

Abstract
Over the past two decades, potent and selective analgesics have been developed from endogenous opioid peptides. Glycosylation provides an important means of modulating interaction with biological membranes, which greatly affects the pharmacodynamics and pharmacokinetics of the resulting glycopeptide analogues. Furthermore, manipulation of the membrane affinity allows penetration of cellular barriers that block efficient drug distribution, including the blood–brain barrier. Extremely potent and selective opiate agonists have been developed from endogenous peptides, some of which show great promise as drug candidates.
The success rate of CNS drug development is lower than that of other therapeutic areas [1]. There are multiple reasons for this dearth of new drugs, including the sheer complexity of the brain and its neuropathologies; a propensity for CNS drugs to cause CNS-mediated side effects that limit dosing and compliance; a lack of validated biomarkers to inform whether a given neurotherapeutic agent is engaging the target in sufficient concentrations to modulate the CNS target; and the presence of the blood–brain barrier (BBB), across which CNS agents need to penetrate. Among these challenges, the BBB is considered to be most problematic for peptide or protein-based therapies [2–4]. However, peptides and proteins do offer distinct advantages for developing efficacious and well-tolerated treatments for CNS diseases, such as chronic pain, Alzheimer's disease and Parkinson's disease. These advantages include intrinsic affinity and selectivity of the peptide for native receptors and the metabolism of the peptide into smaller fragments and amino acids (versus the variety of active metabolites seen with small molecules). In addition, peptides and proteins can bind to multiple sites across a receptor protein, offering greater opportunity to fine tune the receptor–effector response [5,6]. Historical challenges for peptide and protein drug development are being addressed, including modifications that increase stability, techniques that increase yields and lower total synthetic costs, and technology to improve tissue targeting, including access to the CNS. A number of monographs have been written since the beginning of this century that describe formulations and articulate solutions of increasing sophistication to address the problems of peptide-based drugs [7–9].
Opioid receptors
The classical opioid receptors [10] are divided into three subtypes, the μ receptor (MOR or MOP), the δ receptor (DOR or DOP) and the κ receptor (KOR or KOP). Another receptor, the nociceptin or orphanin receptor (NOP or ORL1), is widely distributed in the CNS, and is clearly related to the opioid receptors in terms of its molecular biology but is generally not regarded as an opioid receptor as it does not respond to classical opioid agonists or antagonists. IUPHAR recommends use of the terminology MOP, DOP, KOP and NOP, replacing the older recommendation for OP1 and OP2, for example [201]. All of the opioid receptors have been cloned from various species, including mouse, rat and human. Opioid receptors are GPCRs that consist of highly homologous seven-transmembrane helical domains, and are linked with extracellular peptide loops of very limited size (Figure 1) [11,12].
Figure 1. Opioid receptors.

The μ-opioid receptor G-protein-coupled receptor derived from bovine rhodopsin by homology modeling [12].
The MOP receptor [13] is distributed presynaptically in various brain regions, including the limbic structures, the brainstem (i.e., the periaqueductal grey area) and in the superficial dorsal horn of the spinal cord. It was initially characterized in functional smooth muscle preparations of the guinea pig ileum (GPI). This receptor remains the principal target of opioid analgesics currently in clinical use, with the μ referring to morphine. The MOP receptor is widely distributed in other areas of the brain, as well as non-CNS tissues, most relevantly in the immune system, and the cardiovascular and gastrointestinal systems. Pharmacological activation of MOP not only modifies the transmission and perception of nociceptive stimuli, but also a host of other effects, including reduced respiratory drive in response to increased levels of CO2, opioid-induced bowel dysfunction, abuse liability and pruritus (itching). Repeated or prolonged MOP activation results in adaptations that manifest as tolerance and physical dependence, further complicating management of chronic pain sufferers and patients with substance abuse disorders. Mixed opioid agonists/ antagonists and partial MOP agonists have been used to limit some of the opioid side effects with a mixed degree of success. Clearly, opioid pharmacology is a complex issue [14].
The DOP receptor [15] was originally characterized using the mouse vas deferens smooth muscle tissue preparation (mouse vas deferens [MVD] assay) [16]. The receptor is widely distributed anatomically, being found in the same general anatomical areas as MOP. DOP also contributes to a variety of physical and emotional effects, including initiation of movement [17], regulation of pain and reward circuitry, as well as other complex CNS behaviors (mood/ affect and anxiety). The issues of cellular co-localization of DOP and MOP, as well as the formation of functional ‘hetero dimers’ continue to be the subject of considerable interest [18–20]. Despite the progress that has been made in the localization of opioid receptors [21], the precise localization and neuronal and cellular pathways through which these three receptor types work remains incompletely understood [22,23]. Although DOP receptors contribute to analgesia, euphoria and physical dependence, DOP agonists may be able to produce broad spectrum analgesic efficacy with reduced propensity to produce classic opioid side effects [24].
The KOP receptor, named after the κ-agonist ketocyclazocine, is not as well studied as MOP and DOP, although it has attracted interest from pharmaceutical companies, as either a stand-alone drug [25,26] or in conjunction with agonism of other opioid receptors [27]. Stimulation of CNS KOP receptors is generally associated with dysphoria and psychomimetic effects, along with limited analgesic efficacy, especially in men [28]. Peripherally active KOP agonists produce antinociception in animal models of pain [29] and are being explored as targets for analgesia in several chronic pain states [18,30,31]. KOP antagonists are also being investigated as treatments for addiction and depression [32].
The diversity of the endogenous neuropep-tides and their receptors provide many opportunities for drug discovery [33]. If there were a straightforward methodology for converting endogenous neuropeptides into useful CNS drugs, then a new and potentially sea-changing pharmacopeia would be available for the treatment of CNS disorders.
Endogenous opioid peptide agonists
Since the discovery of the two endogenous pentapeptides, Met-enkephalin and Leu-enkephalin in the 1970s, perhaps as many as 300 endogenous neuropeptides have been identified in widespread locations throughout the CNS Table 1. Endogenous opioid peptides and their receptors undergo modulation in response to various physiological conditions, such as inflammation, tissue injury, pain and other stressors [34].
Table 1.
Endogenous opioid peptides†.
| Peptide | Sequence | Subtype |
|---|---|---|
| Enkephalins | ||
| Leu-enkephalin | YGGFL | δ receptor/μ receptor |
| Met-enkephalin | YGGFM | μ receptor/δ receptor |
| Metorphamide | YGGFMRRV-NH2 | δ receptor/μ receptor |
| Peptide E | YGGFMRRVGRPEWWMDYQKRYGGFL25 | μ receptor/κ receptor |
| Endorphins | ||
| β-endorphin | YGGFMTSEKSQTPLVTLFKNAIIKNAYKKGE31 | μ receptor/δ receptor |
| γ-endorphin | YGGFMTSEKSQTPLVTL17 | μ receptor/unknown |
| α-endorphin | YGGFMTSEKSQTPLVT16 | μ receptor/unknown |
| Dynorphins | ||
| Dynorphin A | YGGFLRRIRPKLKWDNQ17 | κ receptor (μ receptor) |
| Dynorphin B | YGGFLRRQFKVVT13 | κ receptor (μ receptor, δ receptor) |
| Dynorphin1–8 | YGGFLRRQ8 | κ receptor (μ receptor, δ receptor) |
| α-neoendorphin | YGGFLRKYPK | κ receptor (μ receptor, δ receptor) |
| β-neoendorphin | YGGFLRKYP | κ receptor (μ receptor, δ receptor) |
| Nociceptin/orphanin FQ | ||
| Nociceptin | FGGFTGARKSARKLANQ | ORL1 |
| Endomorphins | ||
| Endomorphin-1 | YPWF-NH2 | μ receptor |
| Endomorphin-2 | YPFF-NH2 | μ receptor |
| Dermal peptides | ||
| Dermorphin | YaFGYPS-NH2 | μ receptor |
| Deltorphin A | YmFHLMD | δ receptor |
| Deltorphin C | YaFDVVG-NH2 | δ receptor |
A few of the opioid peptides isolated from the mammalian CNS and from the skin of amphibians are illustrated. Their common names, sequences and suggested receptor binding activities are shown. Weaker binding activities are in parentheses.
The three classes of endogenous opioid peptides (enkephalins, endorphins or dynorphins) are typically assigned to the three types of opioid receptors (DOP, MOP and KOP, respectively). This approach is misleading since the absolute selectivity of each peptide is limited, and it neglects the fact that there are many cleavage variants of the neuropeptides, splice variants for the receptors and, likely, variations in their glycoforms. The endogenous peptides are not orthogonal and neuropeptide receptors might, for example, just as easily be thought of as ‘metorphamide receptors’ or ‘enkephalin receptors’ [35]. Moreover, α-endorphin and γ-endorphin have been found to be inactive at sites that are sensitive to β-endorphin [36] and other sites have been found that are sensitive to γ-endorphin, which have been referred to as ‘non-opioid’ in nature [37,38].
Schwyzer's membrane compartment concepts
Schwyzer articulated critical roles for the membrane in peptide–receptor binding events (Figure 2) [39]. Although his ‘membrane compartment theory’ may have overstated the influence of the membrane in differentiating receptor selectivity (μ vs δ vs κ), it is clear that the membrane environment does play critical roles in pre-organizing the peptide conformation prior to binding, as well as the peptide–receptor binding event itself [40]. We view binding as a three-step process:
■ Adsorption of the peptide ligand to the membrane. This promotes receptor binding by reducing a 3D search for the receptor to a faster 2D search. Surface-assisted ‘reduction-of-dimensionality’ calculations, performed by Polya in 1921, were examined by Max Delbrück in which he quantitatively demonstrated the viability of this theory [41,42];
■ Conformational changes in the peptide induced by the asymmetric environment of the membrane. Amphipathicity of the peptide is believed to reorganize the peptide–membrane aggregates into minimal energy states [43,44];
■ The binding event itself. GPCR binding and activation is a complex, multifaceted phenomenon. It is, however, beyond the scope of this review.
Figure 2. Membrane hopping.

Endogenous opioids associate with the membranes (kon >> koff) and bind to one or more of the opioid receptors via a membrane-bound conformation (Fisher's lock and key). Studies show that active (folded) conformations are favored in the membrane and inactive (random coil) conformations are favored in the absence of a membrane. Incorporation of glycosides, represented by the 270° arc, can shift the kon/koff equilibrium to facilitate ‘membrane hopping’.
The neurovascular unit & the BBB
In order to enter the brain and CNS, pharmaceutical agents must first penetrate the BBB (Figure 3). There are several strategies available to make a peptide metabolically stable but the transport of a peptide across BBB is still remains a major hurdle in developing CNS drugs [45]. Contrary to the belief that small molecules with molecular weights under 400 readily cross the BBB, it has been observed that almost 98% of small molecules do not readily cross the BBB. Further, evidence has accumulated that peptides can penetrate the CNS by several different mechanisms [46–49]. These observations clearly indicate molecule size is not the primary factor, but rather the overall physiochemical characteristics of the molecule is critical for BBB transport. Since the BBB is composed of endothelial membranes, many research groups focus on designing peptides with increased lipid solubility. Simply increasing the lipid solubility of a drug molecule may have undesirable effects, such as decreasing solubility and bioavailability, and increasing plasma protein binding.
