

Abstract
Aims
The goal was to terminate atrial fibrillation (AF) by targeting atrioventricular differences in ionic properties.
Methods and results
Optical mapping was used to record electrical activity during carbachol (0.25–0.5 μM)-induced AF in pig hearts. The atrial-specific current, IKur, was blocked with 100 μM 4-aminopyridine (4-AP) or with 0.5 μM DPO-1. Hearts in AF and ventricular fibrillation (VF) were also subjected to increasing levels of extracellular K+ ([K+]o: 6–12 mM), compared with controls (4 mM). We hypothesized that due to the more negative steady-state half inactivation voltage for the atrial Na+ current, INa, compared with the ventricle, AF would terminate before VF in hyperkalaemia. Mathematical models were used to interpret experimental findings. The IKur block did not terminate AF in a majority of experiments (6/9 with 4-AP and 3/4 with DPO-1). AF terminated in mild hyperkalaemia ([K+]o ≤ 10.0 mM; N = 8). In contrast, only two of five VF episodes terminated at the maximum ([K+]o: 12 mM [K+]o). The IKur block did not terminate a simulated rotor in cholinergic AF because its contribution to repolarization was dwarfed by the large magnitude of the acetylcholine-activated K+ current (IK,ACh). Simulations showed that the lower availability of the atrial Na+ current at depolarized potentials, and a smaller atrial tissue size compared with the ventricle, could partly explain the earlier termination of AF compared with VF during hyperkalaemia.
Conclusion
IKur is an ineffective anti-arrhythmic drug target in cholinergic AF. Manipulating Na+ current ‘availability’ might represent a viable anti-arrhythmic strategy in AF.
Keywords: Atrial fibrillation, IKur, Hyperkalaemia, INa availability
1. Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia and enhances the risk of stroke and heart failure.1 Although non-pharmacological options such as ablation and surgery are increasingly used, anti-arrhythmic drugs constitute the main treatment option.1 However, their effectiveness has been unsatisfactory and, in some cases, harmful due to side effects.2 An additional problem that is associated with drugs is that in some cases they precipitate fatal ventricular arrhythmias.2 As a result, extensive research has been directed towards finding drug targets that are exclusively present in the atria and not in the ventricles.3
It is now clear that distinct differences exist between the atria and the ventricles in terms of ion channel currents and their underlying transcripts in many species including humans.4 A K+ current that is important in human atrial repolarization, but absent in the ventricle, i.e. the ultra-rapid delayed rectifier current, IKur, is currently a target for the development of newer anti-arrhythmic drugs to treat AF.2,3 However, the results thus far have been highly controversial, with studies suggesting that blockade of IKur can either be useful in terminating AF5 or actually be pro-arrhythmic.6 Furthermore, recent studies have also identified important differences between the biophysical characteristics of the Na+ channel (INa) between the atria and ventricles, with the atrium displaying more negative steady-state half inactivation membrane voltage properties.7 This study has opened up another interesting avenue for exploiting atrio-ventricular differences and targeting AF.8
Our objective was to study acute, carbachol-induced AF in isolated pig hearts and to probe for the usefulness of both the above-mentioned ion-channel-based anti-arrhythmic approaches. In experiments, we studied the effectiveness of IKur block in AF via selective blockers of this current: 4-aminopyridine (4-AP) and DPO-1. We also postulated that the inherent differences in atrio-ventricular Na+ channel steady-state inactivation properties could be exploited by manipulating the concentration of extracellular K+ ions ([K+]o). We hypothesized that for mild increases in [K+]o (hyperkalaemia), the atrial INa would be more depressed than its ventricular counterpart as a result of its more negative half-inactivation value, resulting in the termination of acute AF, but not ventricular fibrillation (VF). To interpret our experimental results, we also utilized concomitant computer simulations. We reformulated the equations for IKur that now take into account its rate-dependent attenuation in magnitude.9–11 The updated ionic model of an atrial cell12 was incorporated in a two-dimensional sheet to simulate spiral waves that underlie functional re-entry (rotors) and to study the ionic mechanisms as in a previous study.13
Our results indicate that IKur is not a viable anti-arrhythmic target for stopping acute, cholinergic, high-frequency AF in pigs. In contrast, mild hyperkalaemia effectively terminates AF, but not VF in this model.
2. Methods
2.1. Experiments
This investigation conformed to US NIH Guidelines for the Care and Use of Laboratory Animals (NIH publication no. 85-23, revised 1996). Male pigs (20–25 kg) were anaesthetized initially by a combination of xylazine (2.2 mg/kg) and telazol (6 mg/kg), followed by thiopental (10 mg/kg) or propofol (6 mg/kg). Hearts were excised and Langendorff-perfused with Tyrode's solution whose composition in millimolar was: NaCl, 130; KCl, 4.0; MgCl2, 1; CaCl2, 1.8; NaHCO3, 24; NaH2PO4, 1.2; glucose, 5.6; and albumin 0.04 g/L. Tyrode's solution was gassed with 95% O2–5% CO2, filtered using a Liliput hollow fibre oxygenator (COBE Cardiovascular, Arvada, CO, USA), and perfused at a constant flow rate of 150–200 mL/min at 36 ± 1.0°C. After an initial perfusion period of 15 min, blebbistatin (5–10 μM) was added to the perfusate to abolish motion of the heart. AF was then induced via electrical burst pacing (50–100 ms cycle length and ∼10 s duration) or a battery in the presence of carbachol (0.25–0.5 μM) with 100% success, and the frequency of excitation was mapped for 10 or 20 min (control), before adding 4-AP (100 μM) to selectively block IKur (for another 10 min). The perfusate containing the carbachol and/or 4-AP was never re-cycled. Movies of di-4-ANNEPS (10 μM) fluorescence during AF were obtained from the left atrium (LA) and the right atrium (RA) using a Little Joe CCD camera (Scimeasure; 80 by 80 pixels, 500–1000 frames/s, area of field of view ∼3 cm2). Additionally, two electrograms were simultaneously recorded in the LA and RA to ascertain the presence of either a normal sinus rhythm or AF. Dominant frequency (DF) maps during AF (in control in the presence of carbachol or carbachol + 4-AP) were constructed as in previous studies,14 and the maximum DF (DFmax) was quantified as a function of time. The DF analysis allows for the quantification of how fast the functional re-entrant activity is at each point on the heart surface in the time domain during fibrillation, and DFmax measures the fastest fibrillatory activity. In an additional series of experiments, we investigated the effect of another selective blocker of IKur, DPO-1.15 We first studied the effect of DPO-1 on the atrial and ventricular action potential durations (APDs) during pacing and also its effect on carbachol-induced AF and VF dynamics.
