

Abstract
Objective
To evaluate changes of nuclear factor-kappa B (NF-κB) during radioiodine 131 (131I) therapy and whether NF-κB inhibition could enhance 131I-induced apoptosis in differentiated thyroid cancer (DTC) cells in a synergistic manner.
Methods
Three human DTC cell lines were used. NF-κB inhibition was achieved by using a NF-κB inhibitor (Bay 11-7082) or by p65 siRNA transfection. Methyl-thiazolyl-tetrazolium assay was performed for cell viability assessment. DNA-binding assay, luciferase reporter assay, and Western blot were adopted to determine function and expression changes of NF-κB. Then NF-κB regulated anti-apoptotic factors XIAP, cIAP1, and Bcl-xL were measured. Apoptosis was analyzed by Western blot for caspase 3 and PARP, and by flow cytometry as well. An iodide uptake assay was performed to determine whether NF-κB inhibition could influence radioactive iodide uptake.
Results
The methyl-thiazolyl-tetrazolium assay showed significant decrease of viable cells by combination therapy than by mono-therapies. The DNA-binding assay and luciferase reporter assay showed enhanced NF-κB function and reporter gene activities due to 131I, yet significant suppression was achieved by NF-κB inhibition. Western blot proved 131I could increase nuclear NF-κB concentration, while NF-κB inhibition reduced NF-κB concentration. Western blot also demonstrated significant up-regulation of XIAP, cIAP1, and Bcl-xL after 131I therapy. And inhibition of NF-κB could significantly down-regulate these factors. Finally, synergism induced by combined therapy was displayed by significant enhancements of cleaved caspase 3 and PARP from Western blot, and of Annexin V positively staining from flow cytometry. The iodine uptake assay did not show significant changes when NF-κB was inhibited.
Conclusion
We demonstrated that 131I could induce NF-κB activation, which would attenuate 131I efficacy in DTC cells. NF-κB inhibition by Bay 11-7082 or by p65 siRNA transfection was effective in suppressing NF-κB regulated anti-apoptotic changes and in combined regimen apoptosis was achieved synergistically.
Introduction
Thyroid nodule is a very common clinical problem and thyroid cancer is increasingly prevalent nowadays [1]. Differentiated thyroid cancer (DTC), including papillary and follicular thyroid cancer, comprises the majority of all thyroid cancers. Although the overall prognosis for DTC is good if total thyroidectomy and radioiodine 131 (131I) therapies are applied [2], patients with 131I-refractory metastases could only achieve a 10% 10-year survival rate [3], [4]. And for some cases, even 131I-avid lesions could not be successfully controlled by 131I therapy alone [5]. Therefore, development of novel anti-cancer methods is urgently needed for thyroid cancer.
In recent years, a number of culprit molecular targets have been identified in DTC carcinogenesis [6], [7], [8]. Among these, an emerging body of evidence shows that nuclear factor-kappa B (NF-κB) plays a crucial role in thyroid cancer, including cancer development and progression [6], [7], [9], [10], [11], [12], [13], [14]. It is also demonstrated that NF-κB induction by chemotherapy or radiotherapy could attenuate therapeutic efficacies. And NF-κB inhibition could promote thyroid cancer cell apoptosis, and to achieve synergistic effects [8], [9], [10], [11], [12]. However, to our knowledge, there has been no study investigating the relationship between NF-κB and 131I therapy in DTC, despite the importance of 131I treatment in DTC management.
Therefore, the purpose of the present research was to evaluate changes of NF-κB during 131I therapy. And we also aimed to determine whether combination with a NF-κB inhibitor or small interference RNA (siRNA) transfection could enhance 131I-induced apoptosis in DTC in a synergistic manner.
