

Abstract
The kinase pathway comprising RAS, RAF, mitogen-activated protein kinase kinase (MEK) and extracellular signal regulated kinase (ERK) is activated in most human tumours, often through gain-of-function mutations of RAS and RAF family members1. Using small-molecule inhibitors of MEKand an integrated genetic and pharmacologic analysis, we find that mutation of BRAF is associated with enhanced and selective sensitivity to MEK inhibition when compared to either ‘wild-type’ cells or cells harbouring a RAS mutation. This MEK dependency was observed in BRAF mutant cells regardless of tissue lineage, and correlated with both downregulation of cyclin D1 protein expression and the induction of G1 arrest. Pharmacological MEK inhibition completely abrogated tumour growth in BRAF mutant xenografts, whereas RAS mutant tumours were only partially inhibited. These data suggest an exquisite dependency on MEK activity in BRAF mutant tumours, and offer a rational therapeutic strategy for this genetically defined tumour subtype.
Activating RAS and BRAF mutations typically demonstrate mutual exclusivity in tumours1–3. This suggests an epistatic relationship whereby either mutation is sufficient to deregulate a common effector pathway such as the MEK–ERK kinase cascade. If so, tumours arising as a result of mutation to either RAS or BRAF should harbour similar downstream dependencies that might represent useful therapeutic targets. To test this hypothesis, we examined the consequences of MEK–ERK pathway inhibition in a collection of cancer cell lines that exhibited differing mechanisms of MAP kinase pathway deregulation. Cell lines containing the NRAS(Q61R) or BRAF(V600E) mutations (present in ~15% and ~50% of melanomas, respectively) were analysed alongside a panel of cancer cell lines that lacked both mutations (hereafter referred to as RAS/BRAF-WT). Several of these RAS/BRAF-WT cell lines exhibit levels of ERK phosphorylation comparable to those observed in the setting of RAS or RAF mutation.
MEK1/2 are dual-specificity kinases that phosphorylate and activate ERK, the classical MAP kinase4. To inhibit MEK–ERK, we used the potent and selective MEK inhibitor CI-1040 (ref. 5). CI-1040 is a non-competitive inhibitor of MEK1/2 with a Ki of 300nM in vitro5,6. The only other known CI-1040 target is the MEK5 kinase; however, its inhibition occurs at a 100-fold greater concentration than that required for inhibition of MEK1/2 (ref. 7). Because ERK is the only known MEK substrate, we reasoned that selective MEK inhibition might clarify the role of the MAP kinase pathway in differing genetic contexts.
CI-1040 inhibited MEK (as measured by phosphorylated ERK (p-ERK) levels) with a half-maximal inhibitory concentration (IC50) of 100–500nM in all cell lines tested (Fig. 1 and data not shown). In contrast, the IC50 for growth inhibition by CI-1040 differed markedly in a manner that correlated with the mechanism of ERK activation (Fig. 1a). Whereas RAS/BRAF-WT cells exhibited resistance to CI-1040 even at concentrations in vast excess of those required for ERK inhibition, cells harbouring a BRAF mutation were exquisitely sensitive, with IC50 values of 0.024–0.111 µM (Fig. 1a). Surprisingly, RAS mutant cells did not demonstrate the same sensitivity despite effective inhibition of p-ERK (Fig. 1b and data not shown). These data raised the possibility that RAS and BRAF mutant cancer cells might be differentially dependent on signalling mechanisms that involve MEK, despite their known epistatic relationship in human cancers.
Figure 1. The BRAF(V600E) mutation confers sensitivity to the MEK inhibitor CI-1040.