Figure 3. The neurovascular unit.

The neurovascular unit forms the blood–brain barrier that prevents the passage of most peptides and other polar substances from the capillaries into the brain.
AE: Astrocyte endprocess; BL: Basolateral membrane; EC: Endothelial cell; NU: Neutrophil; P: Pericyte; TJ: Tight junction.
The BBB is composed of endothelial membranes that function as a continuous lipid barrier that protects the brain from toxic substances by preventing their entrance from bloodside to CNS. The BBB is also an enzymatic barrier that poses additional challenges in developing peptide/protein-based CNS drugs. Further, it has been viewed recently as a regulatory interface between the CNS and circulation with nutritional, homeostatic and communication functions. Understanding of the principles and physiology of BBB has improved a great deal in the past decade. Moreover, evidence is accumulating that many peptides and proteins cross the BBB in amounts sufficient to affect CNS function. It is now clear that the BBB is not an absolute physical barrier but a regulatory tool that controls the delivery of the substances to the CNS. Strategies based on this principle are proving to be very successful [50]. Other strategies using ‘molecular umbrellas’ [51], ‘Trojan horses’ [52] and BBB ‘shuttles’ [53] have been proposed by various groups. We have successfully applied glycosylation as a strategy to improve the BBB penetration, as well as the stability and systemic availability of enkephalins and the larger endorphin-like peptides.
Glycopeptide synthesis
Initially, O-linked glycopeptides were considered to be exotic substances, and were difficult to evaluate as drugs simply because the synthetic methods required to produce them in tangible amounts were lacking. In the last 40 years this situation has changed, however, glycopeptides are still significantly more difficult to produce than simple peptides, even if the requisite Fmoc–amino acid glycosides are commercially available, and particularly so if the glycosides require synthesis [54,55]. Enzymatic approaches have been incorporated into chemical methods for serine and threonine glycosides [56]. The ideal methodology should produce high yields of pure diastereomers, typically β-anomers for the highest stability. Many classical methods produce high yields of the desired anomers, but require the production of labile glycosyl donors or very reactive (e.g., unstable) promoters [57].
Production of O-linked glucosides and lactosides of enkephalins and endorphins requires the corresponding acetate-protected glycosides of Fmoc–serine or Fmoc–threonine. The benzophenone Schiff base appeared to be a very good protection for amino groups of serine and threo-nine for glycoside formation [58]. O-glycosides of these amino acids with different monosaccharides, aminosugars and deoxysugars were obtained with excellent yield and very high stereoselectivity [59]. Glycoside peracetates have been used as building blocks for solid-phase glycopeptide synthesis, and significant improvements have been made in the production of the glycoside building blocks [60]. An even more efficient and direct approach to these precursors has now been developed that proceeds directly from Fmoc–serine or Fmoc–threonine as glycosyl acceptors, and either β-d-glucose per-acetate or β-lactose peracetate in the presence of ‘minimally competent’ Lewis acids, such as indium(III)bromide (Figure 4) [61].
Figure 4. Minimally competent Lewis acids as glycosidation promotors.

Lewis acids such as InBr3 can dissociate (lower pathway) from the displaced acetate to form acetic acid and regenerate the Lewis acid catalyst. Stronger Lewis acids remain associated with the acetate (upper pathway) to produce a Brønsted acid and, generally, require a full equivalent of the Lewis acid.
The glycopeptides can be synthesized manually based on established solid-phase N-fluorenylmethoxycarbonyl methods (Fmoc chemistry) (Figure 5). The side chain-protected amino acids used by our research group were Fmoc–Lys(Boc)-OH, Fmoc–Glu(OtBu)-OH, Fmoc–Asn(Trt)-OH, Fmoc–D-Thr(But)-OH, and Fmoc–Tyr(But)-OH. For support we have used Rink amide MBHA 1% DVB resin, with substitution typically ranging from 0.2– 0.8 meq/g. Best results (e.g., highest purities of the crude glycopeptides) were obtained by coupling well below the resin capacity and then capping the excess capacity with acetic anyhydride (Ac2O). Coupling of the Fmoc–amino acids was achieved using manual coupling methods with or without microwave heating or, typically, using mechanization, with extended reaction times or heating only for coupling of the glycosidic residue and the residue following, which may be regarded as an extremely hindered case of amino acid coupling. Use of NMP as a solvent rather than the more polar DMF also aided these couplings. Manual coupling reactions and critical couplings performed during mechanized coupling were typically monitored using Kaiser's ninhydrin test.
Figure 5. Glycopeptide assembly.

MBHA-functionalized Rink polystyrene resin was used to provide the C-terminal amides upon cleavage after classical Fmoc construction of the glycopeptides. Treatment with hydrazine hydrate (H2NNH2•H2O) in methanol (CH3OH) was required to remove the acetates from the glycoside moiety prior to cleavage from the Rink resin. A Boc-protected amino acid may be used for the final amino acid (O-tBu-Tyr), which is cleaved with the TFA cocktail.
The Fmoc group was removed from the N-terminus of the growing glycopeptide chain using a mixture of 3% piperidine and 2% diaza-1,3-bicyclo[5.4.0]-undecane in DMF for 10 min with argon bubbling as agitation. The acetyl protecting groups of the glycosides and the N-terminal Fmoc could be removed with 80% hydrazine hydrate (H2NNH2•H2O) in CH3OH with argon agitation 3X for approximately 2 h. A superior method used Boc protection for the last amino acid, which survived the hydrazine treatment but was removed by TFA. The synthetic glycopeptides were cleaved from the Rink resin with a ‘TFA cocktail’, F3CCOOH:Et3Si H:H2O:PhOCH3:CH2Cl2 (8:0.5:0.5:0.05:1), which also removed the side chain protection, and the N-terminal Boc group, if that method is employed. The crude glycopeptides were precipitated in cold diethylether, redissolved in H2O and then lyophilized prior to reverse-phase HPLC. A preparative scale C18 column (Phenomenex® 250 × 22 mm, 250 × 55 mm or equivalent) was used, with an acetonitrile–water (CH3CN-H2O) gradient containing 0.1% TFA to obtain glycopeptides of greater than 97% purity. Homogeneity of the pure glycopeptides was confirmed by analytical reverse-phase HPLC and MS.
Glycopeptide analgesics based on enkephalins
Our initial attempts at improving the CNS bioavailability of opioid peptides came from collaborations between the Chemistry and Pharmacology Departments at the University of Arizona led by Victor Hruby [62]. In 1983 this group successfully synthesized and characterized a series of cyclic penicillamine containing enkephalin analogues (e.g., DPDPE) that had higher affinity and selectivity for DOP [63]. Incorporation of an unnatural amino acid (D-penicillamine) and the cyclic constraint into the peptide enhanced both its stability and DOP selectivity. One hypothesis was that by increasing the lipophilicity of an already quite lipophilic DPDPE, BBB penetration could be increased, which was confirmed in an in vitro BBB model that used bovine brain microvessel endothelial cells [64].
Concurrently, we tested an alternative hypothesis, which, in retrospect, was naive and incorrect, whereby attachment of a glucose molecule to the modified enkephalin peptide would make the overall ligand a substrate for the Glut-1 transporter [65]. In an effort to enhance CNS bioavailability, we synthesized a series of enkephalin analogues (Tables 2–4). It was predicted that CNS delivery of the enkephalin molecule across the BBB would be increased, and that we would observe antinociceptive activity following systemic administration. While the Glut-1 hypothesis eventually proved to be incorrect, the enkephalin glycosides did penetrate the BBB very effectively and produced potent and long-lasting antinociception in mice after intravenous (iv.) or intraperitoneal (ip.) injection [66].
Table 2.
Cyclic disulfides related to DPDPE†.
| Compound | Structure | IC50 (nM) |
|||
|---|---|---|---|---|---|
| δ receptor | μ receptor | MVD | GPI | ||
| 1 |
|
6.1–6.4 | 30–43 | 5.5 | 26 |
| 2 |
|
3900 | 7700 | 520 | 3700 |
| 3 |
|
9.9 | 42 | 24 | 110 |
| 4 |
|
26–46 | 45–53 | 13 | 60 |
| 5 |
|
85 | 48,000 | 560 | 40,000 |
| 6 |
|
32 | 19 | ||
| 7 |
|
10 | 53.3 | ||
| 8 |
|
48 | 9 | ||
Study of a series of cyclic disulfide glycosides of enkephalin showed that placement of the glycoside was critical to maintain opioid activity and that δ receptor selectivity was reduced in this series of compounds.
GPI: Guinea pig ileum; MVD: Mouse vas deferens.
Table 4.
Linear glycopeptide amides as μ receptor/δ receptor opioid agonists.
| Compound | Structure | Ki (nM) |
A50 |
|||
|---|---|---|---|---|---|---|
| δ receptor | μ receptor | κ receptor | Intracerebroventricular (nmol) | Intravenous (μmol/kg) | ||
| Morphine |
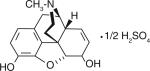
|
290 ± 38 | 0.79 ± 0.12 | 12.0 ± 1.3 | 2384 | 7.84 |
| 17 |
|
9.20 ± 1.70 | 5.00 ± 0.65 | 42.0 ± 5.0 | 0.018 | 3.20 |
| 18 |
|
6.48 | 41.9 | ND | 0.034 | 2.16 |
| 19 |
|
13.0 ± 0.55 | 4.5 ± 0.12 | 31.0 ± 0.3 | ND | ND |
| 20 |
|
9.86 | 30.8 | ND | 0.062 | 6.82 |
| 21 |
|
25.0 | 56.7 | ND | 0.061 | 10.9 |
| 22 |
|
ND | ND | ND | 0.380 | 140.8 |
| 23 |
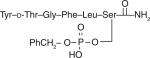
|
3.3 ± 0.33 | 8.7 ± 0.93 | 130 ± 6.2 | 0.093 | 30% at 32.0 mg/kg |
| 24 |

|
7.9 ± 0.73 | 3.9 ± 0.19 | 310 ± 66 | 0.34 | 54.1 |
| 25 |
|
14.0 ± 0.34 | 1100 ± 13 | 3% at 10.0 μM | 0.22 | 5.63 |
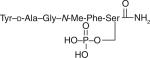
Glycopeptide disaccharides, and phosphates based on δ receptor-preferring enkephalin sequences developed by BP Roques, DtGFLS (DTLES) and DsGFLT (DSLET).
ND: Not determined.