Materials and Methods
Cell culture
The human papillary thyroid carcinoma cell lines KTC-1, TPC-1 and follicular thyroid carcinoma cell line WRO were kindly provided by Dr. Shunichi Yamashita and Dr. Norisato Mitsutake (Department of Molecular Medicine, Atomic Bomb Disease Institute, Nagasaki University Graduate School of Biomedical Sciences, Nagasaki, Japan). DTC cells were grown in Dulbecco's minimum essential medium (GIBCO BRL, NY, USA) supplemented with 5% fetal bovine serum (GIBCO BRL, NY, USA), 1% (w/v) penicillin/streptomycin (Sigma-Aldrich, MO, USA) and 1 mU/mL thyrotropin (Sigma-Aldrich, MO, USA) in a 5% CO2 humidified atmosphere at 37°C.
Transfection with siRNA
SMARTpool NF-κB p65 siRNAs was commercially designed by Dharmacon (Dallas, TX, USA). The pool of siRNAs contained the p65-specific sequences. Scrambled oligonucleotides (sequence AATTCTCCGAACGTGTCACGT) were chemically synthesized by SBS GeneTech (Beijing, China) and it was analyzed in a BLAST search to exclude homology to p65 or other genes [15]. When DTC cells were at 50%–60% confluence, transfection of p65 SMARTpool siRNAs (100 nmol/L), scrambled oligonucleotides (100 nmol/L) or no oligonucleotide (control) was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for 6 hours at 37°C in 6-well plates. Following transfection, cells were grown in new mediums and treated or collected as indicated.
Methyl-thiazolyl-tetrazolium (MTT) assay
DTC cells (100 µl, 6000 cells per well) were seeded to 96-well plates and incubated for 48 hours before any treatment. Then new mediums were changed, and 131I (Beijing Atom HighTech, Beijing, China) or Bay 11-7082 (Sigma-Aldrich, MO, USA) were added in order to achieve final 131I activity concentrations of 5, 10, 20, 50, 100 and 200 MBq/ml or final Bay 11-7082 concentrations of 1, 2, 5, 10 and 20 µmol/L in each well. Cells were exposed to treatments for 48 hours, and 6 replicates were used for each concentration of either drug. In the control wells DMSO was added, and final concentration of DMSO did not exceed 0.2% in any well. After incubation, the cells were treated with MTT (50 µg/well, Sigma-Aldrich, MO, USA) for 1 hour at 37°C. The generated formazan was solubilized with 150 µl/well of DMSO, and the optical densities of the wells were measured at 450 nm with a Multiskan MS Plate Reader (Labsystems, Helsinki, Finland).
Proliferation of DTC cell lines after siRNA transfection was also measured by MTT assay. After transfection with p65 siRNA, scrambled oligonucleotides or no oligonucleotide control, cells were transferred as 6 replicates to 96-well plates at a concentration of 6000 cells per well. After 48 hours of incubation, cells were treated with or without 20 MBq/ml of 131I for 48 hours. Then MTT was performed as above.
Preparation of cell extracts
After different treatments, cells were lysed in high salt buffer [9]. For total cell protein, harvested cells were suspended in 100 µl of lysis buffer (20 mM Tris-HCl pH 7.5, 1 mM EDTA, 150 mM NaCl, 0.5% Triton-X, 50 mM NaF, 10 mM sodium pyrophosphate, 1 mM sodium orthovanadate and 2 mM phenylmethyisulfonylfluoride) for 20 minutes on ice. Then lysate was centrifuged for 15 minutes at 14,000 rpm, and the supernatant was stored at −80°C until use. For nuclear protein, harvested cells were suspended in 400 µL of lysis buffer (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonylfluoride and 0.1% Nonidet P-40) for 20 minutes on ice. Then lysate was centrifuged for 5 minutes at 14,000 rpm. The pellets were resuspended in 40 µL of nuclear extract buffer (20 mM HEPES pH 7.9, 420 mM NaCl, 1.5 mM MgCl2, 1 mM dithiothreitol, 0.2 mM EDTA, 0.5 mM phenylmethylsulfonylfluoride and 20% glycerol) for 20 minutes on ice. After centrifuge for 15 minutes at 14,000 rpm, the supernatant was collected and stored at −80°C until use. Protein concentrations were determined with a bicinchoninic acid assay reagent kit (Sigma-Aldrich, MO, USA).