A second set of experiments studying the effect of hyperkalaemia (6 or 8 mM of K+) ([K+]o; control 4 mM) on carbachol-induced AF was carried out after completing the 4-AP study, by perfusing hearts for 30 min with normal, non-recycled carbachol-free Tyrode's solution (N = 4) to washout all drugs (carbachol, 4-AP).
In a third series of experiments (N = 5), carbachol-induced AF and VF were simultaneously mapped in the LA and the left ventricle (LV). The hearts were subsequently perfused with increasing values of [K+]o, either 6, 8, 10, or 12 mM for approximately 10 min at each concentration (compared with 4 mM in controls).
All data are shown as mean ± SEM. For comparing APDs, repeated two-way analysis of variance with Bonferroni post-test was used. For comparing dominant frequencies for AF/VF in control and drug conditions, unpaired Student's t-test was used; P < 0.05 was considered statistically significant.
2.2. Simulations
To investigate the effects of blocking IKur and changing [K+]o on re-entry dynamics, we used a modified human atrial cell ionic model12 incorporated in a two-dimensional sheet to simulate a rotor, as in a previous study.13 Research in the last two decades has firmly established that transient or stable spiral waves, also called rotors, sustain cardiac fibrillation and represent a useful way to study the underlying ionic properties.16 The specific model modifications and simulations are discussed in detail in the next section.
3. Results
3.1. Experimental
We induced acute AF in nine hearts in the presence of carbachol. We recorded the activation frequencies for 10 min under control conditions, before perfusing 100 μM 4-AP for another 10 min (in three hearts, we ascertained that AF was sustained continuously and thus stable for 20 min, i.e. for the duration of most of our 4-AP experiments). Figure 1Adepicts a representative DF map during acute AF in the pig heart. Similar to previous studies in sheep,17 we found that the DFmax was larger in the LA compared with the RA; this was the case in the majority of the DF maps constructed (∼80%). The 1 s single-pixel recordings from the LA and RA shown below the maps also demonstrate faster activation of the electrical excitation in the LA. The variation of DFmax with time under control conditions, and in the presence of the drug, 100 μM 4-AP, is shown in Figure 1B for six experiments (each set of symbols represents one experiment) in which AF did not terminate. Statistical analyses of all the movies in these six hearts showed that there was a very small but significant increase in the DFmax after the addition of the drug compared with control frequencies (control: 20.97 ± 0.26 Hz, n = 79, N = 6; drug 4-AP: 21.98 ± 0.35 Hz, n = 65, N = 6; P< 0.05). In the remaining three experiments, AF did terminate in two of three after the addition of 4-AP; however, in one experiment, sustained AF could be re-induced even in the presence of 4-AP.
Figure 1.
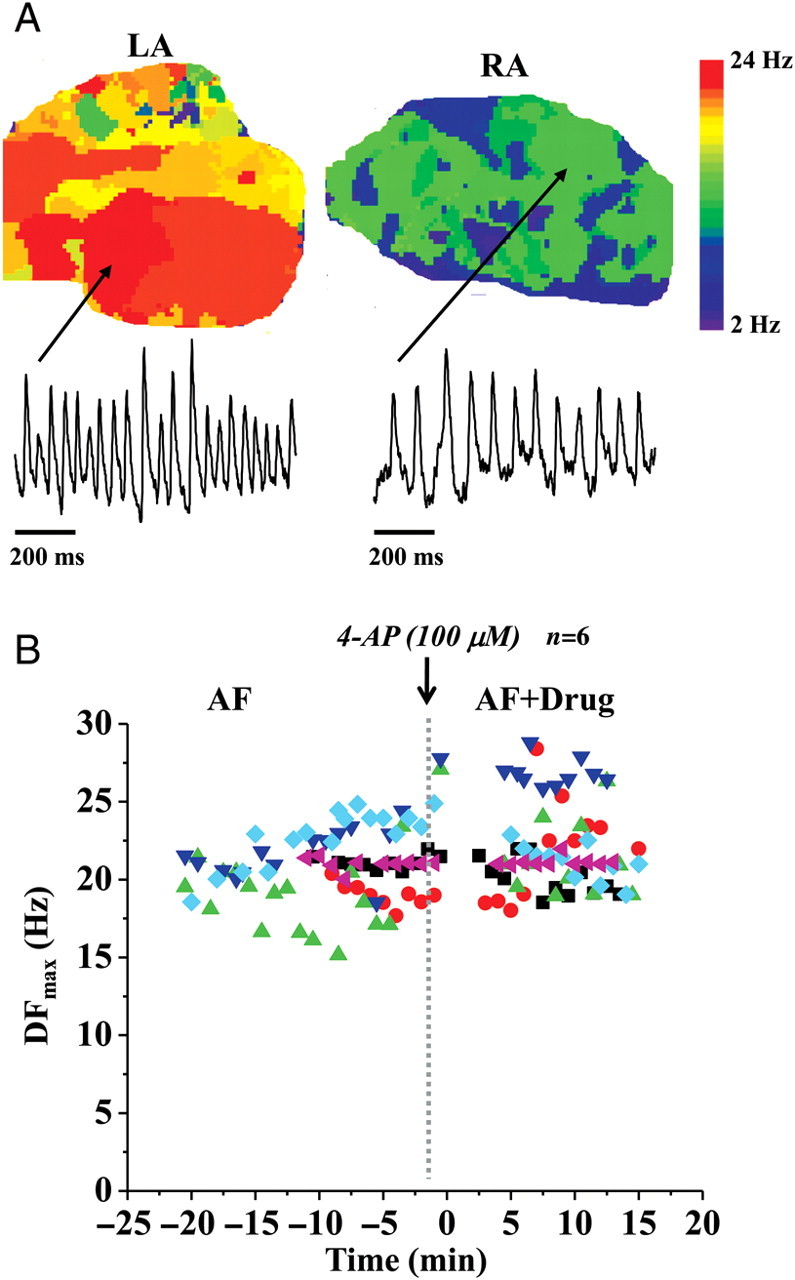
Experiments in acute atrial fibrillation and in the presence of IKur blockade. (A) Representative dominant frequency maps and single-pixel recordings during acute atrial fibrillation in the left atrium and the right atrium. (B) Plot of the maximum dominant frequency (DFmax) in acute atrial fibrillation experiments in pig hearts vs. time. Time before the introduction of the drug to selectively block IKur (100 μM 4-AP) is assigned negative values.