Table 2 highlights some of the initial enkephalin glycopeptides that were synthesized [67]. Glycoside placement proved critical for affinity as determined by radioligand binding studies, and efficacy, as determined by GPI and MVD assays. Glycosylation sites close to the N-terminus resulted in reduced affinity for both DOP and MOP. Extension of the modified enkephalin peptide at the C-terminus allowed for glycosylation while preserving opioid receptor affinity, with some compounds retaining moderate DOP selectivity while others had approximately equal affinity for DOP and MOP. Two glycopeptides, β-glucoside 3 and 4 (Table 2) were tested in mice for their ability to produce CNS-mediated antinociception after systemic administration, and were compared with the unglycosylated peptide control 1. Both glycopeptides 3 and 4 produced dose- and time-related antinociception following ip. injection into mice, whereas the unglycosylated control peptides did not produce any measurable effects. The primary obstacles for better characterization of these glycopeptides were the somewhat tedious synthesis of the cyclic disulfides and the relatively low potency of the compounds (~30 mg/kg A50 values).
Larger quantities of a linear enkephalin glycoside based on Roques's so-called ‘delta-enkephalin’ or DTLET (YtGFLT, Tyr-d-Thr-Gly-Phe-Leu-Thr) were produced. The parent unglycosylated peptides, 9 and 10, retained high affinity for both DOP and MOP, relatively weak binding to KOP, and displayed a slight preference (~tenfold) for functional activity in the mouse MVD assay versus GPI assay [68]. Both compounds were extremely potent (<0.1 nmol A50 values) following intra cerebroventricular (i.c.v.) administration but required very large doses iv. to produce any antinociception in the mouse 55°C tail-flick. The addition of a glucoside to a serine in the sixth position of the peptide resulted in retention of modest selectivity for DOP over MOP in functional MVD/ GPI tissue assays and in receptor-binding studies [69]. Both the parent peptide (9) and glycosylated analogue (12) were extremely potent in the mouse tail-flick assay following intra cerebroventricular injection. However, glycopeptide 12 was significantly more potent following systemic routes of administration (iv., ip. and sc.). In situ BBB studies in rats also indicated, despite the increase in MW and increased water solubility, that the glycopeptide penetrated the BBB more effectively than its unglycosylated peptide counterpart 9 [70]. When compared with morphine, glycopeptide 12 resulted in lower levels of physical dependence as indicated by naloxone-precipitated withdrawal.
In an effort to further explore the structure-activity of glycosylation [71], a number of glycopeptides were synthesized to determine if the type of monosaccharide altered the transport characteristics and systemic potency of the lead peptide pharmacophore; if di- or tri-saccharides provided any additional benefit to pharmacokinetic and pharmacodynamic properties; and if bis- or tris-monosaccharides were viable alternative strategies for improving BBB transport and systemic potency [72]. In addition, several other modifications were made to explore the geometry of the attachment point (D vs L amino acid) of the glycoside and to see if the more sterically hindered threonine attachment differed from serine in its effects on activity. It should be noted that we stayed with the linear enkephalin parent peptide as it had roughly equal affinity for DOP and MOP receptors. This was important in assessing potential effects of glycosylation on preferential biasing for DOP or MOP.
The initial Roques-based linear peptides tested had either an l-Ser or l-Thr added to the sixth position of the peptide. The geometry of the glucoside attachment did not impact functional potency/efficacy in the in vitro or in vivo assays (l-Ser vs d-Ser or l-Thr vs d-Thr). For the monosaccharides, the β-xylose was approximately two-times more potent than β-glucose or α-mannose following iv. administration. The three disaccharides (β-lactose, β-maltose and β-melibiose) were all more potent than the best monosaccharide tested, with the β-melibioside being the most potent of the three.
Based on these results, we synthesized additional glycopeptides that incorporated a trisaccharide (β-maltotriose) to see if additional size/ bulk of the carbohydrate moiety would lead to further increases in iv. potency. The experimen iv. potency. The experimental data indicated a modest fall off in binding affinity and potency in the in vitro and in vivo functional assays. We also extended the hexa-peptide to include one to two additional Ser or Thr attachment points with β-glucose (bis- and tris-monosaccharides) to more fully explore the structure–activity relationship. In all cases, the additional glycosyl bulk reduced potency following i.c.v. administration, and the one compound tested iv. was significantly less potent than the original glycopeptide (β-glucoside and l-serine attachment).
Additional studies confirmed aspects of the in vivo studies [67]. Larger carbohydrates reduced octanol:saline partitioning (logD value), indicating greater water solubility (parent peptide < monosaccharide < disaccharide < trisccharide). Serum and brain stability of the glycopeptides also increased with these substitutions. In an in situ model of BBB transport the disaccharide proved to be the most readily transported with the trisaccharide having a reduced RBr value (though still superior to the unglycosylated control). We extended the in vivo work by adding additional pain assays to assess efficacy. The disaccharide 17 produced potent antinociception in the formic acid, acetic acid and carrageenan assays following systemic administration (all of these pain assays have an inflammatory component).
Based on this modest library of glycopeptides, we chose the β-lactoside (17) as the lead molecule to pursue more advanced in vivo characterization. While not the most potent of the disaccha-rides, the compound was much easier and less costly to synthesize compared with melibiose. Glycopeptide 17 also had some additional desirable characteristics, including being highly water soluble (>50 mg/ml). Based on these findings, we advanced 17 into a more complete characterization of its antinociceptive efficacy and side-effect profile.
As expected, 17 produced full efficacy in the GTPγS assay with a modest selectivity for DOP over MOP. This profile was similar to what was observed in the functional MVD and GPI tissue assays. We confirmed this profile in vivo by pre-treating mice with various opioid antagonists. The general antagonist naloxone completely blocked the actions of 17 in the 55°C tail-flick assay in mice. In contrast, the peripherally selective antagonist naloxone methiodide did not alter the agonist actions of the compound. Subtype selective DOP (naltrindole) and MOP (β-FNA) antagonists each partially blocked the antinociceptive actions of 17 and, when combined, they completely eliminated the agonist actions. The KOP-selective antagonist nor-BNI was without effect.
As a lead molecule, 17 was also tested in several rat models of pain to determine how broad an antinociceptive spectrum the compound might have. The first assay used was a post-surgical incision model of the hind paw [73]. Morphine and 17 both produced dose-related reversal of the tactile allodynia associated with the injury [D Giuvelis et al., Unpublished Data]. On a μmol/kg basis, 17 was almost equal to morphine in terms of potency. Similar results were seen in a subchronic inflammatory pain model induced by complete Freund's adjuvant, although, in this case, 17 had greater potency than morphine, possibly due to enhanced DOP signaling under inflammatory conditions. Finally, 17 was compared with gabapentin in a rat spinal nerve ligation model of neuropathic pain. Glycopeptide 17 produced potent reversal of the tactile allodynia and thermal hyperalgesia post-ligation, whereas gabapentin only reversed tactile allodynia at the doses examined [D Giuvelis et al., Unpublished Data]. Collectively, the data indicate that a mixed DOP/MOP agonist has a broad spectrum of antinociceptive effects in acute and chronic pain models, including ones that have inflammatory and/or neuropathic pain components.
One of the initial screens for side effect was to inject increasing doses of 17 or morphine and collect locomotor data in an automated open field assay. Morphine and other MOP agonists stimulate forward locomotion in imprinting control region mice. This effect becomes pronounced at near maximal and supramaximal antinociceptive doses. The mixed MOP/DOP agonist 17 produced an initial and transient decrease in forward locomotion that was replaced by a very mild stimulation of activity at later time points. We further investigated the initial inhibition of locomotor activity by pretreating mice with naloxone methiodide or nor-BNI. Both pretreatments attenuated the effects of 17 on locomotion and completely eliminated both effects when the two opioid antagonists were administered simultaneously. This indicated that stimulation of peripheral opioid receptors can produce a transient decrease in exploratory locomotor behavior and there may be a modest κ-agonist effect of the compound in the CNS that contributes to reduced stimulation of locomotor activity but does not contribute to the antinociceptive effects in the 55°C tail-flick assay.
Interestingly, one of the other gross observable differences between 17 and morphine is a lack of Straub tail and muscular rigidity with 17. We quantified this effect in dose–response curves versus antinociception with both morphine and the mixed agonist 17. The potential of morphine to produce both effects overlapped, whereas it took much higher doses of 17 to produce the muscular rigidity and Straub tail compared with its antinociceptive effects.
Based on the mixed DOP/MOP profile of 17, we were interested in evaluating the tolerance and physical dependence liability of the compound relative to morphine. For tolerance studies, we used a common paradigm involving twice-daily injections of the approximate A90 doses of the agonist (or vehicle) for 3 days. On the morning of day 4, full dose–response curves were constructed for each compound in the agonist- and vehicle-treated animals. Repeated doses of morphine resulted in an approximately 13-fold rightward shift in the A50 value indicating substantial development of antinociceptive tolerance. Equivalent doses of 17 (in terms of analgesia) resulted in a significantly reduced rightward shift (<fivefold). Morphine and glycopeptide 17 have similar durations of action and AUC values, making the comparisons more straightforward.
For assessment of physical dependence liability, we used both an acute (single high-dose administration of agonist) and chronic (twice-daily injections for 3 days) dependence protocol. In both cases, injection of the general opioid antagonist naloxone was used to precipitate withdrawal and several indices of withdrawal were recorded (vertical jumps and paw tremors, for example). The level of physical dependence/severity of withdrawal was consistently lower with the 17 exposure compared with equivalent exposures of morphine [74]. The working hypothesis for explaining these results is that the antinociceptive effects of the mixed DOP/MOP compound synergize at the cellular or network level, whereas the processes that drive tolerance and/or physical dependence are additive or subadditive. A predominantly MOP-selective agonist, on the other hand, requires significant occupation of the MOP receptors at sites both responsible for antinociception and tolerance/dependence. Other explanations are possible, including the formation of heterodimers with the glycopeptide (17) versus the small molecule (morphine) that lead to activation of different signaling pathways.
To further characterize the side-effect profiles of the glycopeptide, two commonly used assays for assessing MOP effects were used (gastrointestinal transit and respiratory depression). We had predicted that the DOP/MOP profile would have a reduced effect on these parameters compared with equivalent doses of morphine. This was not the case. Glycopeptide 17 also inhibited upper-gastrointestinal transit and suppressed respiratory response to elevations in CO2 on the minute ventilation parameter. The former may have been due to the apparent higher concentrations of glycopeptide 17 in the peripheral circulation compared with CNS, thus, overwhelming the MOP populations in the enteric nervous system. These values were estimated from the i.c.v. versus iv. potency ratios to produce antinociception for 17 versus morphine (more formal pharmacokinetic measures are currently being conducted). The respiratory depression observations indicate that a slight preference for DOP over MOP is not sufficient to differentiate from a MOP selective agonist.