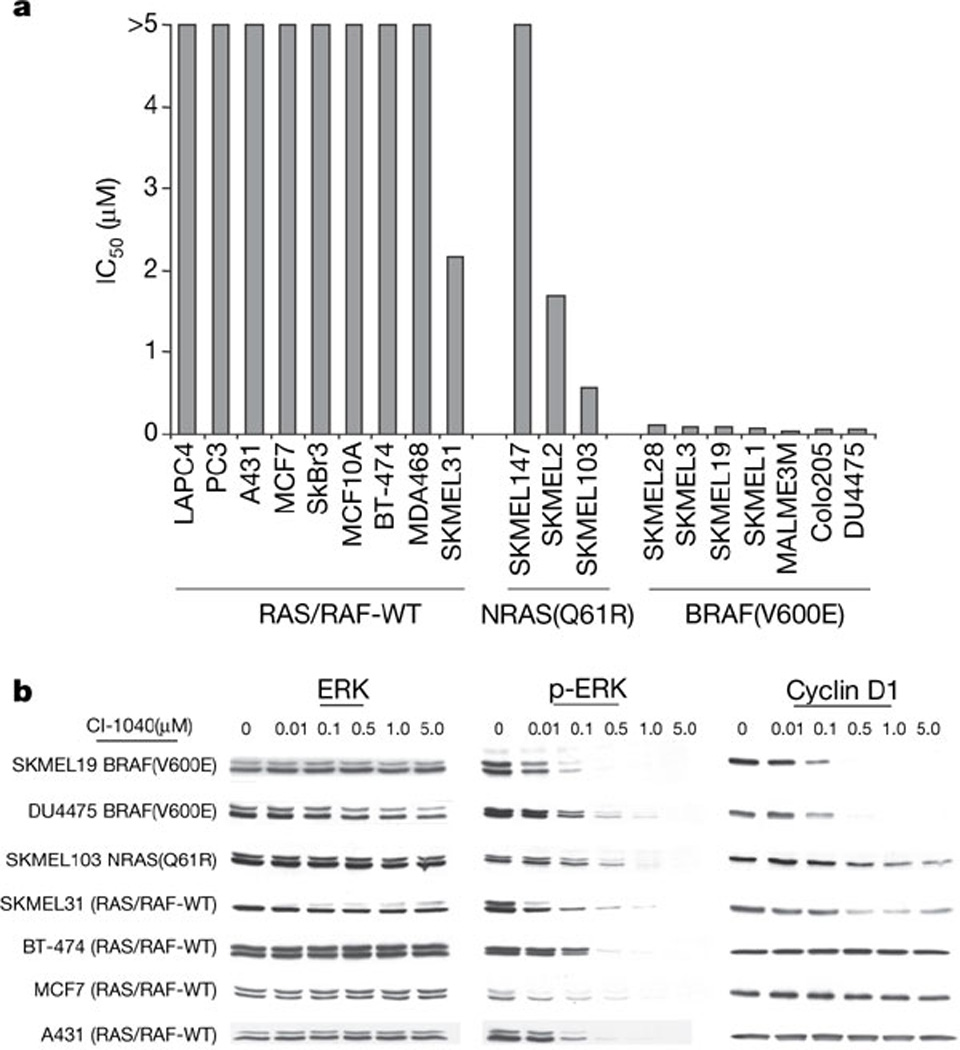
a, CI-1040 IC50 values as a function of BRAF and NRAS mutational status. b, Immunoblot of p-ERK and total ERK, demonstrating that CI-1040 inhibits MAPK activity with IC50 values ranging from 100 to 500 nM. Cells were treated for 24 h. MEK inhibition caused profound downregulation of cyclin D1 expression in BRAF mutant tumour cells. In contrast, cyclin D1 declined only modestly in SKMEL103 melanoma cells with the NRAS(Q61R) mutation and in SKMEL31 RAS/BRAF-WT cells. Cyclin D1 expression was unaffected by CI-1040 in non-melanoma cells with wild-type RAS and BRAF.
To explore this hypothesis in an unbiased manner, we interrogated the large-scale chemical sensitivity data available for the NCI60 cancer cell lines8 using supervised learning methods previously applied to the analysis of gene-expression data. NCI60 cell lines were partitioned into two classes according to the presence or absence of the BRAF(V600E) mutation. We then performed a supervised analysis9 where the mean −log10(GI50) values for each compound in the BRAF(V600E) and non-mutant classes were compared using a variance fixed t-test metric and ranked according to T-score (the GI50 is the concentration that inhibits cell growth by 50%). Thirty-six compounds exhibited significantly increased potency against the BRAF(V600E) class distinction (Fig. 2a and Supplementary Table S1; false discovery rate (FDR) = 0.25, nominal P value <3 × 10−4). The top-scoring compound against the BRAF(V600E) class was hypothemycin (a resorcylic acid lactone, the homologues of which possess potent and selective MEK inhibitory activity), which was found to inhibit p-ERK at a potency comparable to CI-1040 (Supplementary Fig. S1)10,11. Additional top-scoring compounds included protein LF (anthrax lethal factor), a zinc metalloproteinase known to inactivate MEK through enzymatic cleavage12, and PD98059 (ref. 13), a well-characterized MEK inhibitor. Thus, at least three of the most potent compounds against the BRAF(V600E) class distinction appeared to exert their effects through MEK inhibition. These results were consistent with the CI-1040 analysis and suggest that BRAF mutation might confer a preferential sensitivity to MEK inhibition in human cancer cells.
