google Scholar]
- 7.Peters MG, Terrault NA. Alcohol use and hepatitis C. Hepatology. 2002;36:S220–5. doi: 10.1053/jhep.2002.36811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donato F, Gelatti U, Limina RM, Fattovich G. Southern Europe as an example of interaction between various environmental factors: a systematic review of the epidemiologic evidence. Oncogene. 2006;25:3756–70. doi: 10.1038/sj.onc.1209557. [DOI] [PubMed] [Google Scholar]
- 9.Hassan MM, et al. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology. 2002;36:1206–13. doi: 10.1053/jhep.2002.36780. [DOI] [PubMed] [Google Scholar]
- 10.Yuan JM, Govindarajan S, Arakawa K, Yu MC. Synergism of alcohol, diabetes, and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the U.S. Cancer. 2004;101:1009–17. doi: 10.1002/cncr.20427. [DOI] [PubMed] [Google Scholar]
- 11.Lai MS, Hsieh MS, Chiu YH, Chen TH. Type 2 diabetes and hepatocellular carcinoma: A cohort study in high prevalence area of hepatitis virus infection. Hepatology. 2006;43:1295–302. doi: 10.1002/hep.21208. [DOI] [PubMed] [Google Scholar]
- 12.Moriya K, et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001;61:4365–70. [PubMed] [Google Scholar]
- 13.Korenaga M, et al. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem. 2005;280:37481–8. doi: 10.1074/jbc.M506412200. [DOI] [PubMed] [Google Scholar]
- 14.Koike K, et al. Molecular basis for the synergy between alcohol and hepatitis C virus in hepatocarcinogenesis. J Gastroenterol Hepatol. 2008;23 (Suppl 1):S87–91. doi: 10.1111/j.1440-1746.2007.05292.x. [DOI] [PubMed] [Google Scholar]
- 15.Testro AG, Visvanathan K. Toll-like receptors and their role in gastrointestinal disease. J Gastroenterol Hepatol. 2009;24:943–54. doi: 10.1111/j.1440-1746.2009.05854.x. [DOI] [PubMed] [Google Scholar]
- 16.Mandrekar P, Bala S, Catalano D, Kodys K, Szabo G. The opposite effects of acute and chronic alcohol on lipopolysaccharide-induced inflammation are linked to IRAK-M in human monocytes. J Immunol. 2009;183:1320–7. doi: 10.4049/jimmunol.0803206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Machida K, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci U S A. 2009;106:1548–53. doi: 10.1073/pnas.0807390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roskams TA, et al. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739–45. doi: 10.1002/hep.20130. [DOI] [PubMed] [Google Scholar]
- 19.Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818–22. doi: 10.1038/sj.onc.1209558. [DOI] [PubMed] [Google Scholar]
- 20.Burke ZD, Thowfeequ S, Peran M, Tosh D. Stem cells in the adult pancreas and liver. Biochem J. 2007;404:169–78. doi: 10.1042/BJ20070167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatch HM, Zheng D, Jorgensen ML, Petersen BE. SDF-1alpha/CXCR4: a mechanism for hepatic oval cell activation and bone marrow stem cell recruitment to the injured liver of rats. Cloning Stem Cells. 2002;4:339–51. doi: 10.1089/153623002321025014. [DOI] [PubMed] [Google Scholar]
- 22.Alison MR. Liver stem cells: implications for hepatocarcinogenesis. Stem Cell Rev. 2005;1:253–60. doi: 10.1385/SCR:1:3:253. [DOI] [PubMed] [Google Scholar]
- 23.Zender L, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125:1253–67. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang Y, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A. 2008;105:2445–50. doi: 10.1073/pnas.0705395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Craig CE, et al. The histopathology of regeneration in massive hepatic necrosis. Semin Liver Dis. 2004;24:49–64. doi: 10.1055/s-2004-823101. [DOI] [PubMed] [Google Scholar]
- 26.Yang ZF, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–66. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 27.Ma S, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–56. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 28.Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A. 1981;78:7634–8. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loh YH, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–40. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–8. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- 31.Rao S, Orkin SH. Unraveling the transcriptional network controlling ES cell pluripotency. Genome Biol. 2006;7:230. doi: 10.1186/gb-2006-7-8-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007;17:42–9. doi: 10.1038/sj.cr.7310125. [DOI] [PubMed] [Google Scholar]
- 33.Chambers I, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–55. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 34.Hoei-Hansen CE, et al. Stem cell pluripotency factor NANOG is expressed in human fetal gonocytes, testicular carcinoma in situ and germ cell tumours. Histopathology. 2005;47:48–56. doi: 10.1111/j.1365-2559.2005.02182.x. [DOI] [PubMed] [Google Scholar]
- 35.Hart AH, et al. The pluripotency homeobox gene NANOG is expressed in human germ cell tumors. Cancer. 2005;104:2092–8. doi: 10.1002/cncr.21435. [DOI] [PubMed] [Google Scholar]
- 36.Santagata S, Ligon KL, Hornick JL. Embryonic stem cell transcription factor signatures in the diagnosis of primary and metastatic germ cell tumors. Am J Surg Pathol. 2007;31:836–45. doi: 10.1097/PAS.0b013e31802e708a. [DOI] [PubMed] [Google Scholar]
- 37.Ezeh UI, Turek PJ, Reijo RA, Clark AT. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer. 2005;104:2255–65. doi: 10.1002/cncr.21432. [DOI] [PubMed] [Google Scholar]
- 38.Gibbs CP, et al. Stem-like cells in bone sarcomas: implications for tumorigenesis. Neoplasia. 2005;7:967–76. doi: 10.1593/neo.05394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–58. doi: 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, et al. Expression of Nanog gene promotes NIH3T3 cell proliferation. Biochem Biophys Res Commun. 2005;338:1098–102. doi: 10.1016/j.bbrc.2005.10.071. [DOI] [PubMed] [Google Scholar]
- 41.Machida K, et al. Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol. 2006;80:866–74. doi: 10.1128/JVI.80.2.866-874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–53. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 43.Bernstein BE, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–26. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 44.Gan Q, Yoshida T, McDonald OG, Owens GK. Concise review: epigenetic mechanisms contribute to pluripotency and cell lineage determination of embryonic stem cells. Stem Cells. 2007;25:2–9. doi: 10.1634/stemcells.2006-0383. [DOI] [PubMed] [Google Scholar]
- 45.Lin CH, et al. Genome-wide hypomethylation in hepatocellular carcinogenesis. Cancer Res. 2001;61:4238–43. [PubMed] [Google Scholar]
- 46.Gaudet F, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–92. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 47.Rajasekhar VK, Begemann M. Concise review: roles of polycomb group proteins in development and disease: a stem cell perspective. Stem Cells. 2007;25:2498–510. doi: 10.1634/stemcells.2006-0608. [DOI] [PubMed] [Google Scholar]
- 48.Qian C, Zhou MM. SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cell Mol Life Sci. 2006;63:2755–63. doi: 10.1007/s00018-006-6274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]