DNA-binding assay
The multi-well colorimetric assay for active NF-κB was performed [9]. Briefly, equal amount of nuclear extracts were incubated in 96-well plates coated with immobilized oligonucleotide containing a NF-κB consensus binding site. NF-κB binding to the target oligonucleotide was detected with primary antibody specific to p65 subunit and HRP-conjugated secondary antibody. For quantification of activity, optical densities were measured at 450 nm with a Multiskan MS Plate Reader.
Luciferase assay
DTC cells were seeded in 24-well plates at 1×105 cells per well. The cells were co-transfected with 400 ng of pNF-κB-luc (Clontech, Mountain View, CA, USA) and 4 ng of pRL-SV40 (Promega, Madison, WI, USA) using Lipofectamine 2000. The pRL-SV40 plasmid with a cDNA encoding Renilla luciferase was used as an internal control in each experiment [16]. Cells were rested for 12 hours after transfection, then incubated with or without 131I, or with combined therapy of 131I plus Bay 11-7082 for 6 hours. Activities of firefly and Renilla luciferases were determined sequentially from a single sample with the Dual-luciferase Reporter Assay system (Promega, Madison, WI, USA) using a Lumat LB 9507 luminometer (Bethold Technologies, Bad Wildbad, Germany).
DTC cells were co-transfected with pNF-κB-luc and pRL-SV40 at 24 hours after p65 siRNA or scramble transfection [15]. After 12 hours, the cells were treated with or without 20 MBq/ml of 131I for 6 hours. Then the reporter gene activities were assayed as described above.
Western blot
Equal amounts of protein were electrophoresed by SDS-PAGE in 10% or 15% polyacrylamide gels. Proteins were transferred onto nitrocellulose membrane (Amersham Biosciences, NJ, USA) by semidry blotting. Membranes were blocked with Tris-buffered saline/0.1% Tween 20 (TBST) containing 5% milk for 60 minutes at room temperature, and then immune-blotted with appropriately diluted primary antibodies at 4°C overnight. After washing three times with TBST, the blots were incubated with HRP-conjugated secondary antibody for 60 minutes at room temperature. Then the complexes were visualized in iChemi XR imaging system (Syngene, MD, USA) by using chemiluminescence reagents (Millipore, MA, USA). Semi-quantification was performed by Quantity One Software Version 4.6.2 (Bio-Rad, CA, USA). Antibodies for NF-κB p65, X-linked inhibitor of apoptosis (XIAP), B-cell lymphoma extra large (Bcl-xL), cleaved caspase 3, poly-ADP-ribose polymerase (PARP) and β-actin were from Cell Signaling Technology (Beverly, MA, USA). Antibody for cellular inhibitor of apoptosis 1 (cIAP1) was from R&D Systems (Minneapolis, MN, USA). And antibody for proliferating cell nuclear antigen (PCNA) was from BD Transduction Laboratories (San Jose, CA, USA).
Flow cytometry analysis of apoptotic cells
After 24 hours of different treatments, adherent cells were harvested by trypsinization, and 4×105 cells were double stained with FITC-conjugated annexin V and propidium iodide for 15 minutes at room temperature in a Ca2+-enriched binding buffer (BD Pharmingen, CA, USA) and then subjected to FACSAria™ flow cytometer (BD Biosciences, CA, USA). FITC and propidium iodide emissions were detected in FL-1 and FL-3 channels, respectively. Analysis was done with Cell Quest software (BD Biosciences, CA, USA).
Iodide uptake assay
To determine whether NF-κB inhibition can influence radioactive iodide uptake, iodide uptake assay was performed as described by Weiss et al. [17] with some modifications. KTC-1 cells were seeded in 6-well plates and treated with or without 5 µmol/L Bay 11-7082 for 24 hours. Then cells were cultured with 1 mL new medium per well, containing 37 KBq Na125I (Beijing Atom HighTech, Beijing, China), for 1 hour. Afterwards, the 125I-containing medium was decanted. Then cells were washed twice with PBS and trypsinized for total cell number counting. Finally radioactivity of the cells was measured with a γ counter (LKB 1261 Gamma Counter, Wallac, Turku, Finland).