Figure 2. Chemical sensitivity associated with mutant BRAF and RAS class distinctions.
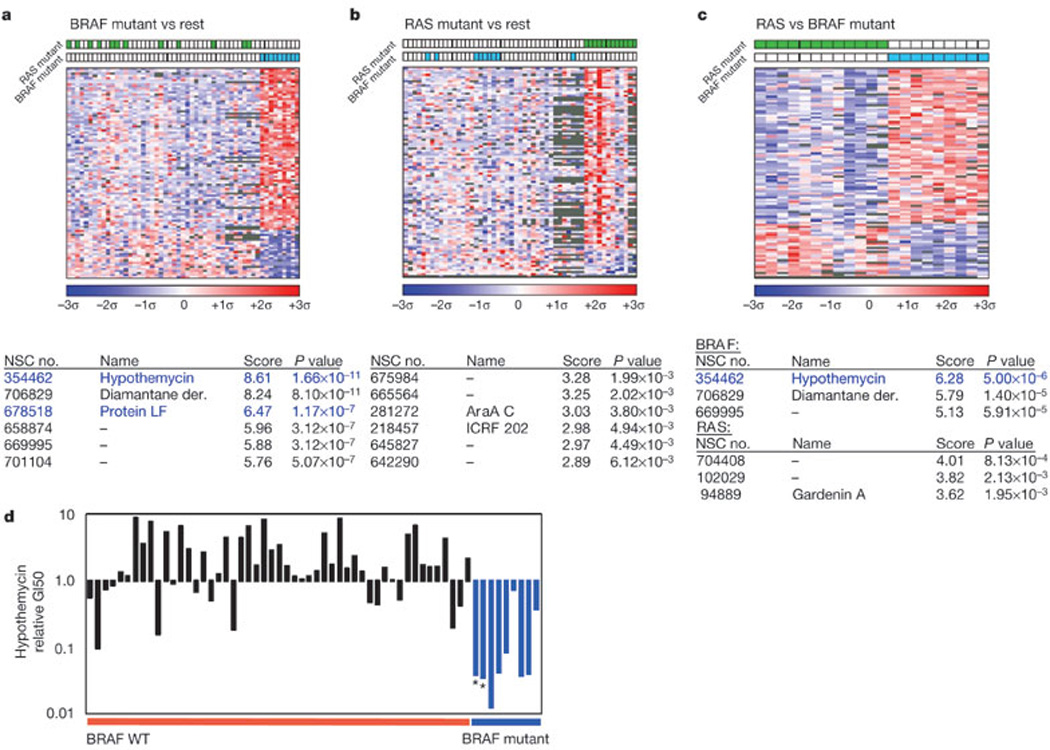
a–c, Colourgrams show BRAF mutant (a) or RAS mutant (b) versus the remaining NCI60 cells, or for mutant RAS versus mutant BRAF lines (c). Columns denote NCI60 cell lines; rows denote compounds; colour denotes the number of standard deviations above (red) or below (blue) the mean for all cell lines (top 100 compounds for each class distinction shown; see Methods). NSC numbers, names, variance-fixed T-scores (absolute values; see Methods) and asymptotic P values are shown for top-scoring compounds. Blue font indicates known MEK inhibitors. d, Relative GI50 values for the MEK inhibitor hypothemycin in non-haematological NCI60 cell lines. Black bars indicate BRAF wild-type cells; blue bars indicate BRAF(V600E) cells; asterisks indicate non-melanoma cell lines with the BRAF(V600E) mutation.