Additional studies were conducted with 17 with respect to its abuse liability. As mentioned previously, the level of locomotor stimulation with 17 was markedly reduced compared with morphine. The stimulation of forward locomotion is generally interpreted as an activation of mesolimbic dopamine systems and an indicator of abuse liability. Our group has also conducted preliminary studies using conditioned place preference and iv. drug self administration in rodents. In the conditioned place preference studies, morphine produced a significant place preference whereas antinociceptive equivalent doses of 17 did not (similar to vehicle). In rat self-administration studies 17 maintained significantly lower numbers of infusions than the more MOP selective agonists morphine and fentanyl. In addition, the cumulative latency to delivery of the first three infusions of 17 (at the peak of the dose–effect curve) was significantly longer than morphine and fentanyl [Stevenson et al. Manuscript in Preparation]. These experiments suggest that the reinforcing effects of 17 are less than the prototypical MOP agonists morphine and fentanyl. The reinforcing effects of 17 were also evaluated in rhesus monkeys [75]. Under the conditions examined 17 did not support self-administration in rhesus across a series of doses/concentrations, although the results are more difficult to interpret due to species differences with respect to pharmacokinetics.
Current studies with DOP-selective peptides
With respect to our analgesic drug-development efforts, the prior work with 17 (mixed DOP/ MOP agonist) indicated to us that greater DOP selectivity might be needed in order to differentiate a lead candidate from currently available MOP analgesics. This was based not only on the extensive characterization we had done with 17, but also the literature demonstrating involvement of DOP receptors in neuropathic and other chronic pain states, and the further improvement in side-effect profiles [76–78]. We have also been interested in exploiting potential differences between the small-molecule DOP agonists, such as BW373U86 and SNC80, and the larger peptide-based deltorphin II analogues [79–82]. The former are ineffective in acute nociceptive assays that have high stimulus intensities, whereas the later are effective. This may be due to differences in ligand/receptor biasing or interactions with unique homo- or hetero-dimers of the DOP [83].
A series of glycosylated deltorphin analogues were synthesized that retained their high affinity, selectivity and efficacy at the DOP. Through an in vitro screening process, two to three lead compounds emerged, including glycopeptide 25. (Table 4) This compound exhibits a low nM EC50 value in the GTPγS assay and is very potent when injected i.c.v. (<1 nmol A50 value) (Kitsos et al. Mauscript in Preparation). Glycopeptide 25 is also systemically bioavailable following iv. and p.o. dosing, although the oral dose requires a proprietary co-formulation technology developed by Unigene. We are currently conducting further assessments of efficacy and side effects of glycopeptide 25 and comparing them to morphine and to 17. These preliminary data suggest that we the efficacy of the δ-agonist 25 is at least equal to the mixed μ/δ-agonist 17 with a side-effect profile on gastrointestinal transit that is superior to morphine and 17.
DAMGO - based MOP agonists
To further explore and exploit the biousian hypothesis [84], the classical μ-agonist (Ki: 0.53 nM) DAMGO [85,86] was used as a lipophilic peptide message [87], and additional moieties added to provide a water soluble address to produce a series of MOP-selective ligands. Their pharmacology was assessed in vitro and in vivo [88,89]. It was believed that the exploration of the biousian hypothesis [84] within the context of a pure MOP agonist would simplify interpretation of the results (Figure 6). The binding and antinociceptive effects of 26–29 are shown in Table 5 with values for morphine sulfate and DAMGO included for comparison. Binding was determined in Chinese hamster ovary cell membranes as before. Antinociception (A50 values) was determined after i.c.v. or iv. administration using the mouse 55°C tail-flick assay.
Figure 6. Antinociception studies indicate a U-shaped or V-shaped curve when the A50 potency values are correlated with predicted amphipathicity.

The hydrodynamic values (glucose units) or Connolly-derived amphipathicity values are plotted along the X-axes, and A50 values derived from mouse intravenous tail-flick data are plotted on the Y-axis. Both analyses produce a U-shape or V-shape, as predicted by the biousian hypothesis [84]. The amphipathicity values were calculated using the formula A = e–Awater/Alipid, where Awater = the Connolly surface area of the hydrophilic moiety (Å2) and Alipid = the Connolly surface area of the rest of the lipophilic peptide message segment YaG(N-MeF).
Table 5.
Glycopeptides based on DAMGO.
| Compound | Structure | Ki (nM) |
A50 |
|||
|---|---|---|---|---|---|---|
| δ receptor | μ receptor | κ receptor | Intracerebroventricular (nmol) | Intravenous (μmol/kg) | ||
| Morphine |
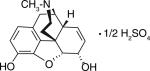
|
290 ± 38 | 0.79 ± 0.12 | 12.0 ± 1.3 | 2384 | 7.84 |
| DAMGO |
|
990 ± 35 | 0.56 ± 0.006 | 270 ± 9.3 | 30 | 1.88 |
| 26 |
|
600 ± 44 | 0.68 ± 0.02 | 190 ± 9.3 | 2.0 | 0.20 |
| 27 |
|
730 ± 66 | 1.30 ± 0.16 | 160 ± 10 | 2.0 | 0.27 |
| 28 |
|
54% at 10 μM | 1.30 ± 0.14 | 270 ± 2.5 | 19 | 0.72 |
| 29 |
|
1600 ± 129 | 0.66 ± 0.05 | 350 ± 51 | 2.0 | 1.15 |
| 30 |
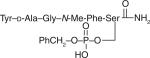
|
55 ± 4.6 | 4.2 ± 0.34 | 570 ± 9.8 | 33 | 30% at 38.1 mg/kg |
| 31 |
|
1400 ± 180 | 3.8 ± 0.73 | 31% at 10 μM | 42 | 1.5 |
Glycopeptide disaccharides and phosphates based on the μ receptor-preferring sequences.
Since the binding affinities and receptor preferences of the μ-selective DAMGO derivatives are similar, the analgesic potencies of the glycopeptides are largely determined by their ability to penetrate the BBB by transcytosis [67], which in turn depends on the biousian character of the drugs [84]. One may consider two extremes that result in poor delivery of a peptide drug:.
■ The peptide binds tightly to biological membrane, effectively removing it from solution;
■ The peptide remains in aqueous solution, effectively preventing it from binding to biological membranes.
Thus, the goal in producing glycopeptides that are capable of effective CNS delivery, binding and GPCR activation, is to balance the degree of glycosylation, which effectively determines the amount of time the glycopeptide spends on the endothelial membrane of the BBB, as well as other membranes that the glycopep-tide is likely to encounter. Affinity for the membrane is still required for effective binding and activation of the GPCR but a certain amount of membrane hopping is required for effective drug transport. Thus, a plot of the BBB transport or antinociceptive A50 values versus the membrane affinity produced a U-shaped or V-shaped curve (Figure 6).
Unpublished studies in mice showed that disaccharide 29 produced behaviors (Straub tail and hyperlocomotion) suggestive of ‘narcotic intoxication’ at very low doses and had an extreme addiction liability as indicated by naloxone precipitated withdrawal studies. While μ-agonists, such as peptide 26 or glycopeptide 27, could provide some useful clinical features that that morphine and other μ-selective anal-gesics do not possess, there does not seem to be much appetite for adding drugs to the pharmacopeia with side-effect profiles worse than morphine, and that are likely to be extremely addictive.
Kaiser's pioneering studies on the structure of β-endorphin
The late Emil Kaiser led a group of researchers at the Rockefeller Institute (USA) in studies of the structure and function of naturally occurring peptides, focusing their pioneering efforts on endogenous opioid hormones of mammalian origin including β-endorphin [90] and other seemingly diverse peptides from arthropods such as bee venom, or melittin (Figure 7)[91]. In fact, both β-endorphin and melittin interact strongly with biological membranes. It is in understanding the similarities and differences in exactly how these two peptides interact with membranes that will provide a rationale and opportunity for improving peptide drug delivery and effective design of CNS drugs. Our group has incorporated a design strategy for BBB penetration that exploits amphipathic α-helices that can interact strongly with cellular membranes to enhance endocytotic events. Critically, this biousian approach also preserves a degree of hydrophilicity for the overall peptide, especially when it is not interacting with the phospholipid membrane [84].
Figure 7. Representations of the amphipathic helical region in β-endorphin.

(A) Human β-endorphin [12–28] represented both as an α-helical net projection (left) and as a π-helical net projection (right) [90]. The lipophilic (hydrophobic) residues are circled. (B) Human β-endorphin [12–30] represented as an axial projection of a π-helix [91].
All of our glycosylated enkephalins display well-defined secondary structures in sodium dodecyl sulfate (SDS) micelles, irrespective of the identity of the glycoside attached to the peptide, which represent the simplest model for a biological membrane. In contrast, no defined structure is observed in aqueous media (Figure 8a) [92]. Similarly, the longer glycopeptides related to β-endorphin tend to adopt amphipathic helical structures in SDS micelles as well as phospholipid bicelles (Figure 8b) [93]. These larger glycopeptides displayed an ensemble of random coil conformations in aqueous solvent despite they are 17 residues in length. It is evident from these observations that amphipathicity of the glycopeptide (not simply hydrophilic or hydrophobic nature alone) is essential for membrane transport. This biousian behavior (i.e., random coil state in aqueous environment and highly folded state in membrane environment) appears essential for the effective transport of these larger peptides across the BBB [84].
Figure 8.

Micelle-bound structures of glycopeptide analogues determined by NMR related to (A) enkephalins and (B) endorphins [92,93].
Biousian behavior has also been observed for other endogenous GPCR peptide ligands [84,94]. Many, if not all peptide ligands that activate GPCRs lack a well-defined structure in aqueous buffer, but tend to fold into largely α-helical conformations in the presence of organic solvents [95] and lipid micelles [96], and in crystals [97,98]. Inooka and coworkers have been able to demonstrate that micelle-bound conformation of a peptide ligand is closely related to the actual receptor-bound conformation, that is, the α-helical region is similar in both cases [99].
Glycopeptide opioids based on endorphins
Initially, we focused on the importance of the glycoside in the context of the short enkephalin-based glycopeptides to enhance membrane hopping and BBB penetration, as evidenced by peptide 35 compared with glycopeptide (Table 6) [35]. The much larger endorphin-based peptides did not always require glycosylation in order to achieve effective membrane hopping rates to produce effective drug transport (Figure 9). In fact, the glycosylation could have no effect (32 vs 33), or even a deleterious effect on iv. potency (32 vs 34). We think that the flexibility of the linker (proline vs GABA vs DAVA) may play an important role as well, satisfying both the GPCR and the membrane binding requirements. More importantly although, the intrinsic stability of the membrane bound helix can decisively affect drug transport, as evidenced by iv. potency in vivo.
Table 6.