We also tested the effect of DPO-1 on pig cardiac electrophysiology. First, the effect of DPO-1 on APDs at 50 and 75% repolarization (APD50 and APD75) was measured at a pacing cycle length of 300 ms (in the absence of carbachol). Representative atrial (Figure 2A) and ventricular action potentials (Figure 2B) are shown in control (solid) and in the presence of 0.5 μM DPO-1 (dashed), respectively. The average effects of DPO-1 (0.5, 1.0, and 5.0 μM) on APD50,75 in LA (Figure 2C) and LV (Figure 2D) tissues are shown. The results show that at 0.5 μM concentration, the pig atrial APD is significantly prolonged (control: APD75 = 114.95 ± 6.125 ms; DPO-1: APD75 = 136.27 ± 5.24 ms; N = 3, P< 0.05). An additional increase in concentration (1.0 and 5.0 μM) did not increase the atrial APD any further (APD75: 141.53 ± 10.33 and 143.86 ± 8.87 ms, respectively). The ventricular APDs were not significantly altered by DPO-1 (control: APD75 = 203.12 ± 1.95 ms; DPO-1, 0.5 μM: APD75 = 209.43 ± 3.15 ms; DPO-1, 1.0 μM: APD75 = 205.09 ± 2.7 ms; N = 3, P= NS). We then tested the effect of 0.5 μM DPO-1 on carbachol-induced AF (Figure 2E) and VF (Figure 2F); AF was not terminated in 3/4 experiments, whereas VF did not terminate in 4/4 experiments. In the three experiments in which AF did not terminate, DPO-1 (0.5 μM) decreased the DFmax slightly but significantly (control: 23.06 ± 0.72 Hz, n = 62, N = 3; drug DPO-1: 20.61 ± 0.69 Hz, n = 32, N = 3; P< 0.05). There was no difference in VF frequencies (control: 14.11 ± 0.22 Hz, n = 40; drug DPO-1: 13.68 ± 0.12 Hz, n = 81; N = 4; P= NS). In additional experiments, we subjected hearts in AF/VF to higher concentrations of DPO-1, i.e. 1.0 and 5.0 μM (Figure 2G and H). At the highest concentration of DPO-1 (5.0 μM), AF terminated in 4/4 experiments, whereas VF did not. Our analyses show no effect of DPO-1 on atrial conduction velocities at all concentrations (data not shown).Therefore, these results suggest that at higher concentrations, DPO-1 also blocks the acteylcholine-dependent K+ channel, IK,ACh (activated by carbachol).
Figure 2.

Experiments in acute atrial fibrillation and ventricular fibrillation and in the presence of IKur blockade (via DPO-1). (A) Representative pig optical atrial action potential in control (solid) and in the presence of 0.5 μM DPO-1 (dashed) at 300 ms pacing. (B) Representative pig optical ventricular action potential in control (solid) and in the presence of 0.5 μM DPO-1 (dashed) at 300 ms pacing. (C) Average atrial action potential durations at 50% (APD50) and 75% (APD75) repolarization in control and in the presence of 0.5, 1.0, and 5.0 μM DPO-1. (D) Average ventricular action potential durations at 50% (APD50) and 75% (APD75) repolarization in control and in the presence of 0.5 and 1.0 μM DPO-1. (E) Plot of DFmax in acute atrial fibrillation experiments in control and in the presence of 0.5 μM DPO-1. (F) Plot of DFmax during ventricular fibrillation in control and in the presence of 0.5 μM DPO-1. (G) Plot of DFmax in acute atrial fibrillation experiments in control and in the presence of 1.0 and 5.0 μM DPO-1. (H) Plot of DFmax during ventricular fibrillation in control and in the presence of 1.0 and 5.0 μM DPO-1.
In a second series of experiments, hearts in AF were subjected to mild hyperkalaemic conditions, with [K+]o values increased to either 6 or 8 mM, compared with 4 mM (controls). In four of five experiments, AF terminated spontaneously, and in two experiments, AF converted to an atrial tachycardia before termination (data not shown).
To study the effect of hyperkalaemia on cardiac fibrillation more systematically, in our last series of experiments, both AF and VF were induced and mapped simultaneously. The pig hearts were perfused with Tyrode's solution containing varying [K+]o, viz., 4, 6, 8, 10, and 12 mM. Figure 3A presents representative DF maps, along with DFmax values in LA and LV, different values of [K+]o. VF was relatively stable at each individual [K+]o, and the DFmax decreased gradually with increasing [K+]o. The DFmax in AF was relatively stable at 4 and 6 mM [K+]o. However, it decreased rapidly during the 10 min perfusion of 8 mM [K+]o, as illustrated by DF maps in the initial, middle, and final periods at that [K+]o. In addition, AF became more organized, converting to atrial tachycardia (ATach) in the final stage and subsequently terminating during transition from 8 to 10 mM [K+]o. Composite data from all five experiments in which AF and VF were mapped simultaneously at varying [K+]o are quantified in Figure 3B. The rate of decrease in DFmax as a function of [K+]o is seen to be different between AF and VF, with a much steeper slope in the former. Overall, when the hyperkalaemia experiments were pooled together, the value of [K+]o for AF termination was always ≤10 mM (N = 8). In comparison, VF only terminated in two of five cases at 12 mM [K+]o; in three of five cases, VF did not terminate even at 12 mM [K+]o.