NCI60 cell lines that harbour RAS mutations are non-overlapping with respect to the BRAF(V600E) mutation, supporting the notion of a redundant pathway function8. To explore this further, supervised analysis of the NCI60 data was repeated, using the class distinction RAS mutant versus wild-type RAS. Surprisingly, and in contrast to the results observed for the BRAF(V600E) class distinction, no compound surpassed the Bonferroni significance threshold in the RAS mutant class (Fig. 2b). Conceivably, RAS and BRAF mutations might elicit similar dependencies despite these results, and our failure to identify compounds in the RAS analysis might reflect confounding genetic heterogeneity. Thus, we performed an additional supervised analysis that directly compared only the BRAF and RAS mutant lines. If these classes do indeed manifest common genetic dependencies, compounds that target the relevant mechanisms (for example, the MEK–ERK pathway) should fail to score by this class distinction. However, hypothemycin again distinguished BRAF and RAS mutant cells (Fig. 2c); protein LF also retained a high rank.
Because most BRAF(V600E) cell lines analysed were melanoma-derived, the enhanced sensitivity to MEK inhibition may have reflected a melanocytic lineage effect independent of the BRAF(V600E) mutation; however, several lines of evidence rendered this possibility unlikely. First, in the NCI60 analyses all BRAF(V600E) cell lines exhibited markedly reduced hypothemycin GI50 values relative to the mean across the sample set, regardless of tissue type (Fig. 2d). Colo205, an NCI60 colon cancer line with the BRAF(V600E) mutation, was also found to exhibit sensitivity to CI-1040 at an equivalent level to the melanoma cells (Fig. 1a). In addition, the two melanoma lines lacking a BRAF mutation were clearly indifferent to the effects of hypothemycin. Finally, only one of the breast/prostate cell lines demonstrated similar sensitivity to the drug: the breast cancer line DU-4475 (IC50 24 nM). Notably, sequencing of BRAF in this cell line showed that it also contained a V600E mutation. Thus, the sensitivity of cancer cell lines to MEK inhibition correlated most closely with BRAF mutation status.
In many cell types, RAS–RAF–MEK–ERK signalling is required for both D-cyclin expression and assembly of the cyclin D–cdk4 complex14. The marked sensitivity of BRAF mutant cells to MEK inhibitors allowed us to examine the functional consequences of MAP kinase blockade in this context. Treatment of BRAF mutant cell lines with CI-1040 caused a marked decline in D-cyclin protein levels (Figs 1b and 3a, d). In the SKMEL28 cell line, this decline was followed by loss of RB phosphorylation and a profound G1 cell cycle arrest (Fig. 3). G1 arrest was accompanied by apoptosis in several BRAF mutant cell lines (Fig. 3c, d), suggesting that MEK inhibition in a BRAF-mutant context exerts both cytocidal and cytostatic effects. In contrast, CI-1040 concentrations that completely inhibited p-ERK had no effect on cyclin D1 protein expression in the vast majority of RAS/BRAF-WT cells, as shown in Fig. 1b for the MCF7, BT-474 and A431 cell lines. BT-474 and A431 exhibited robust MAPK activity driven by HER2/neu and EGFR, respectively, suggesting that cyclin D1 expression and G1 progression are driven by MEK/ERK-independent mechanisms in certain RAS/BRAF-WT cells.
Figure 3. MEK inhibition causes loss of D-cyclin expression, RB hypophosphorylation and G1 arrest in BRAF mutant cancer cells.

a, Immunoblot showing the kinetics of change in p-ERK, D-cyclin expression and RB in SKMEL28 cells treated with 1 µM CI-1040. b, CI-1040 induced a G1 growth arrest in BRAF mutant tumour cells but not in RAS/BRAF-WT breast cancer cells (BT-474 shown). c, d, CI-1040 induces apoptosis in some but not all cancer cell lines with the BRAF(V600E) mutation as measured by FACS analysis (c) and PARP cleavage (d).
To determine whether the differential sensitivity to MEK inhibition observed for BRAF mutant cancer cells was re-capitulated in vivo, mice harbouring xenograft tumours were treated with the MEK inhibitor PD0325901. PD0325901 is a derivative of CI-1040 that has improved oral bioavailability and induces a longer duration of target suppression15. The effects of PD0325901 on tumour cells in vitro are qualitatively identical to those of CI-1040, including the marked selectivity for BRAF mutant cell lines, but occur at 100-fold lower concentrations (Supplementary Fig. S2).