The amphipathic helix address†.
| Compound | Message sequence | Link | Helical address sequence | Ki (nM) |
A50 |
|||
|---|---|---|---|---|---|---|---|---|
| δ receptor | μ receptor | κ receptor | Intracerebroventricular (nmol) | Intravenous (μmol/kg) | ||||
| 32 | YtFGL | P | NLBEKALKSL-NH2 | 9.6 | 2.3 | 2.8 | 0.57 | 0.32 |
| 33 | YtFGL | P | NLBEKALKS*L-NH2 | 3.6 | 2.6 | 2.3 | 0.58 | 0.36 |
| 34 | YtFGL | P | NLBEKALKS**L-NH2 | 12.0 | 8.2 | 5.7 | 0.11 | 1.06 |
| 35 | YtFGL | GABA | SL-NH2 | 16 | 11 | 38 | 0.064 | (>> 10) |
| 36 | YtFGL | GABA | S*L-NH2 | 39 | 16 | 56 | 0.059 | 2.29 |
| 37 | YtFGL | GABA | NLBEKALKSL-NH2 | 29 | 31 | 49 | 0.143 | 1.12 |
| 38 | YtFGL | GABA | NLBEKALKS*L-NH2 | 4.2 | 4.3 | 11 | 0.046 | 0.28 |
| 39 | YtFGL | GABA | NLBEKALKS**L-NH2 | 4.3 | 0.97 | 11 | 0.165 | 1.13 |
| 40 | YtFGL | DAVA | NLBEKALKSL-NH2 | 32 | 37 | 64 | 0.162 | 1.41 |
For endorphin analogues 32, 33 and 34, the glycosylation state showed only minor effects on binding and intravenous potency. The addition of a carbohydrate (35 vs 36) clearly shows a dramatic effect on intravenous potency, presumably by affecting transport, and combining the two features into the address region (e.g., 38) provides the best delivery.
S = OH; S* = Glc; S** = Lact.
DAVA: Delta amino valeric acid; GABA: Gamma amino butyric acid; P: Proline.
Figure 9. Biousian behavior in a helix context.

Modulation of amphipathic helix stability should modulate interactions with biological membranes and ‘searching’ for the receptor.
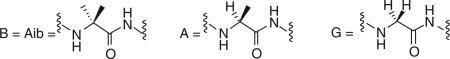
To determine the effect of helix stability a series of opioid C-terminal amide peptide 16-mers 41–47 were prepared using seven different helical segments with a minimal perturbation of the structure. Two amino acid residues (position 8 or 12) were substituted with one of three amino acids; helix-weakening glycine (Gly or G); helix-preferring L-alanine (Ala or A); or strongly helix-preferring α-aminoisobutyric acid (Aib or B) to produce seven different peptides of decreasing helix stability (B~B, B~A, A~B, A~A, A~G, G~A and G~G). Each series of helices was prepared in three different glycosylation states; one peptide series with unglycosylated l-serine (S°); a series of monosaccharides bearing a single β-O-d-glucose (Glc) on l-serine (S*); and a series of disaccharide glycopeptides bearing β-O-lactose (Lact) on l-serine (S**). These closely related seven peptides and 14 glycopeptides were characterized by high field (600 MHz) NMR in the presence of SDS and D2O/ H2O, and by circular dichroism in H2O, H2O/F3CCH2OH (data not shown here), and SDS/H2O (Figure 10) [100,101]. The degree of helicity was conveniently expressed as%-helicity per residue [102,103]. Note that helicity was essentially absent in aqueous media (Table 7). Peptide 41 was not soluble in H2O. However, in the presence of SDS micelles there was a clear trend, (Table 8) with B~B strongly favoring helical conformations, G~G disfavoring helical conformations, and intermediate levels of helicity for the others in relatively smooth, although not monotonic, stepwise fashion (Figure 11). The most helical compound, 41, was not soluble in water, but readily dissolved in the presence of SDS.
Figure 10. Circular dichroism to measure helicity.

Modulation of the amphipathic helix stability can be directly measured by examining the circular dichroism behavior in (A) sodium dodecylsulfate, (B) trifluoroethanol and (C) water, and measuring the elipticity at 222 nm.
Table 7.
Intrinsic helix stability in H2O†.
| Series | Message sequence | Link | Helical address sequence | Helicity per residue in H2O buffer (%) |
||
|---|---|---|---|---|---|---|
| S° = OH | S* = Glc | S** = Lact | ||||
| 41 | YtFGL | P | NLBEKBLKSo/*/**L-NH2 | Not soluble | 4.5 | 0.7 |
| 42 | YtFGL | P | NLBEKALKSo/*/**L-NH2 | 12.2 | 7.5 | 1.0 |
| 43 | YtFGL | P | NLAEKBLKSo/*/**L-NH2 | 7.4 | 7.0 | 1.3 |
| 44 | YtFGL | P | NLAEKALKSo/*/**L-NH2 | 7.2 | 6.9 | 0.1 |
| 45 | YtFGL | P | NLAEKGLKSo/*/**L-NH2 | 8.7 | 0.0 | 0.0 |
| 46 | YtFGL | P | NLGEKALKSo/*/**L-NH2 | 7.1 | 0.8 | 0.0 |
| 47 | YtFGL | P | NLGEKGLKSo/*/**L-NH2 | 8.1 | 0.7 | 0.0 |

| ||||||
Variation in the intrinsic helicity was achieved with minimal impact on other properties by altering only two amino acid residues with B (Aib), A (Ala) or G (Gly). Water solubility could be affected by altering the glycosylation state of the S.
Except for the extremely helical peptides without glycosylation, all of these compounds were highly water soluble, and showed only random coil behavior in aqueous solution.
Aib: α-aminoidobutyric acid; P: Proline; S: Serine.
Table 8.
Amphipathic helix stability in the presence of micelles†.
| Compound | Message sequence | Link | Helical address sequence | Helicity per residue in sodium dodecyl sulfate micelles (%) | ||
|---|---|---|---|---|---|---|
| So = OH | S* = Glc | S** = Lact | ||||
| 41 | YtFGL | P | NLBEKBLKSo/*/**L-NH2 | 70 | 56.7 | 40.8 |
| 42 | YtFGL | P | NLBEKALKSo/*/**L-NH2 | 60.8 | 44.9 | 35.7 |
| 43 | YtFGL | P | NLAEKBLKSo/*/**L-NH2 | 58.5 | 35.7 | 33.6 |
| 44 | YtFGL | P | NLAEKALKSo/*/**L-NH2 | 54.4 | 35.7 | 31.8 |
| 45 | YtFGL | P | NLAEKGLKSo/*/**L-NH2 | 37.8 | 24.6 | 13.3 |
| 46 | YtFGL | P | NLGEKALKSo/*/**L-NH2 | 33.0 | 14.5 | 4.5 |
| 47 | YtFGL | P | NLGEKGLKSo/*/**L-NH2 | 11.0 | 6.4 | 2.8 |
| ||||||
Helix amphipath stability was greatly altered in the presence of sodium dodecyl sulfate by altering the two amino acid residues with B (Aib), A (Ala) or G (Gly). Water solubility and the degree of helix formation (%-helix/residue) was also affected by altering the glycosylation state of the S.
Aib: α-aminoidobutyric acid; P: Proline; S: Serine.
Figure 11.

Helicity in sodium dodecyl sulfate as a function of peptide address sequence and glycosylation state.
All 21 compounds were characterized in terms of δ/μ/κ-binding, and subjected to the 55°C tail-flick test in mice, both after i.c.v. administration (Tables 9–11). Administration with iv. bolus injection is currently being studied, and the data (not shown) is only preliminary. Although there were only modest variations in the binding selectivity, and in the A50 values obtained after i.c.v. administration (bypassing the BBB), there are large variations in the A50 values obtained after iv. injection, ranging from little or no antinociceptive activity observed at 32 mg/kg down to extremely potent activities with A50 values below 500 μg/kg.
Table 9.
Binding and intracerebroventricular antinociception as a function of helix stability: unglycosylated peptides.
| Compound | Message sequence | Link | Helical address sequence | Ki (nM) |
A50 |
||
|---|---|---|---|---|---|---|---|
| δ receptor | μ receptor | κ receptor | Intracerebroventricular (nmol) | ||||
| 41* | YtFGL | P | NLBEKBLKSL-NH2 | 16 | 8.7 | 27 | 1.66 |
| 42*/32 | YtFGL | P | NLBEKALKSL-NH2 | 9.6 | 2.3 | 2.8 | 0.57 |
| 43* | YtFGL | P | NLAEKBLKSL-NH2 | 9.7 | 10 | 28 | 0.18 |
| 44* | YtFGL | P | NLAEKALKSL-NH2 | 3.5 | 2.8 | 5.5 | 0.55 |
| 45* | YtFGL | P | NLAEKGLKSL-NH2 | 15 | 17 | 49 | 0.79 |
| 46* | YtFGL | P | NLGEKALKSL-NH2 | 14 | 21 | 23 | 0.96 |
| 47* | YtFGL | P | NLGEKGLKSL-NH2 | 19 | 14 | 17 | 1.92 |
S = OH; S* = Glc; S** = Lact.
P: Proline; S: Serine.
Table 11.
Binding, intracerebroventricular and intravenous antinociception as a function of helix stability: lactosides
| Compound | Message sequence | Link | Helical address sequence | Ki (nM) |
A50 |
A50 |
||
|---|---|---|---|---|---|---|---|---|
| δ receptor | μ receptor | κ receptor | Intracerebroventricular (nmol) | Intravenous (μmol/kg) | ||||
| 41** | YtFGL | P | NLBEKBLKS**L-NH2 | 5.9 | 1.2 | 2.5 | 0.57 | 4.3 |
| 42**/34 | YtFGL | P | NLBEKALKS**L-NH2 | 12 | 8.2 | 5.7 | 0.11 | 1.1 |
| 43** | YtFGL | P | NLAEKBLKS**L-NH2 | 14 | 3.2 | 3.5 | 0.21 | 1.2 |
| 44** | YtFGL | P | NLAEKALKS**L-NH2 | 2.6 | < 1 | 3.5 | 0.76 | 4.1 |
| 45** | YtFGL | P | NLAEKGLKS**L-NH2 | 4.7 | 0.97 | 4.3 | 0.14 | 8.7 |
| 46** | YtFGL | P | NLGEKALKS**L-NH2 | 13 | 4.4 | 6.6 | 0.50 | |
| 47** | YtFGL | P | NLGEKGLKS**L-NH2 | 4.5 | 1.4 | 2.6 | 0.23 | > 40 |
S = OH; S* = Glc; S** = Lact.
P: Proline; S: Serine.
Future perspective
While it would be premature to state that we have a complete understanding of the role of amphipathicity, biousian behavior [84] and membrane affinity in the transport of opioid peptides and glycopeptides, it is clear that the transport of these compounds across biological membranes of interest is not only possible, but quite efficient. It seems clear that the observed transport phenomena will not be limited to this class of neuropeptides, as researchers are exploring glycosylated endomorphins [104], dermorphins [105], dermorphins [106] and other peptides [107] with good results. Thus, we remain optimistic that interest in glycopeptide drugs derived from neuropeptides will increase and that all major pharmaceutical companies will establish research programs in the area.
Executive summary.