Figure 3.
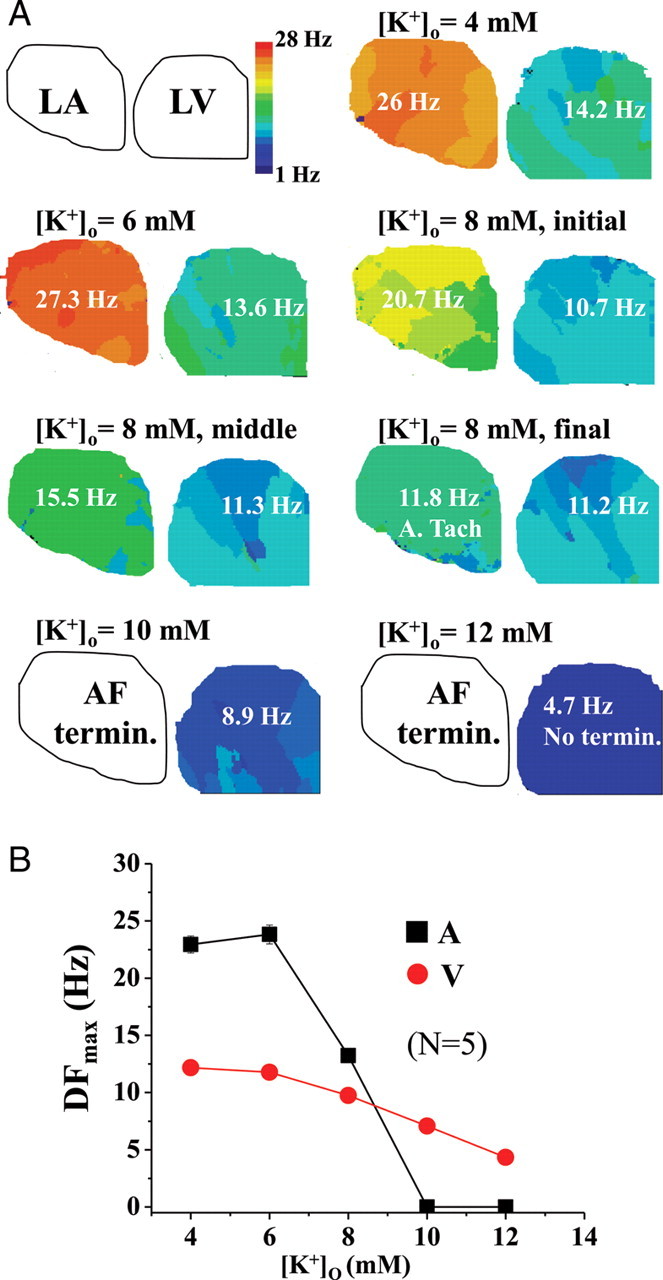
Experiments in acute atrial and ventricular fibrillation simultaneously during hyperkalaemia (6–12 mM [K+]o). (A) Representative plots of the dominant frequency maps in acute atrial (in the left atrium) and ventricular fibrillation (in the left ventricle) in the same heart that was perfused at different concentrations of [K+]o at 4, 6, 8, 10, and 12 mM, for 10 min at each concentration. DFmax of atrial fibrillation changed substantially during perfusion of 8 mM [K+]o; hence, three maps at the initial, middle, and final stages of the 10 min perfusion period are shown. Atrial fibrillation converted to atrial tachycardia (ATach) in the final stages and subsequently terminated during transition from 8 to 10 mM [K+]o. In this experiment, ventricular fibrillation did not terminate even at 12 mM [K+]o. (B) Comparison of average DFmax values during atrial and ventricular fibrillation at different concentrations of [K+]o.
3.2. Numerical
For simulations, IKur inactivation in the human atrial model16 was re-formulated to include a weighted inactivation of fast and slow gating variables as follows:
| (1) |
where  (ns/pF) is the maximal channel conductivity;
(ns/pF) is the maximal channel conductivity; 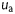 ,
, 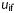 , and
, and 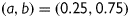 the activation and fast and slow inactivation gating variables, respectively; a and b the weighting coefficients; V (mV) the transmembrane voltage; and EK the potassium reversal potential. Classification of inactivation into fast and slow components was performed to reproduce the bi-exponential recovery of IKur that was observed in porcine atrial cells.15 The following set of equations and parameters was used for the calculation of Eq. (1):
the activation and fast and slow inactivation gating variables, respectively; a and b the weighting coefficients; V (mV) the transmembrane voltage; and EK the potassium reversal potential. Classification of inactivation into fast and slow components was performed to reproduce the bi-exponential recovery of IKur that was observed in porcine atrial cells.15 The following set of equations and parameters was used for the calculation of Eq. (1):
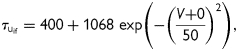 |
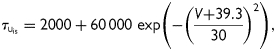 |
(2) |
and 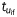 .
.
Figure 4A shows the fast and slow inactivation rate constants as a function of voltage. The dashed line represents the calculated values as per Eq. (2), and the filled circles show experimental data.13 The rate dependence of the normalized IKur is shown in Figure 4B, using a pacing protocol adapted from Feng et al.9 (their Figure 5). We employed 50 ms long pacing pulses rather than the 100 or 200 ms pulses that were originally used to account for the shorter APD in swine atrial myocytes compared with human cells.11 An exponential decay in the relative current (i.e. current magnitude following the test pulse divided by the maximal current magnitude at the lowest frequency of 0.1 Hz) is demonstrated, with the current magnitude decreasing to about 10% at 19 Hz compared with its 0.1 Hz pacing value. Additional modifications that were employed to adapt the human atrial ionic model to swine kinetics were the inclusion of a more detailed calcium handling,18 the modification of IK1 conductance by [K+]o,19 and the replacement of the transient outward current, Ito, by a calcium-activated Ito2 that was observed in pigs.10 The resulting characteristic action potential of the adapted ionic kinetics model is shown in Figure 4C for steady-state 1 Hz pacing in normal (continuous) and blocked IKur (dotted) conditions, exhibiting resemblance to measured swine action potentials in cellular experiments.11
Figure 4.