Daily treatment with PD0325901 at doses of 5 and 25 mg kg−1 completely suppressed the growth of SKMEL28 and Colo205 BRAF(V600E) mutant xenografts (Fig. 4a and Supplementary Fig. S3; P < 0.01 for both 5 and 25 mg kg−1 versus control, P = 0.16 for 5 versus 25 mg kg−1). Growth suppression was associated with loss of D-cyclin expression, induction of p27 and hypophosphorylation of RB (Fig. 4e and Supplementary Fig. S4). In contrast, PD0325901 treatment of SKMEL103 (NRAS(Q61R)), SKMEL30 (NRAS(Q61R)) and SKMEL31 (RAS/BRAF-WT) xenografts at a dose of 5 mg kg−1 only delayed tumour growth, with complete growth suppression requiring 25 mg kg−1 (Fig. 4b, c and Supplementary Fig. S3b; P < 0.01 for 5 versus 25 mg kg−1, and 5 and 25 mg kg−1 versus control). BT-474 xenografts (BRAF/RAS-WT) were completely insensitive to PD0325901 (Fig. 4d). PD0325901 treatment at the doses studied was non-toxic and resulted in profound p-ERK inhibition in all xenograft models studied; however, RB phosphorylation, cyclin D expression and proliferation as measured by Ki67 were unaffected by MEK inhibition in PD0325901-resistant BT-474 xenografts (Fig. 4e, f), and there was no correlation between basal p-ERK levels and PD0325901 sensitivity (Supplementary Fig. S4). Thus, the MEK dependency characteristic of BRAF mutant tumour cells in vitro was also apparent in vivo.
Figure 4. PD0325901 completely suppresses the growth of BRAF(V600E) mutant xenografts.
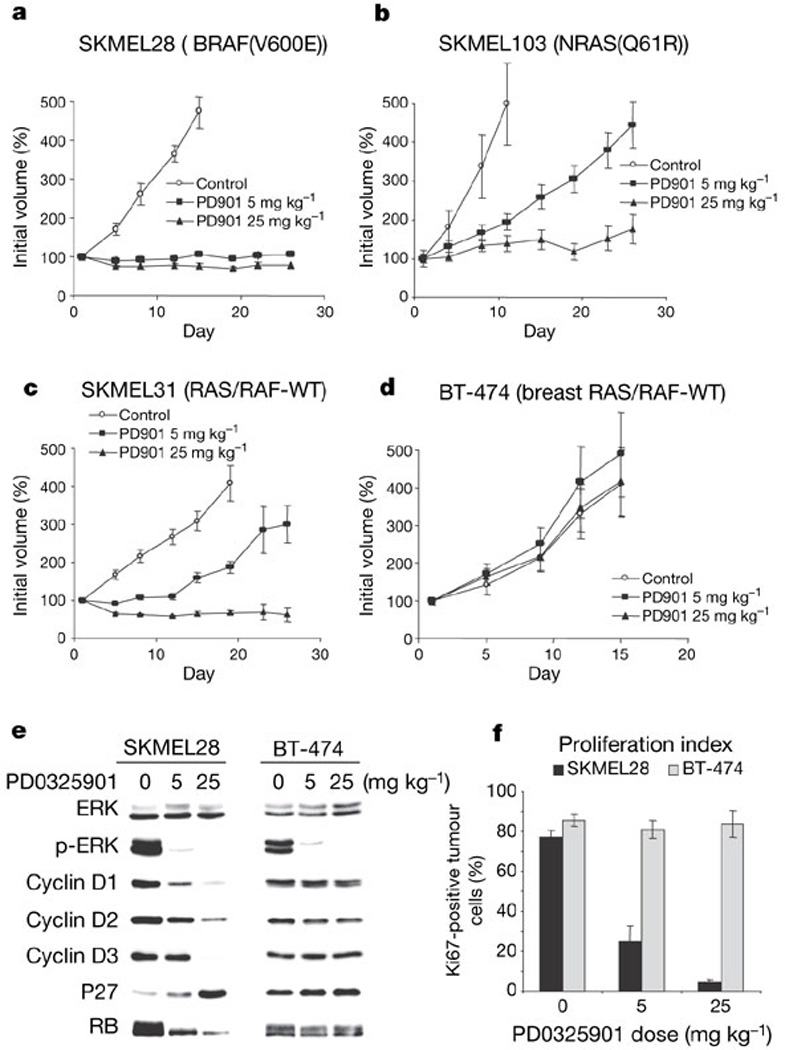
a, PD0325901 suppressed the growth of SKMEL28 (BRAF(V600E)) xenografts at both the 5 and 25 mg kg−1 dose levels. b, c, In contrast, 5 mg kg−1 PD0325901 only delayed the growth of SKMEL103 (RAS(Q61R)) and SKMEL31 (RAS/RAF-WT) xenografts, with complete growth suppression requiring the higher dose. d, BT-474 xenografts (RAS/RAF-WT) were refractory to MEK inhibition (n = 10 mice per group). e, f, PD0325901 reduced p-ERK levels in both SKMEL28 and BT-474 xenograft tumours but caused downregulation of D-cyclins, induction of p27 and hypophosphorylation of RB, and a decline in the proliferative index only in the SKMEL28 xenografts. Tumour lysates were derived from mice euthanized 8 h after the final treatment. All error bars indicate standard error.