To measure iodide uptake changes after siRNA transfection, KTC-1 cells transfected with p65 siRNA, scrambled oligonucleotides or no oligonucleotide control were transferred to 6-well plates. Then cells were incubated in the present of Na125I for 1 hour. Then cell number was counted, and radioactivity was measured.
Statistical Analysis
All data were presented as mean ± SD. Statistics were performed with SPSS 15.0 (SPSS Incorporated, IL, USA). Differences between groups were analyzed by one-way analysis of variance (ANOVA) or independent samples T-Test. Least significant difference (LSD) test was used for multiple comparisons among groups. P value not exceeding 0.05 was considered statistically significant.
Results
Cytotoxic effect of 131I, Bay 11-7082 and siRNA therapies
MTT assay was done after 48 hours of different treatments, and cell viabilities were calculated based on the total number of cells grown without any therapeutic intervention. As illustrated in Figure 1A and 1B, survival rates of all three cell lines showed inverse relationship to 131I activity concentrations and Bay 11-7082 dosage concentrations. For the following in vitro experiments, 131I activity concentration and Bay 11-7082 dosage concentration were determined as 20 MBq/ml and 5 µmol/L, respectively. Figure 1C showed that, by ANOVA, different treatment engendered different growth-inhibitory effects (P<0.01). And LSD revealed significant decrease of viable cells by combination therapy than by either mono-therapy (P<0.01).
Figure 1. Cytotoxic effects of 131I, Bay 11-7082 and siRNA therapies on thyroid cancer cells.

After exposure to indicated concentrations of Bay 11-7082 (A), activity concentrations of 131I (B) or combined therapies (C) for 48 hours, cell viabilities were determined by MTT assay. After DTC cells were treated with no oligonucleotide control, scrambled oligonucleotides or p65 siRNA transfection, cells were transferred to 96-well plates. Then cells were treated with or without 131I (20 MBq/ml) for 48 hours, and MTT was performed (D). Similar results were obtained in three independent experiments.
Figure 1D showed that scrambled oligonucleotides transfection did not inhibit DTC cell proliferation, while siRNA transfection could achieve obvious inhibitory effects (P<0.05). Moreover, combination therapy could significantly induce stronger cell death than either 131I therapy or siRNA transfection (P<0.01).
Effects of different therapies on NF-κB
To examine the effects of 131I on NF-κB, we performed DNA-binding assay using nuclear extracts from 131I-treated DTC cells for 6, 24 and 48 hours with untreated cells as control. NF-κB function increased during the time course of 131I mono-therapy, reaching peaks at 24-hour time point for KTC-1 and WRO cells. However, combination with Bay 11-7082 could significantly reduced NF-κB function (Figure 2A). In the above three time points, significant NF-κB inhibition was shown by T-Test in the combined therapies than 131I mono-therapies in all three cell lines (P<0.01). DTC cells were also transfected with p65 siRNA, scrambled oligonucleotides or no oligonucleotide control. Then three groups of all the cell lines were exposed to 131I for 24 hours. Although NF-κB function was strongly induced by 131I in both control and scramble groups, significant suppression could be obtained by p65 siRNA transfection (Figure 2B) in all three cell lines (P<0.01).
Figure 2. 131I-induced NF-κB activation can be reduced by Bay 11-7082 or p65 siRNA treatments.

(A) DTC cells were treated either with 131I (20 MBq/ml) or in combination with Bay 11-7082 (5 µmol/L) for 6, 24 and 48 hours. Nuclear extracts were prepared, and DNA-binding assay was done. (B) DTC cells were treated with no oligonucleotide control, scrambled oligonucleotides or p65 siRNA transfection. Then cells were exposed to 131I for 24 hours, and DNA-binding assay was performed. Similar results were obtained in three independent experiments.