New numerical formulation of IKur. (A) Plots of model time constants for the fast and slow time constants of the inactivation for IKur in the human atrial mathematical model (Courtemanche), adjusted according to data for inactivation in the pig atrium. (B) This plot shows the frequency dependence of IKur; this current is reduced in magnitude in response to pulses at higher frequencies. (C) Plot of the model action potential at 1 Hz under control conditions (to mimic the pig atrial action potential) and the plot of the action potential when IKur was completely blocked at 1 Hz.
Figure 5.
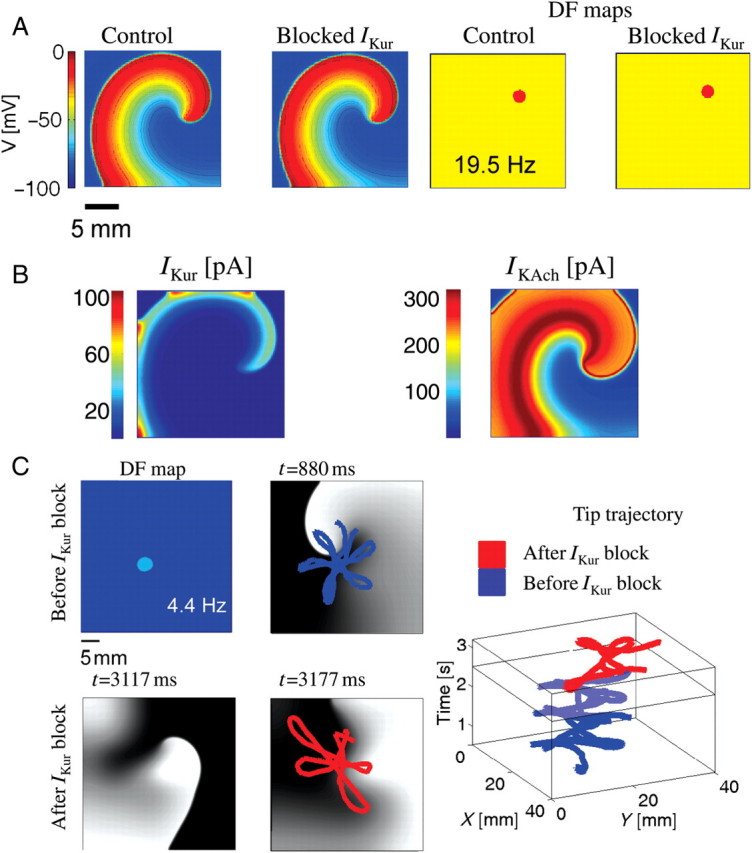
Simulations of IKur block on rotor. (A) Left two panels depict representative snapshots of a rotor under control conditions, and when IKur was blocked completely, in carbachol-induced atrial fibrillation; right panels show respective dominant frequency maps. (B) Snapshot of spatial distribution of IKur and IK,ACh currents in a rotor during atrial fibrillation. (C) Top two left panels depict a dominant frequency map and a rotor snapshot when atrial fibrillation was induced in the absence of carbachol. The rotor tip trajectory before IKur block is plotted in blue. Bottom two left panels depict rotor snapshots in simulated atrial fibrillation (in the absence of IK,ACh) when IKur was blocked. The two snapshots are given at t = 3117 ms and just before termination of re-entry at t = 3177 ms. The rightmost panel depicts rotor tip meander before and after IKur block.
A simulated re-entry was initiated via cross-field stimulation, in which two appropriately timed line stimuli (S1 and S2) were given from adjacent edges of a two-dimensional sheet, such that S2 was perpendicular to S1, and the coupling interval between the two stimuli allowed for the interaction between the wave front of S2 and the recovering tail of S1 to generate a rotor.13 The simulations were run for 5 s The leftmost panels in Figure 5A show a snapshot of a rotor (at a particular time) in terms of the absolute membrane voltage in controls and when IKur was blocked. The DF maps for these rotors are shown in the two right panels of Figure 5A; the rotor frequencies were similar in both conditions (19.5 Hz). To understand whether the IKur blockade had no effect on rotational frequency due to its slow recovery kinetics, we performed an additional numerical experiment, in which IKur fast and slow inactivation time constants (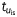 and
and  ) were made very similar; however, this also did not affect the rotor frequency when IKur was blocked during carbachol-induced AF (data not shown). We then compared the magnitude of IKur during re-entry with the largest active K+ conductance, i.e. IK,ACh. Figure 5B shows the distribution of IKur (left panel) and IK,ACh (right panel) underlying the rotor. IK,ACh is approximately seven times larger in magnitude than IKur in the rotors. These simulations suggest that IK,ACh masks any effect of IKur changes and thus have minimal effect on rotor frequency in carbachol-induced AF. However, to test whether the IKur block is effective in terminating re-entry initiated in the absence of an active IK,ACh, we conducted additional simulations of re-entry in a 4×4 cm tissue (Figure 5C). A stable re-entry (with a large core size of ∼2 × 2 cm) and a DF = 4.4 Hz were observed (top two left panels; Figure 5C). At 2.5 s post-stimulation, IKur was blocked, resulting in the annihilation of the rotor at the boundary at 3177 ms (i.e. after 677 ms). Two snapshots of rotors during IKur block (at t = 3117 ms) and annihilation (at t = 3177 ms) are shown (bottom two left panels; Figure 5C). The right panel depicts the spiral wave trajectory before and after IKur block (Figure 5C, right panel). Thus, in the absence of IK,ACh, at a re-entry frequency of ∼5 Hz, the block of IKur terminates re-entry.