Excess MAP kinase pathway activation occurs commonly in human tumours. In melanoma and other solid tumours, mutation of BRAF and RAS occurs frequently and tends to exhibit mutual exclusivity, suggesting that each mutation confers a similar selective advantage1. However, our findings suggest that tumour cells carrying BRAF mutations are much more reliant on MEK–ERK signalling than are RAS mutant cells, or cells that activate MAP kinase by other means. Thus, BRAF mutant cancer cells may harbour a critical dependency on MEK–ERK that renders them highly sensitive to pharmacological MEK inhibition.
BRAF mutations occur at a high frequency in melanomas, but are also observed in colon, lung and several other tumour types1,2. Expression of BRAF(V600E) in non-transformed melanocytes leads to constitutive ERK activation and tumorigenicity in mice, and depletion of BRAF but not A-RAF or C-RAF in BRAF(V600E) mutant melanoma cells reduces ERK activity16,17. Our data suggest that D-cyclin expression is also deregulated and ERK-dependent in BRAF-mutant tumours. Cyclin D downregulation may therefore mediate at least some of the anti-proliferative effects observed after MEK inhibition. On the other hand, MEK inhibition had little effect on D-cyclin expression in most BRAF/RAS-WT tumour cells. In these cells, mutations in the PTEN or phosphatidylinositol-3-OH kinase (PI(3)K) genes, or activation of other pathways, may drive D-cyclin expression in an ERK-independent fashion18,19. Our results are also consistent with a model in which ERK regulates G1 progression only in certain lineages (for example, melanocytes); presumably, such lineage differences in cell growth control contribute to the imbalanced frequency of RAS and BRAF mutations observed across tumour types.
RAS-dependent transformation has been found previously to require activation of cyclin D1 (refs 20–23). As both oncogenic RAF and activated ERK also induce cyclin D1 expression24,25, it has been presumed that in human tumours with RAS mutation, cyclin D1 expression was controlled by RAS-mediated MEK–ERK activation. However, our results suggest that in certain genetic contexts, including some tumours with RAS mutation, ERK signalling may be dispensable for cyclin D1 expression and cell proliferation. RAS family members have multiple other targets, such as PI(3)K and RalGDS; these may exert more prominent oncogenic effects in certain tumour subtypes, thereby reducing the requirement for MEK–ERK activation26,27. Our findings therefore raise the possibility that single-agent therapeutic strategies may prove insufficient in RAS mutant tumours. Instead, direct RAS inhibitors or combinatorial strategies may be required.
Thus far, the use of BRAF inhibitors in clinical trials has met with mixed results. On the other hand, the favourable therapeutic index and selectivity of MEK inhibitors may provide an appealing therapeutic strategy for BRAF mutant cancers. We therefore propose clinical trials of MEK inhibitors in which patients are stratified based on BRAF mutational status.
METHODS
Cell culture
CI-1040 and PD0325901 were obtained from Pfizer Global Research and Development. Drugs were dissolved in DMSO to yield 10 mM stock solutions and stored at −20 °C. All SKMEL lines were obtained from A. Houghton and P. Chapman with the remainder obtained from the ATCC. Cells were maintained in either RPMI or a 1:1 mixture of DMEM:F12 medium supplemented with 2 mM glutamine, 50 U ml−1 each of penicillin and streptomycin, and 10% heat-inactivated fetal bovine serum, and incubated at 37 °C in 5% CO2.