■ The three opiate receptors, μ, δ and κ receptors, and their endogenous peptide neurotransmitters, the enkephalins and endorphins, play numerous roles in the CNS. Principal among these roles is the modulation of acute and chronic pain states. The endogenous peptide neurotransmitters are highly amphipathic, spending the bulk of their independent existence after cleavage from larger precursor proteins and prior to release stored in vesicles contained within the presynaptic membrane. Upon release they float in the postsynaptic membrane where they bind and activate opiate receptors. The effects of the endogenous peptide ligands are local in nature due to their poor transport properties.
■ Glycosylation of the relatively short enkephalins allows them to leave the membrane environment to engage in ‘membrane hopping’. The key is to add a water soluble moiety to the peptide in such a way that it enhances water solubility without interfering with its interaction with the membrane. We have dubbed this effect ‘biousian behavior’ in which the glycopeptide may exist either in a relatively constrained membrane bound state, or an aqueous state as a ‘random coil’ ensemble. The degree of glycosylation can be adjusted to optimize the biological transport rates of the enkephalin-based glycopeptides to provide drugs based on the endogenous enkephalins.
■ The biousian behavior permits the glycopeptides and related serine phosphates to penetrate the blood–brain barrier, probably via transcytosis.
■ The larger endorphins have a message–linker–address peptide format. The message exists as a turn structure and is largely responsible for receptor binding and activation. The address exists as an amphipathic helix and is largely responsible for binding to the membrane. A flexible linker domain allows the receptor binding requirements and the membrane binding requirements to be met simultaneously. The address domain can be altered in order to optimize the biological transport rates of the endorphin-based glycopeptides based on the endogenous endorphins.
■ Peptide sequences can be varied to produce glycopeptide drugs that favor any of the three opiate receptors, or various combinations, such as μ/κ receptors and mixed μ/δ-agonists. Side-effect profiles of the analgesics can be manipulated by altering the agonist features of the enkephalins or the endorphin message segments.
■ Glycopeptide-based drugs have been developed that exert analgesic effects in mice far in excess of the classical narcotics, such as morphine, Demerol® and Oxycontin®. We are optimistic that interest in glycopeptide drugs derived from neuropeptides will increase, and that all major pharmaceutical companies will establish research programs in the area. We expect that the applications will not be limited to opiate agonists and that a large number of the 250 plus endogenous neuropeptides can be converted into glycopeptide drugs capable of penetrating the blood–brain barrier.
Table 3.
Linear glycopeptide amides as δ receptor/μ receptor opioid agonists.
| Compound | Structure | Ki (nM) |
A50 |
|||
|---|---|---|---|---|---|---|
| δ receptor | μ receptor | κ receptor | Intracerebroventricular (nmol) | Intravenous (μmol/kg) | ||
| Morphine |
|
290 ± 38 | 0.79 ± 0.12 | 12.0 ± 1.3 | 2384 | 7.84 |
| 9 |
|
4.1 ± 41 | 1.4 ± 0.08 | 34.0 ± 2.2 | 0.068 | 46.4 |
| 10 |
|
9.71 | 11.7 | ND | 0.038 | 32.6 |
| 11 |
|
46.0 | 65.8 | ND | 0.092 | 9.45 |
| 12 |
|
7.0 ± 1.2 | 2.4 ± 0.017 | 49 ± 4.3 | 0.023 | 11.4 |
| 13 |
|
23.0 | 15.2 | ND | 0.033 | 16.7 |
| 14 |
|
16.8 | 39.8 | ND | 0.022 | ND |
| 15 |
|
54.4 | 297.8 | ND | 0.035 | ND |
| 16 |
|
24.5 | 31.8 | ND | 0.040 | 22.0 |
Glycopeptide monosaccharides based on DOP-preferring enkephalin sequences developed by BP Roques, DtGFLS (DTLES) and DsGFLT (DSLET).
ND: Not determined.
Table 10.
Binding and intracerebroventricular antinociception as a function of helix stability: glucopeptides.
| Compound | Message sequence | Link | Helical address sequence | Ki (nM) |
A50 |
||
|---|---|---|---|---|---|---|---|
| δ receptor | μ receptor | κ receptor | Intracerebroventricular (nmol) | ||||
| 41* | YtFGL | P | NLBEKBLKS*L-NH2 | 11 | 9.1 | 6.9 | 1.13 |
| 42*/33 | YtFGL | P | NLBEKALKS*L-NH2 | 3.6 | 2.3 | 2.6 | 0.58 |
| 43* | YtFGL | P | NLAEKBLKS*L-NH2 | 14 | 6.1 | 12 | 1.46 |
| 44* | YtFGL | P | NLAEKALKS*L-NH2 | 7.9 | 6.2 | 8.9 | 1.44 |
| 45* | YtFGL | P | NLAEKGLKS*L-NH2 | 6.3 | 2.2 | 6.3 | 1.47 |
| 46* | YtFGL | P | NLGEKALKS*L-NH2 | 5.7 | 2.9 | 11 | 2.54 |
| 47* | YtFGL | P | NLGEKGLKS*L-NH2 | 7.1 | 3.5 | 4.4 | 2.12 |
S = OH; S* = Glc; S** = Lact.
P: Proline; S: Serine.
Acknowledgements
We also thank D Guivalis, L St Louis, B Anglin and L Szabò for assistance with the manuscript.
We thank the Office of Naval Research (N00014-05-01-0807 and N00014-02-01-0471), the National Science Foundation (CHE-607917) and the National Institutes of Health (NINDS-NS-052727) for financial support. R Polt and EJ Bilsky own founder's stock in Biousian Biosystems, Inc., which holds intellectual property related to glycopep-tide 25.
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- 1.Borsook D, Hargreaves RJ, Becerra L. Can functional magnetic resonance imaging improve success rates in central nervous system drug discovery? Expert Opin. Drug Disc. 2011;6:597–617. doi: 10.1517/17460441.2011.584529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banks WA. Delivery of peptides to the brain: emphasis on therapeutic development. Biopolymers. 2008;90:589–594. doi: 10.1002/bip.20980. [DOI] [PubMed] [Google Scholar]
- 3.Begley DJ. Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol. Therap. 2004;104:29–45. doi: 10.1016/j.pharmthera.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Pardridge WM. Brain Drug Targeting. Cambridge University Press; Cambridge, UK: 2001. [Google Scholar]
- 5.Gruber CW, Muttenthaler M, Freissmuth M. Ligand-based peptide design and combinatorial peptide libraries to target G protein-coupled receptors. Curr. Pharm. Des. 2010;16(28):3071–3088. doi: 10.2174/138161210793292474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibáñez CF. Beyond the cell surface: new mechanisms of receptor function. Biochem. Biophys. Res. Commun. 2010;396(1):24–27. doi: 10.1016/j.bbrc.2010.01.136. [DOI] [PubMed] [Google Scholar]
- 7.Cleland JL, Langer R. Formulation and Delivery of Proteins and Peptides. American Chemical Society; Washington, DC, USA: 1994. [Google Scholar]
- 8.Taylor MD, Amidon GL. Peptide-Based Drug Design. American Chemical Society; Washington, DC, USA: 1995. [Google Scholar]
- 9.Van der Walle C. Peptide and Protein Delivery. Academic Press; London, UK: 2011. [Google Scholar]
- 10.Snyder SH. Opiate receptors and beyond: 30 years of neural signaling research. Neuropharmacology. 2004;47:274–285. doi: 10.1016/j.neuropharm.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Lagerström MC, Schiöth HB. Structural diversity of G protein coupled receptors and significance for drug discovery. Nature Rev. Drug Disc. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 12.Tyndall JDA, Pfeiffer B, Abbenante G, Fairlie DP. Over one hundred peptide-activated G protein-coupled receptors recognize ligands with turn structure. Chem. Rev. 2005;105:793–826. doi: 10.1021/cr040689g. [DOI] [PubMed] [Google Scholar]
- 13.Martin WR, Eades CG, Thompson JA, et al. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976;197:517–532. [PubMed] [Google Scholar]
- 14.Evans CJ. Secrets of the opium poppy revealed. Neuropharmacology. 2004;47:293–299. doi: 10.1016/j.neuropharm.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 15.Chang K-J, Porreca F, Woods J. The Delta Receptor. Marcel Dekker, Inc.; NY, USA: 2004. [Google Scholar]
- 16.Lord JAH, Waterfield AA, Hughes J, Kosterlitz HW. Endogenous opioid peptides: multiple agonists and receptors. Nature. 1977;267:495–499. doi: 10.1038/267495a0. [DOI] [PubMed] [Google Scholar]
- 17.Yue X, Falk T, Zuniga LA, et al. Effects of the novel glycopeptide opioid agonist MMP-2200 in preclinical models of Parkinson's disease. Brain Res. 2011;1413:72–83. doi: 10.1016/j.brainres.2011.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neumeyer JL, Zhang A, Xiong W, et al. Design and synthesis of novel dimeric morphinan ligands for kappa and micro opioid receptors. J. Med. Chem. 2003;46:5162–5170. doi: 10.1021/jm030139v. [DOI] [PubMed] [Google Scholar]
- 19.Jordan BA, Devi L. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhushan RG, Sharma SK, Xie Z, et al. A bivalent ligand (KDN-21) reveals spinal delta and kappa opioid receptors are organized as heterodimers that give rise to delta(1) and kappa(2) phenotypes. Selective targeting of delta-kappa heterodimers. J. Med. Chem. 2004;47:2969–2972. doi: 10.1021/jm0342358. [DOI] [PubMed] [Google Scholar]
- 21.Simantov R, Kuhar MJ, Uhl GR, et al. Opioid peptide enkephalin: Immunohistochemical mapping in rat central nervous system. Proc. Natl Acad. Sci. USA. 1977;74:2167–2171. doi: 10.1073/pnas.74.5.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scherrer G, Imamachi N, Cao Y-Q, et al. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell. 2009;137:1148–1159. doi: 10.1016/j.cell.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schiller PW. Bi- or multifunctional opioid peptide drugs. Life Sci. 2010;86:598–603. doi: 10.1016/j.lfs.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pradhan AA, Befort K, Nozaki C, et al. The delta opioid receptor: an evolving target for the treatment of brain disorders. TIPS. 2011;32:581–590. doi: 10.1016/j.tips.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patkar KA, Murray TF, Aldrich JV. The effects of C-terminal modifications on the opioid activity of [N-benzyltyr1]dynorphin A-(1–11) analogues. J. Med. Chem. 2009;52:6814–6821. doi: 10.1021/jm900715m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Binder W, Walker JS. Effect of the peripherally selective β-opioid agonist, asimadoline, on adjuvant arthritis. Br. J. Pharmacol. 1998;124:647–654. doi: 10.1038/sj.bjp.0701874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stevenson GW, Folk JE, Linsenmayer DC, et al. Opioid interactions in rhesus monkeys: effects of δ + μ and δ + κ agonists on schedule-controlled responding and thermal nociception. J. Pharm. Exp. Therap. 2003;307:1054–1064. doi: 10.1124/jpet.103.056515. [DOI] [PubMed] [Google Scholar]