) were made very similar; however, this also did not affect the rotor frequency when IKur was blocked during carbachol-induced AF (data not shown). We then compared the magnitude of IKur during re-entry with the largest active K+ conductance, i.e. IK,ACh. Figure 5B shows the distribution of IKur (left panel) and IK,ACh (right panel) underlying the rotor. IK,ACh is approximately seven times larger in magnitude than IKur in the rotors. These simulations suggest that IK,ACh masks any effect of IKur changes and thus have minimal effect on rotor frequency in carbachol-induced AF. However, to test whether the IKur block is effective in terminating re-entry initiated in the absence of an active IK,ACh, we conducted additional simulations of re-entry in a 4×4 cm tissue (Figure 5C). A stable re-entry (with a large core size of ∼2 × 2 cm) and a DF = 4.4 Hz were observed (top two left panels; Figure 5C). At 2.5 s post-stimulation, IKur was blocked, resulting in the annihilation of the rotor at the boundary at 3177 ms (i.e. after 677 ms). Two snapshots of rotors during IKur block (at t = 3117 ms) and annihilation (at t = 3177 ms) are shown (bottom two left panels; Figure 5C). The right panel depicts the spiral wave trajectory before and after IKur block (Figure 5C, right panel). Thus, in the absence of IK,ACh, at a re-entry frequency of ∼5 Hz, the block of IKur terminates re-entry.
We conducted additional simulations to study re-entry dynamics during hyperkalaemia. We hypothesized that increased levels of [K+]o may terminate re-entry activity via reduced sodium availability.14 The left panel in Figure 6A shows the voltage dependence of the steady-state sodium inactivation (i.e. the product of h and j gates). We incorporated a left-shifted inactivation curve in the atrium compared with the ventricle, as was shown experimentally in dogs.7 (Data are currently unavailable pertaining to INa differences between pig atrial and ventricular cells.) As [K+]o increases, the resting membrane potential will be depolarized according to the Nernst equation. Due to the differences in inactivation curves between the atrium and the ventricle, the availability of sodium channels (hj) will therefore be significantly decreased in the atrium as [K+]o increases, compared with the ventricle, as shown in Figure 6A, right panel. In Figure 6B, we simulated the effects of hyperkalaemia on AF re-entry in a 3 × 3 cm2 atrial tissue model. We investigated the consequences of increasing [K+]o from 5 to 5.5, 6.0, and 6.2 mM every 1000 ms on rotor stability (four snapshots in Figure 6B, left panels). We noticed that as [K+]o was increased, the meandering of the spiral wave became more significant, increasing the core size. When [K+]o reached a level of 6.2 mM, the spiral wave drifted rapidly to the boundary and annihilated. This process is demonstrated by the time-space plot of the spiral tip location that is given in the right panel of Figure 6B. For reference, we repeated these simulations with the sodium inactivation curve relevant to ventricular tissue, for both the same tissue size of 3 × 3 cm2 (Figure 6C, upper panels) and an enlarged tissue of 5 × 5 cm2 (Figure 6C, lower panels). In both simulations, the tissue sustained spiral wave activity up to much higher [K+]o levels than those with the left-shifted sodium inactivation curve of the atrium. In the 3 × 3 and 5 × 5 cm2 tissue models, simulated re-entrant activity was terminated when [K+]o was higher than 11.0 and 11.2 mM, respectively, by the rotor drifting onto the boundary.
Figure 6.
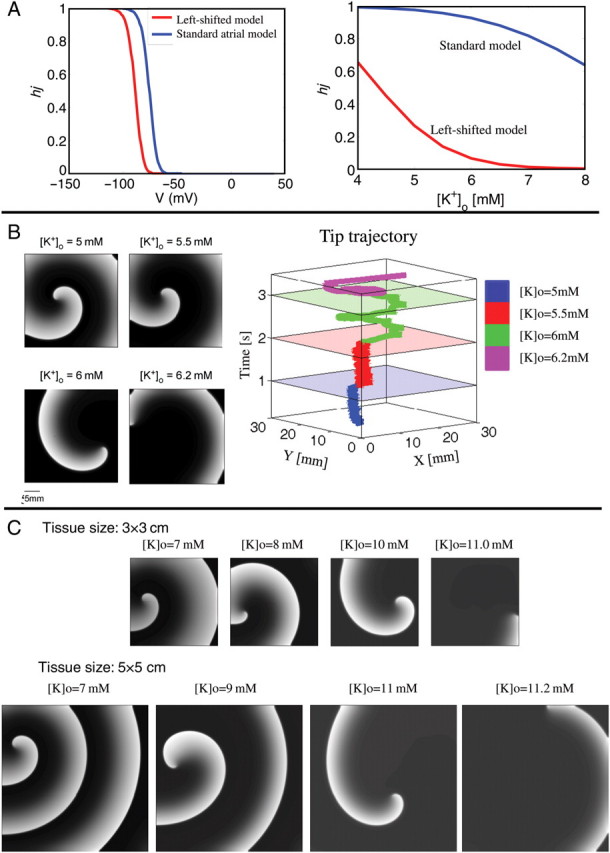
Effect of hyperkalaemia on simulated rotor. (A) The left panel depicts differences between the steady-state inactivation properties of INa in the atrium (red) and ventricle (blue). The inactivation is measured as the product of the fast (h) and the slow (j) sodium channel inactivation gates: hj. The right panel depicts how steady-state inactivation properties of INa vary as a function of increases in [K+]o. (B) The left panels show snapshots of the simulated rotor (with atrial model steady-state Na+ inactivation) at different values of [K+]o. The right panel depicts the trajectory of the rotor tip at different values of [K+]o. The rotor is seen to terminate at 6.2 mM [K+]o. (C) The top panel shows snapshots of the simulated rotor in a 3 × 3 cm tissue (with ventricular model steady-state Na+ inactivation) at different values of [K+]o. The rotor is seen to terminate at 10.6 mM [K+]o. The bottom panel depicts snapshots of the simulated rotor, but now in a 5 × 5 cm tissue (with ventricular INa) at different values of [K+]o. The rotor is now seen to terminate at 11.4 mM [K+]o.
4. Discussion
The main new findings from this study are as follows. (i) The IKur block is not able to terminate carbachol-induced AF in isolated pig hearts. Simulations suggest that this ineffectiveness is mainly due to the smaller contribution of IKur to the net repolarizing current in the presence of a large IK,Ach. (ii) Elevated [K+]o terminates AF more readily and effectively than VF. Numerical simulations indicate that this may be partly due to the steady-state inactivation property differences of INa between the atrium and the ventricle and the larger tissue size of the ventricle.