Alamar blue cell proliferation assay
Cells were plated in 96-well plates at a density of 2,000–5,000 cells per well. After 24 h, cells were treated with a range of drug concentrations prepared by serial dilution. The cells were exposed to Alamar blue (AccuMed International, OH) 3–5 days after drug treatment, and plates were read using a fluorescence spectrophotometer.
Western blot analysis
Treated cells were harvested, washed with PBS and lysed in NP40 lysis buffer (50 mM Tris (pH 7.4), 1% NP40, 150 mM NaCl, 40 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulphonylfluoride, and 10 µg ml−1 each of leupeptin, aprotinin and soybean trypsin inhibitor) for 30 min on ice. Lysates were centrifuged at 13,200 r.p.m. for 10 min and the protein concentration of the supernatant was determined by BCA assay (Pierce). Equal amounts of total protein were resolved by SDS–PAGE and transferred onto nitrocellulose membranes. Blots were probed overnight at 4 °C with antibody raised against the protein of interest. Anti-MAP kinase, phospho-MAP kinase, RB and cleaved PARP antibodies were obtained from Cell Signaling Technology. Anti-cyclin D1, anti-cyclin D2 and anti-cyclin D3, and p27 antibodies, were obtained from Santa Cruz Biotechnology. After incubation with horseradish peroxidase-conjugated secondary antibodies, proteins were detected using chemiluminescence (Amersham).
Apoptosis
After drug treatment, both adherent and floating cells were harvested and stained with ethidium bromide. Detection and quantification of apoptotic cells (sub-G1) were performed by flow cytometric analysis.
Animal studies
Four- to six-week-old nu/nu athymic female mice were obtained from the National Cancer Institute, Frederick Cancer Center and maintained in ventilated caging. Experiments were carried out under an IACUC approved protocol and institutional guidelines for the proper and humane use of animals in research were followed. Tumours were generated by injecting 0.5–1.0 × 107 tumour cells together with reconstituted basement membrane (Matrigel, Collaborative Research). For the BT-474 model, before tumour cell inoculation, 0.72 mg pellet−1 17β-estradiol pellets (Innovative Research of America) were inserted subcutaneously. Before initiation of treatment, mice were randomized to receive PD0325901 at a dose of 5 and 25 mg kg−1 or vehicle only as control. PD0325901 was formulated in 0.5% hydroxypropyl methylcellulose plus 0.2% Tween 80, and administered by oral gavage. Mice were killed by CO2 euthanasia. The average tumour diameter (two perpendicular axes of the tumour were measured) was measured in control and treated groups using a calliper. The data are expressed as the increase or decrease in tumour volume in mm3 (mm3 = π/6 × (larger diameter × (smaller diameter)2). Treatment arms were compared using the Wilcoxon rank sums test. To prepare lysates, tumour tissue was homogenized in 2% SDS lysis buffer and then processed as described above. For immunohistochemical studies, xenograft tumours were fixed overnight in paraformaldahyde followed by dehydration in graded ethanols.
Statistical methods
Pharmacological data (−log10(GI50)) for 42,796 compounds were downloaded from the NCI website (http://dtp.nci.nih.gov/docs/cancer/cancer_data.html). The GI50 data were used to populate a matrix with MATLAB software as previously described28. Briefly, where multiple NCS entries existed, the entry with the largest number of replicates was included; in cases where multiple entries had the same number of replicates, the largest mean (−log10(GI50)) value for the NCI60 data set was selected. Incomplete data were assigned as NaN (not a number) for statistical purposes. These GI50 data from all solid tumour NCI60 cell lines were used in a supervised analysis according to the class distinctions described. BRAF mutant status was determined based on published data1 and by genotyping assays performed by our group28. Because the GI50 data are non-gaussian with many (−log10(GI50)) values at or near 4, a variance-fixed t-test was used to calculate significance. Here, the mean and median standard deviation was calculated for compounds for which the mean −log10(GI50) values across the NCI60 set were between 6 and 7. For both calculations, the standard deviation was near 0.4; thus, this value was used as a minimum threshold standard deviation for the supervised analysis. Compounds with the top variance-fixed T-scores for the relevant class distinctions were selected for additional analysis; in Fig. 2, the absolute values of these scores are indicated. GI50 values and distributions for selected compounds were analysed through the NCI Developmental Therapeutic website.