- 28.Roth BL, Baner K, Westkaemper R, et al. Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc. Natl Acad. Sci. USA. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aldrich JV, McLaughlin JP. Peptide kappa opioid receptor ligands: potential for drug development. AAPS J. 2009;11:312–322. doi: 10.1208/s12248-009-9105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamada H, Shimoyama N, Sora I, et al. Morphine can produce analgesia via spinal kappa opioid receptors in the absence of mu opioid receptors. Brain Res. 2006;1083:61–69. doi: 10.1016/j.brainres.2006.01.095. [DOI] [PubMed] [Google Scholar]
- 31.Giardina G, Clarke GD, Grugni M, et al. Central and peripheral analgesic agents: chemical strategies for limiting brain penetration in kappa-opioid agonists belonging to different chemical classes. Farmaco. 1995;50:405–418. [PubMed] [Google Scholar]
- 32.Carroll I, Thomas JB, Dykstra LA, et al. Pharmacological properties of JDTic: a novel kappa-opioid receptor antagonist. Eur. J. Pharmacol. 2004;501:111–9. doi: 10.1016/j.ejphar.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 33.Hökfelt T, Bartfai T, Bloom F. Neuropeptides: opportunities for drug discovery. Lancet Neurology. 2003;2:463–472. doi: 10.1016/s1474-4422(03)00482-4. [DOI] [PubMed] [Google Scholar]
- 34.Cahill CM, Morinville A, Hoffert C, et al. Up-regulation and trafficking of (opioid receptor in a model of chronic inflammation: implications for pain control. Pain. 2003;101:199–208. doi: 10.1016/s0304-3959(02)00333-0. [DOI] [PubMed] [Google Scholar]
- 35.Weber E, Escht FS, Bohlen P, et al. Metorphamide: isolation, structure, and biologic activity of an amidated opioid octapeptide from bovine brain. Proc. Natl Acad. Sci. USA. 1983;80:7362–7366. doi: 10.1073/pnas.80.23.7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arakawa K, De Jong W, Mulder AH, et al. The electrically stimulated release of [3H] noradrenaline from nucleus tractus solitarii slices in vitro is modulated via β-opioid receptors. Eur. J. Pharm. 1991;192:311–316. doi: 10.1016/0014-2999(91)90057-w. [DOI] [PubMed] [Google Scholar]
- 37.Ronken E, Wiegant VM, Kaspersen FM, et al. Topography and characteristics of specific binding sites for non-opioid β-type endorphins in the rat brain as studied by autoradiography with [35S]Metdesenkephalin-γ-endorphin. Brain Res. 1993;615:63–70. doi: 10.1016/0006-8993(93)91114-8. [DOI] [PubMed] [Google Scholar]
- 38.Navolotskaya E, Kovalitskaya YA, Zolotarev YA, et al. Binding of synthetic fragments of β-endorphin to nonopioid β-endorphin receptor. J. Peptide Sci. 2008;14:1121–1128. doi: 10.1002/psc.1049. [DOI] [PubMed] [Google Scholar]
- 39.Schwyzer R, Sargent DF. Membrane lipid phase as catalyst for peptide receptor interactions. Proc. Natl Acad. Sci. USA. 1986;83:5774–78. doi: 10.1073/pnas.83.16.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamm LK. Protein–Lipid Interactions. From Membrane Domains to Cellular Networks. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2005. [Google Scholar]
- 41.Adam G, Delbrück M. In: Structural Chemistry and Molecular Biology. Rich R, Davidson N, editors. Freeman & Co.; San Francisco, CA, USA: 1968. pp. 198–199. [Google Scholar]
- 42.Saffman PG, Delbrück M. Brownian motion in biological membranes. Proc. Natl Acad. Sci. USA. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segrest JP, Jackson RL. A molecular theory of lipid-protein interactions in the plasma lipoproteins. Morrisett JD et al. FEBS Lett. 1974;38:247–253. doi: 10.1016/0014-5793(74)80064-5. [DOI] [PubMed] [Google Scholar]
- 44.Epand RM, Shai Y, Segrest JP, et al. Mechanisms for the modulation of membrane bilayer properties by amphipathic helical peptides. Biopolymers. 1995;37:319–338. doi: 10.1002/bip.360370504. [DOI] [PubMed] [Google Scholar]
- 45.Pardridge WM. The blood–brain barrier: bottleneck in brain drug development. NeuroRx: J. Am. Soc. Exp. NeuroTher. 2005;2:3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terasaki T, Hirai K, Sato H, et al. Absorptive-mediated endocytosis of a dynorphin-like analgesic peptide, E-2078 into the blood–brain barrier. J. Pharm. Exp. Ther. 1989;251:351–357. [PubMed] [Google Scholar]
- 47.Deguchi Y, Miyakawa Y, Sakurada S, et al. Blood–brain barrier transport of a novel micro 1-specific opioid peptide, H-Tyr-d-Arg-Phe-beta-Ala-OH (TAPA). J. Neurochem. 2003;84:1154–1161. doi: 10.1046/j.1471-4159.2003.01582.x. [DOI] [PubMed] [Google Scholar]
- 48.Kastin AJ, Pan W, Maness LM, et al. Peptides crossing the blood–brain barrier: some unusual observations. Brain Res. 1999;848:96–100. doi: 10.1016/s0006-8993(99)01961-7. [DOI] [PubMed] [Google Scholar]
- 49.Bredeloux P, Cavelier F, Dubuc I, et al. Synthesis and biological effects of c(Lys-Lys-Pro-Tyr-Ile-Leu-Lys-Lys-Pro-Tyr-Ile-Leu) (JMV2012), a new analogue of neurotensin that crosses the blood–brain barrier. J. Med. Chem. 2008;51:1610–1616. doi: 10.1021/jm700925k. [DOI] [PubMed] [Google Scholar]
- 50.Egleton RD, Davis P. Development of neuropeptide drugs that cross the blood–brain barrier. NeuroRx: J. Am. Soc. Exp. NeuroTher. 2005;2:44–53. doi: 10.1602/neurorx.2.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mehiri M, Chen W-H, Janout V, et al. Molecular umbrella transport: exceptions to the classic size/lipophilicity rule. J. Am. Chem. Soc. 2009;131:1338–1339. doi: 10.1021/ja806476t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pardridge WM. Re-engineering biopharmaceuticals for delivery to brain with molecular Trojan horses. Bioconjugate Chem. 2008;19:1327–1338. doi: 10.1021/bc800148t. [DOI] [PubMed] [Google Scholar]
- 53.Malakoutikhah M, Teixidó M, Giralt E. Toward an optimal blood–brain barrier shuttle by synthesis and evaluation of peptide libraries. J. Med. Chem. 2008;51:4881–4889. doi: 10.1021/jm800156z. [DOI] [PubMed] [Google Scholar]
- 54.Meldal M, Hilaire PM. Synthetic methods of glycopeptide assembly and biological analysis of glycopeptide products. Curr. Opin. Chem. Biol. 1997;1:552–563. doi: 10.1016/s1367-5931(97)80052-x. [DOI] [PubMed] [Google Scholar]
- 55.Boons GJ, Polt R. Carbohydrate Chemistry. Blackie Press; London, UK: 1998. The chemistry of O- and N-linked glycopeptides. pp. 223–242. [Google Scholar]
- 56.Eriko T, Yuko N, Yasuhiro K, et al. Chemoenzymatic synthesis of a MUC1 glycopeptide carrying non-natural sialyl TF-beta O-glycan. Biosci. Biotech. Biochem. 2006;70:2515–2522. doi: 10.1271/bbb.60244. [DOI] [PubMed] [Google Scholar]
- 57.Nianhuan Y, Gabriel F, Hamed M, et al. Facile synthesis of glycosylated Fmoc amino acid building blocks assisted by microwave irradiation. Carbohydrate Res. 2010;345:2277–2281. doi: 10.1016/j.carres.2010.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Polt R, Szabò LZ, Treiberg J, et al. General methods for alpha-O-ser/thr or beta-O-ser/thr glycosides and glycopeptides. Solid-phase synthesis of O-glycosyl cyclic enkephalin analogues. J. Am. Chem. Soc. 1992;114:10249–58. [Google Scholar]
- 59.Mitchell SA, Pratt MR, Hruby VJ, Polt R. Solid-phase synthesis of O-linked glycopeptide analogues of enkephalin. J. Org. Chem. 2001;66:2327–42. doi: 10.1021/jo005712m. [DOI] [PubMed] [Google Scholar]
- 60.Keyari CM, Polt R. Serine and threonine Schiff base esters react with β-anomeric peracetates in the presence of BF3•Et2O to produce β-glycosides. J. Carbohydr. Chem. 2010;29:181–206. [Google Scholar]
- 61.Lefever MR, Szabò LZ, Anglin B, et al. Glycosylation of β-amino acids by sugar acetate donors with InBr3. Minimally competent Lewis acids. Carbohydr. Res. 2012;347 doi: 10.1016/j.carres.2012.01.008. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hruby VJ, Toth G, Gehring CA, et al. Topographically designed analogues of [d-Pen2,d-Pen5]enkephalin. J. Med. Chem. 1991;34:1823–1830. doi: 10.1021/jm00110a010. [DOI] [PubMed] [Google Scholar]
- 63.Mosberg HI, Hurst R, Hruby VJ, et al. Bis-penicillamine enkephalins possess highly improved specificity toward delta opioid receptors. Proc. Natl Acad. Sci. USA. 1983;80:5871–5874. doi: 10.1073/pnas.80.19.5871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weber SJ, Abbruscato TJ, Brownson EA, et al. Assessment of an in vitro blood–brain barrier model using several [Met5]enkephalin opioid analogs. J. Pharm. Exp. Ther. 1993;266:1649–1655. [PubMed] [Google Scholar]
- 65.Rodriguez RE, Rodriguez FD, Sacristan MP, et al. New glycosylpeptides with high antinociceptive activity. Neurosci. Lett. 1989;101:89–94. doi: 10.1016/0304-3940(89)90446-1. [DOI] [PubMed] [Google Scholar]
- 66.Polt R, Porreca F, Szabò LZ, et al. Glycopeptide enkephalin analogues produce analgesia in mice: evidence for penetration of the blood–brain barrier. Proc. Natl Acad. Sci. USA. 1994;91:7114–7118. doi: 10.1073/pnas.91.15.7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Egleton RD, Mitchell SA, Huber JD, et al. Improved bioavailability to the brain of glycosylated Met-enkephalin analogs. Brain Res. 2000;881:37–46. doi: 10.1016/s0006-8993(00)02794-3. [DOI] [PubMed] [Google Scholar]