4.1. Is IKur an effective anti-arrhythmic target in atrial fibrillation?
IKur is an attractive target for anti-arrhythmic drug development primarily due to its presence in the atrium and not in the ventricle in humans.2,3 However, some doubts exist regarding the efficacy of blocking IKur to terminate AF due to its frequency-dependent reduction in magnitude.9–11 Furthermore, IKur may be reduced under chronic AF conditions in humans.13 Despite this, in a chronic AF model in goats, AVE0118, a relatively selective blocker of IKur, was successful in cardioverting AF.5 The IKur block also terminated re-entry under simulated chronic AF conditions.13 In contrast, in a canine model of cholinergic AF, blocking IKur with 4-AP did not terminate AF.6 Instead, IKur blockade was pro-arrhythmic.6 Our experimental results in pigs are consistent with this study.6 Furthermore, our simulation results mimic the experimental findings. Therefore, our working hypothesis is that, at least in acute or paroxysmal models of AF, at frequencies >10 Hz, IKur is unlikely to be a useful drug target. However, our current (Figure 5C) and previous simulation results13 also suggest that the IKur block can terminate re-entry at slower re-entry frequencies of 4–6 Hz. Additional experiments in chronic AF with specific IKur blockade (4-AP and DPO-1) will be required to test this hypothesis, since AVE0118 is not a very specific blocker of IKur, blocking additionally Ito and IK,ACh.20
4.2. Hyperkalaemia, preferential termination of atrial fibrillation, and its implications.
Our study also demonstrates that hyperkalaemia superimposed upon fibrillation (AF or VF) tends to be anti-arrhythmic. Much smaller changes in [K+]o are needed to terminate AF when compared with VF. This potentially relates to the differences in the biophysical properties of INa availability between the atrium and the ventricle7 and also partially explains the well-known, but poorly understood, sensitivity of the atrium to hyperkalaemia.21 An interesting aspect to note in this regard is that at ∼8 mM [K+]o where AF seems to terminate, the normal ventricular excitability is expected to be increased, from the well-known biphasic changes in conduction velocity and excitability as a function of [K+]o.22 This has been attributed to the decreased difference between the resting membrane potential and the threshold membrane potential.18 Previous in vivo experiments in dogs showed that both atrial and ventricular tissues demonstrated the biphasic response.21 However, the threshold value for [K+]o at which the intra-ventricular conduction time started to reduce with respect to its own controls was much higher (9.3 mM) than a similar threshold for intra-atrial conduction times (7.5 mM). Simultaneous ECG recordings in that study suggested that the QRS and P wave durations mirrored the changes in the intra-ventricular and intra-atrial conduction times, respectively.21 Thus it is possible to envision a scenario that in a heart with normal ventricular function, small increases in [K+]o may not be pro-arrhythmic and thus be safe for terminating AF. However, whether this will also apply in cases of abnormal ventricular function in hearts with an infarct or even pump failure remains unknown. The use of hyperkalaemia to terminate AF in our experiments demonstrates a ‘proof-of-principle’ experiment that exploits the inherent differences in the Na+ channel availability between the atria and the ventricles to attempt and terminate AF. It raises the intriguing question as to whether it might be possible to design a drug that acts by depolarizing the membrane potential or affects INa channel availability for facilitating the termination of AF, but without compromising safety in the ventricle (i.e. being pro-arrhythmic).
4.3. Limitations
In our Langendorff-perfused experiments, we have used the motion uncoupler Blebbistatin; the use of this drug may adversely affect ion channels in the atrium. In our experiments, the heart was not superfused with Tyrode's solution. This will inscribe some temperature gradients across the surface of the heart. Our results suggest that DPO-1 in addition to blocking IKur at lower concentrations (0.5 μM) may also block IK,ACh (>1μM). Additional patch-clamp experiments will be necessary to systematically determine the IK,ACh blocking properties of DPO-1, including its EC50. We have not yet directly investigated the ionic and molecular differences in INa between the atrium and the ventricle in the pig, including the steady-state inactivation properties in isolated cells. In our computer simulations, we have only used a modified human atrial model (not a detailed pig atrial or ventricular ionic model) and also studied the dynamics of a single rotor, without considering the multiple wavelet theory, or the complex three-dimensional structure of the atria.1 Additionally, to mimic differences between AF and VF seen experimentally, we have only changed the properties of the Na+ current and tissue size. Clearly, many other differences in ion channels, including the inward rectifier K+ current, IK1, exist between the atrium and the ventricle23 and will need to be more accurately modelled to quantitatively reproduce experimental data.
Funding
This study was supported by National Heart Lung and Blood Institute (NHLBI) Grants P01-HL039707, P01-HL070074, and RO1-HL080159 (J.J.); American Heart Association post-doctoral fellowship (S.F.N.); and Scientist Development Grant (S.V.P.).
Acknowledgements
We thank Brian Hooven and Elliot Hwang for their help with some experiments.
Conflict of interest: none declared.