Supplementary Material
Acknowledgements
The authors thank H. Ju, W. L. Wong and H. Tseng for technical assistance. This work was supported by grants from the National Institutes of Health (L.A.G., C.A.P., G.G., T.R.G., W.R.S. and N.R.), the William H. Goodwin and Alice Goodwin Foundation for Cancer Research, the MSKCC Experimental Therapeutics Program (D.B.S. and N.R.), the Waxman Foundation (D.B.S. and N.R.), the Howard Hughes Medical Institute (G.G. and T.R.G.), Golfers Against Cancer (D.B.S. and N.R.) and the American Society of Clinical Oncology (D.B.S. and C.A.P.).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information The authors declare competing financial interests: details accompany the paper at www.nature.com.
References
- 1.Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 2.Brose MS, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- 3.Gorden A, et al. Analysis of BRAF and N-RAS mutations in metastatic melanoma tissues. Cancer Res. 2003;63:3955–3957. [PubMed] [Google Scholar]
- 4.Crews CM, Alessandrini A, Erikson RL. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258:478–480. doi: 10.1126/science.1411546. [DOI] [PubMed] [Google Scholar]
- 5.Sebolt-Leopold JS, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nature Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 6.Ohren JF, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nature Struct. Mol. Biol. 2004;11:1192–1197. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- 7.Mody N, Leitch J, Armstrong C, Dixon J, Cohen P. Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 2001;502:21–24. doi: 10.1016/s0014-5793(01)02651-5. [DOI] [PubMed] [Google Scholar]
- 8.Stinson SF, et al. Morphological and immunocytochemical characteristics of human tumour cell lines for use in a disease-oriented anticancer drug screen. Anticancer Res. 1992;12:1035–1053. [PubMed] [Google Scholar]
- 9.Golub TR, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 10.Zhao A, et al. Resorcylic acid lactones: naturally occurring potent and selective inhibitors of MEK. J. Antibiot. (Tokyo) 1999;52:1086–1094. doi: 10.7164/antibiotics.52.1086. [DOI] [PubMed] [Google Scholar]
- 11.Dombrowski A, et al. Production of a family of kinase-inhibiting lactones from fungal fermentations. J. Antibiot. (Tokyo) 1999;52:1077–1085. doi: 10.7164/antibiotics.52.1077. [DOI] [PubMed] [Google Scholar]
- 12.Chopra AP, Boone SA, Liang X, Duesbery NS. Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J. Biol. Chem. 2003;278:9402–9406. doi: 10.1074/jbc.M211262200. [DOI] [PubMed] [Google Scholar]
- 13.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 14.Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1) Proc. Natl Acad. Sci. USA. 1998;95:1091–1096. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nature Rev. Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 16.Wellbrock C, et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–2342. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- 17.Karasarides M, et al. B-RAF is a therapeutic target in melanoma. Oncogene. 2004;23:6292–6298. doi: 10.1038/sj.onc.1207785. [DOI] [PubMed] [Google Scholar]
- 18.Muise-Helmericks RC, et al. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J. Biol. Chem. 1998;273:29864–29872. doi: 10.1074/jbc.273.45.29864. [DOI] [PubMed] [Google Scholar]
- 19.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filmus J, et al. Induction of cyclin D1 overexpression by activated ras. Oncogene. 1994;9:3627–3633. [PubMed] [Google Scholar]
- 21.Liu JJ, et al. Ras transformation results in an elevated level of cyclin D1 and acceleration of G1 progression in NIH 3T3 cells. Mol. Cell. Biol. 1995;15:3654–3663. doi: 10.1128/mcb.15.7.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albanese C, et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 23.Aktas H, Cai H, Cooper GM. Ras links growth factor signalling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Mol. Cell. Biol. 1997;17:3850–3857. doi: 10.1128/mcb.17.7.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerkhoff E, Rapp UR. Induction of cell proliferation in quiescent NIH 3T3 cells by oncogenic c-Raf-1. Mol. Cell. Biol. 1997;17:2576–2586. doi: 10.1128/mcb.17.5.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem. J. 1997;326:61–68. doi: 10.1042/bj3260061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamad NM, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045–2057. doi: 10.1101/gad.993902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez-Garcia A, et al. RalGDS is required for tumour formation in a model of skin carcinogenesis. Cancer Cell. 2005;7:219–226. doi: 10.1016/j.ccr.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 28.Garraway LA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.