- 68.Zajac J-M, Gacel G, Petit F, et al. Deltakephalin, Tyr-d-Thr-Gly-Phe-Leu-Thr : a new highly potent and fully specific agonist for opiate β-receptors. Biochem. Biophys. Res. Commun. 1983;111:390–397. doi: 10.1016/0006-291x(83)90318-2. [DOI] [PubMed] [Google Scholar]
- 69.Bilsky EJ, Egleton RD, Mitchell SA, et al. Enkephalin glycopeptide analogues produce analgesia with reduced dependence liability. J. Med. Chem. 2000;43:2586–2590. doi: 10.1021/jm000077y. [DOI] [PubMed] [Google Scholar]
- 70.Egleton RD, Mitchell SA, Huber JD, et al. Improved blood–brain barrier penetration and enhanced analgesia of an opioid peptide by glycosylation. J. Pharm. Exp. Therap. 2001;299:967–972. [PubMed] [Google Scholar]
- 71.Horvat S. Opioid peptides and their glycoconjugates: structure-activity relationships. Curr. Med. Chem. – Central Nervous System Agents. 2001;1:133–154. [Google Scholar]
- 72.Elmagbari NO, Egleton RD, Palian MM, et al. Antinociceptive structure–activity studies with enkephalin-based opioid glycopeptides. J. Pharm. Exp. Therap. 2004;311:290–297. doi: 10.1124/jpet.104.069393. [DOI] [PubMed] [Google Scholar]
- 73.Brennan TJ, Vandermeulen EP, Gebhart GF. Characterization of a rat model of incisional pain. Pain. 1996;64:493–501. doi: 10.1016/0304-3959(95)01441-1. [DOI] [PubMed] [Google Scholar]
- 74.Lowery JJ, Raymond TJ, Giuvelis D, et al. In vivo characterization of MMP-2200, a mixed δ/μ opioid agonist, in mice. J. Pharmacol. Exp. Ther. 2011;336:767–78. doi: 10.1124/jpet.110.172866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Do Carmo GP, Polt R, Bilsky R, et al. Behavioral pharmacology of the μ/δ opioid glycopeptide MMP2200 in Rhesus monkeys. J. Pharm. Exp. Therap. 2008;326:939–948. doi: 10.1124/jpet.108.138180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brainin-Mattos J, Smith ND, Malkmus S, et al. Cancer-related bone pain is attenuated by a systemically available delta-opioid receptor agonist. Pain. 2006;122:174–181. doi: 10.1016/j.pain.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 77.Codd EE, Carson JR, Colburn RW, et al. JNJ-20788560 [9-(8-azabicyclo[3.2.1] oct-3-ylidene)-9H-xanthene-3-carboxylic acid diethylamide], a selective delta opioid receptor agonist, is a potent and efficacious antihyperalgesic agent that does not produce respiratory depression, pharmacologic tolerance, or physical dependence. J. Pharmacol. Exp. Ther. 2009;329:241–51. doi: 10.1124/jpet.108.146969. [DOI] [PubMed] [Google Scholar]
- 78.Otis V, Sarret P, Gendron L. Spinal activation of delta opioid receptors alleviates cancer-related bone pain. Neurosci. 2011;183:221–9. doi: 10.1016/j.neuroscience.2011.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bilsky EJ, Calderon SN, Wang T, et al. SNC 80, a selective, nonpeptidic and systemically active opioid delta agonist. J. Pharmacol. Exp. Ther. 1995;273:359–66. [PubMed] [Google Scholar]
- 80.Bilsky EJ, Bernstein RN, Pasternak GW, et al. Selective inhibition of [d-Ala2, Glu4] deltorphin antinociception by supraspinal, but not spinal, administration of an antisense oligodeoxynucleotide to an opioid delta receptor. Life Sci. 1994;55:PL37–PL43. doi: 10.1016/0024-3205(94)90110-4. [DOI] [PubMed] [Google Scholar]
- 81.Wild KD, McCormick J, Bilsky EJ, et al. Antinociceptive actions of BW373U86 in the mouse. J. Pharmacol. Exp. Ther. 1993;267:858–865. [PubMed] [Google Scholar]
- 82.Kreil G, Barra D, Simmaco M, et al. Deltorphin, a novel amphibian skin peptide with high selectivity and affinity for delta opioid receptors. Eur. J. Pharmacol. 1989;162:123–128. doi: 10.1016/0014-2999(89)90611-0. [DOI] [PubMed] [Google Scholar]
- 83.Pradhan AA, Befort K, Nozaki C, et al. The delta opioid receptor: an evolving target for the treatment of brain disorders. TIPS. 2011;32:581–90. doi: 10.1016/j.tips.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roemer D, Buescher HH, Hill RC, et al. A synthetic enkephalin analogue with prolonged parenteral and oral analgesic activity. Nature. 1977;268:547–549. doi: 10.1038/268547a0. [DOI] [PubMed] [Google Scholar]
- 85.Handa BK, Land AC, Lord JA, et al. Analogues of β-LPH61-64 possesing selective agonist activity at β-opiate receptors. Eur. J. Pharmacol. 1981;70:531–540. doi: 10.1016/0014-2999(81)90364-2. [DOI] [PubMed] [Google Scholar]
- 86.Sargent DF, Schwyzer R. Membrane lipid phase as catalyst for peptide receptor interactions. Proc. Natl Acad. Sci. USA. 1986;83:5774–5778. doi: 10.1073/pnas.83.16.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lowery JJ, Yeomans L, Keyari CM, et al. Glycosylation improves the central effects of DAMGO. Chem. Biol. Drug Des. 2007;69:41–47. doi: 10.1111/j.1747-0285.2007.00462.x. [DOI] [PubMed] [Google Scholar]
- 88.Yeomans L, Muthu D, Lowery JJ, et al. Phosphorylation of enkephalins: NMR and CD studies in aqueous and membrane-mimicking environments. Chem. Biol. Drug Des. 2011;78:749–56. doi: 10.1111/j.1747-0285.2011.01203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Egleton RD, Bilsky EJ, Tollin G, et al. Biousian glycopeptides penetrate the blood–brain barrier. Tetrahedron Asymm. 2005;16:65–75. [Google Scholar]
- 90.Taylor JW, Miller RJ, Kaiser ET. Structural characterization of beta-endorphin through the design, synthesis, and study of model peptides. Mol. Pharm. 1982;22:657–666. [PubMed] [Google Scholar]
- 91.Kaiser ET, Kézdy FJ. Amphiphilic secondary structure: design of peptide hormones. Science. 1984;223:249–255. doi: 10.1126/science.6322295. [DOI] [PubMed] [Google Scholar]
- 92.Palian MM, Boguslavsky VI, O'Brien DF, et al. Glycopeptide-membrane interactions: glycosyl enkephalin analogues adopt turn conformations by NMR and CD in amphipathic media. J. Am. Chem. Soc. 2003;125(19):5823–5831. doi: 10.1021/ja0268635. [DOI] [PubMed] [Google Scholar]
- 93.Dhanasekaran M, Alves I, Yeomans L, et al. Glycopeptides related to β-endorphin adopt helical amphipathic conformations in the presence of lipid bilayers. J. Am. Chem. Soc. 2005;127:5435–5448. doi: 10.1021/ja0432158. [DOI] [PubMed] [Google Scholar]
- 94.Neumann J, Couvineau A, Murail S, et al. Class-B GPCR activation: is ligand helix-capping the key? TIBS. 2008;33:314–319. doi: 10.1016/j.tibs.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 95.Gronenborn AM, Bovermann G, Clore GM. A 1H-NMR study of the solution conformation of secretin. Resonance assignment and secondary structure. FEBS Lett. 1987;215:88–94. doi: 10.1016/0014-5793(87)80119-9. [DOI] [PubMed] [Google Scholar]
- 96.Neidigh JW, Fesinmeyer RM, Prickett KS, et al. Exendin-4 and glucagon-like-peptide-1: NMR structural comparisons in the solution and micelle-associated states. Biochemistry. 2001;40:13188–13200. doi: 10.1021/bi010902s. [DOI] [PubMed] [Google Scholar]
- 97.Sasaki K, Dockerill S, Adamiak DA, et al. X-ray analysis of glucagon and its relationship to receptor binding. Nature. 1975;257:751–757. doi: 10.1038/257751a0. [DOI] [PubMed] [Google Scholar]
- 98.Jin L, Briggs Sl, Chandrasekhar S, et al. Crystal structure of human parathyroid hormone 1–34 at 0.9-Å resolution. J. Biol. Chem. 2000;275:27238–27244. doi: 10.1074/jbc.M001134200. [DOI] [PubMed] [Google Scholar]
- 99.Inooka H, Ohtaki T, Kitahara O, et al. Conformation of a peptide ligand bound to its G-protein coupled receptor. Nat. Struct. Biol. 2001;8:161–165. doi: 10.1038/84159. [DOI] [PubMed] [Google Scholar]
- 100.Lefever MR. Ph.D. Dissertation. The University of Arizona; USA: 2010. Design, synthesis and characterization of helical opioid glycopeptides and fluorescent derivatives including optimization of serine glycosylation utilizing sugar acetates. [Google Scholar]
- 101.Li Y. Ph.D. Dissertation. The University of Arizona; USA: 2011. Design, synthesis, and characterization of helical opioid glycopeptide agonists: the study of the structure-activity relationship and transport of glycopeptides related to beta-endorphin and dynorphin. [Google Scholar]
- 102.Job GE, Kennedy RJ, Heitmann B, et al. Temperature- and length-dependent energetics of formation for polyalanine helices in water: assignment of w(Ala)(n,T) and temperature-dependent CD ellipticity standards. J. Am. Chem. Soc. 2006;128:8227–8233. doi: 10.1021/ja060094y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chin DH, Woody RW, Rohl CA, et al. Circular dichroism spectra of short, fixed-nucleus alanine helices. Proc. Natl Acad. Sci. USA. 2002;99:15416–15421. doi: 10.1073/pnas.232591399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Biondi B, Giannini E, Negri L, et al. Opioid peptides: synthesis and biological activity of new endomorphin analogues. Int. J. Peptide Res. Therap. 2006;12:145–151. [Google Scholar]
- 105.Negri L, Lattanzi R, Tabacco F, et al. Dermorphin and deltorphin glycosylated analogues: synthesis and antinociceptive activity after systemic administration. J. Med. Chem. 1999;42:400–404. doi: 10.1021/jm9810699. [DOI] [PubMed] [Google Scholar]
- 106.Biondi L, Filira F, Giannini E, et al. Novel glycosylated [Lys7]-dermorphin analogues: synthesis, biological activity and conformational investigations. J. Pept. Sci. 2007;13:179–189. doi: 10.1002/psc.829. [DOI] [PubMed] [Google Scholar]
- 107.Dangoor D, Biondi B, Gobbo M, et al. Novel glycosylated VIP analogs: synthesis, biological activity, and metabolic stability. J. Pept. Sci. 2008;14:321–328. doi: 10.1002/psc.932. [DOI] [PubMed] [Google Scholar]
- 201.Cox BM, Borsodi A, Caló G, et al. Opioid receptors, introductory chapter. IUPHAR database. www.iuphar-db.org/DATABASE/FamilyIntroductionForward?familyId=50.