References
- 1.Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–226. doi: 10.1038/415219a. doi:10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- 2.Page RL, Roden DM. Drug therapy for atrial fibrillation: where do we go from here? Nat Rev Drug Discov. 2005;4:899–910. doi: 10.1038/nrd1876. doi:10.1038/nrd1876. [DOI] [PubMed] [Google Scholar]
- 3.Ehrlich JR, Nattel S. Atrial-selective pharmacological therapy for atrial fibrillation: hype or hope? Curr Opin Cardiol. 2009;24:50–55. doi: 10.1097/HCO.0b013e32831bc336. doi:10.1097/HCO.0b013e32831bc336. [DOI] [PubMed] [Google Scholar]
- 4.Gaborit N, Le Bouter S, Szuts V, Varro A, Escande D, Nattel S, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol. 2007;582:675–693. doi: 10.1113/jphysiol.2006.126714. doi:10.1113/jphysiol.2006.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaauw Y, Gögelein H, Tieleman RG, van Hunnik A, Schotten U, Allessie MA. Early' class III drugs for the treatment of atrial fibrillation: efficacy and atrial selectivity of AVE0118 in remodeled atria of the goat. Circulation. 2004;110:1717–1724. doi: 10.1161/01.CIR.0000143050.22291.2E. doi:10.1161/01.CIR.0000143050.22291.2E. [DOI] [PubMed] [Google Scholar]
- 6.Burashnikov A, Antzelevitch C. Can inhibition of IKur promote atrial fibrillation? Heart Rhythm. 2008;5:1304–1309. doi: 10.1016/j.hrthm.2008.05.020. doi:10.1016/j.hrthm.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burashnikov A, Di Diego JM, Zygmunt AC, Belardinelli L, Antzelevitch C. Atrium-selective sodium channel block as a strategy for suppression of atrial fibrillation: differences in sodium channel inactivation between atria and ventricles and the role of ranolazine. Circulation. 2007;116:1449–1457. doi: 10.1161/CIRCULATIONAHA.107.704890. doi:10.1161/CIRCULATIONAHA.107.704890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antzelevitch C, Burashnikov A. Atrial-selective sodium channel block as a novel strategy for the management of atrial fibrillation. J Electrocardiol. 2009;42:543–548. doi: 10.1016/j.jelectrocard.2009.07.007. doi:10.1016/j.jelectrocard.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng J, Xu D, Wang Z, Nattel S. Ultrarapid delayed rectifier current inactivation in human atrial myocytes: properties and consequences. Am J Physiol. 1998;275:H1717–H1725. doi: 10.1152/ajpheart.1998.275.5.H1717. [DOI] [PubMed] [Google Scholar]
- 10.Li GR, Sun H, To J, Tse HF, Lau CP. Demonstration of calcium-activated transient outward chloride current and delayed rectifier potassium currents in Swine atrial myocytes. J Mol Cell Cardiol. 2004;36:495–504. doi: 10.1016/j.yjmcc.2004.01.005. doi:10.1016/j.yjmcc.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Ehrlich JR, Hoche C, Coutu P, Metz-Weidmann C, Dittrich W, Hohnloser SH, et al. Properties of a time-dependent potassium current in pig atrium: evidence for a role of kv1.5 in repolarization. J Pharmacol Exp Ther. 2006;319:898–906. doi: 10.1124/jpet.106.110080. doi:10.1124/jpet.106.110080. [DOI] [PubMed] [Google Scholar]
- 12.Courtemanche M, Ramirez RJ, Nattel S. Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am J Physiol. 1998;275:H301–H321. doi: 10.1152/ajpheart.1998.275.1.H301. [DOI] [PubMed] [Google Scholar]
- 13.Pandit SV, Berenfeld O, Anumonwo JM, Zaritski RM, Kneller J, Nattel S, et al. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J. 2005;88:3806–3821. doi: 10.1529/biophysj.105.060459. doi:10.1529/biophysj.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pandit SV, Warren M, Mironov S, Tolkacheva EG, Kalifa J, Berenfeld O, et al. Mechanisms underlying the antifibrillatory action of hyperkalemia in Guinea pig hearts. Biophys J. 2010;98:2091–2101. doi: 10.1016/j.bpj.2010.02.011. doi:10.1016/j.bpj.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lagrutta A, Wang J, Fermini B, Salata JJ. Novel, potent inhibitors of human Kv1.5 K+ channels and ultrarapidly activating delayed rectifier potassium current. J Pharmacol Exp Ther. 2006;317:1054–1063. doi: 10.1124/jpet.106.101162. doi:10.1124/jpet.106.101162. [DOI] [PubMed] [Google Scholar]
- 16.Jalife J, Pandit SV. Ionic mechanisms of wavebreak in fibrillation. Heart Rhythm. 2005;2:660–663. doi: 10.1016/j.hrthm.2005.03.016. doi:10.1016/j.hrthm.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 17.Sarmast F, Kolli A, Zaitsev A, Parisian K, Dhamoon AS, Guha PK, et al. Cholinergic atrial fibrillation: I(K,ACh) gradients determine unequal left/right atrial frequencies and rotor dynamics. Cardiovasc Res. 2003;59:863–873. doi: 10.1016/s0008-6363(03)00540-6. doi:10.1016/S0008-6363(03)00540-6. [DOI] [PubMed] [Google Scholar]
- 18.Viswanathan PC, Shaw RM, Rudy Y. Effects of IKr and IKs heterogeneity on action potential duration and its rate dependence: a simulation study. Circulation. 1999;99:2466–2474. doi: 10.1161/01.cir.99.18.2466. [DOI] [PubMed] [Google Scholar]
- 19.Nygren A, Fiset C, Firek L, Clark JW, Lindblad DS, Clark RB, et al. Mathematical model of an adult human atrial cell: the role of K+ currents in repolarization. Circ Res. 1998;82:63–81. doi: 10.1161/01.res.82.1.63. [DOI] [PubMed] [Google Scholar]
- 20.Gögelein H, Brendel J, Steinmeyer K, Strübing C, Picard N, Rampe D, et al. Effects of the atrial antiarrhythmic drug AVE0118 on cardiac ion channels. Naunyn Schmiedebergs Arch Pharmacol. 2004;370:183–192. doi: 10.1007/s00210-004-0957-y. [DOI] [PubMed] [Google Scholar]
- 21.Arnsdorf MF, Schreiner E, Gambetta M, Friedlander I, Childers RW. Electrophysiological changes in the canine atrium and ventricle during progressive hyperkalaemia: electrocardiographical correlates and the in vivo validation of in vitro predictions. Cardiovasc Res. 1977;11:409–418. doi: 10.1093/cvr/11.5.409. doi:10.1093/cvr/11.5.409. [DOI] [PubMed] [Google Scholar]
- 22.Buchanan JW, Gettes L. Ionic environment and propagation. In: Zipes DP,, Jalife J, editors. Cardiac Electrophysiology: From Cell to Bedside. Philadelphia, PA: W.B. Saunders Company; 1990. pp. 149–156. [Google Scholar]
- 23.Dhamoon AS, Pandit SV, Sarmast F, Parisian KR, Guha P, Li Y, et al. Unique Kir2.x properties determine regional and species differences in the cardiac inward rectifier K+ current. Circ Res. 2004;94:1332–1339. doi: 10.1161/01.RES.0000128408.66946.67. doi:10.1161/01.RES.0000128408.66946.67. [DOI] [PubMed] [Google Scholar]