NF-κB responsive promoter activity was performed as well (Figure 3). We found that 131I significantly enhanced the NF-κB reporter gene activities, to about 2.5, 1.7 and 3.0 folds in KTC-1, TPC-1 and WRO cell lines (P<0.05). Nevertheless, combination with Bay 11-7082 could inhibit NF-κB functions to about 0.16, 0.14 and 0.24 folds of the induced levels in KTC-1, TPC-1 and WRO cell lines (P<0.01). And p65 siRNA transfection could reduce NF-κB functions to nearly 0.14, 0.14 and 0.26 folds of the induced levels respectively (P<0.01).
Figure 3. Repoter gene activity changes after different treatments.

(A) DTC cells were transfected with pNF-κB-luc, and pRL-SV40 served as an internal control. After 12 hours, cells were left untreated or incubated with 131I (20 MBq/ml), or in combination with Bay 11-7082 (5 µmol/L) for 6 hours. NF-κB activation was detected by luciferase reporter assay. (B) At 24 hours after transfection with scrambled oligonucleotides or p65 siRNA, cells were co-transfected with pNF-κB-luc and pRL-SV40. After 12 hours, cells were left untreated or treated with 131I (20 MBq/ml) for 6 hours. Then luciferase reporter assay was performed. Similar results were obtained in three independent experiments.
Next we examined nuclear NF-κB protein levels 6 hours after different treatments by Western blot (Figure 4). The amounts of p65 increased in 131I groups in all the cell lines, but suppressed obviously when Bay 11-7082 was added in the combination therapy. The similar results were obtained by p65 siRNA transfection, while scrambled oligonucleotides transfection could not achieve such effects.
Figure 4. NF-κB protein level changes by different therapies.

DTC cells were left untreated as controls (A), or treated with 20 MBq/ml 131I (B) or in combination with 5 µmol/L Bay 11-7082 (C) for 6 hours. Then nuclear protein extracts were examined for NF-κB p65 by Western blot. In the second set of experiments, control cells were left untreated (D). After no oligonucleotide (E), scrambled oligonucleotides (F) or p65 siRNA (G) transfection, DTC cells were treated with 131I (20 MBq/ml) for 6 hours. Then nuclear protein levels of NF-κB p65 were analyzed. PCNA was used as a loading control for nuclear proteins. Similar results were obtained in three independent experiments.
Effects of different therapies on NF-κB regulated anti-apoptotic factors
As being potent apoptosis inhibitors, XIAP, cIAP1 and Bcl-xL are target genes positively regulated by NF-κB [7], [18]. We tested what influences different treatments could inflict on them by Western blot, using KTC-1 as a representative cell line (Figure 5). Despite certain basal levels of XIAP, cIAP1 and Bcl-xL, 131I increased XIAP to 5.5–5.7 folds, cIAP1 to 4.8–6.7 folds and Bcl-xL to 3.6–5.1 folds, respectively. However, after combination with Bay 11-7082, XIAP, cIAP1 and Bcl-xL protein expressions were markedly reduced to 0.09, 0.10 and 0.17 folds of the enhanced levels respectively. After p65 siRNA transfection, these protein expressions were significantly inhibited to 0.07, 0.06 and 0.10 folds respectively. ANOVA and LSD revealed significant increases of these factors in 131I groups than in control groups (P<0.01), and significant decreases of them in combination groups than in 131I groups (P<0.01).
Figure 5. Effects of different treatments on NF-κB regulated anti-apoptotic factors.

KTC-1 cells were left untreated as controls (A), or treated with 20 MBq/ml 131I (B) or in combination with 5 µmol/L Bay 11-7082 (C) for 24 hours. Then whole cell lysates were examined by Western blot for XIAP, cIAP1 and Bcl-xL. In the second set of experiments, control cells were left untreated (D). After no oligonucleotide (E), scrambled oligonucleotides (F) or p65 siRNA (G) transfection, KTC-1 cells were treated with 131I (20 MBq/ml) for 24 hours. Then protein levels of XIAP, cIAP1 and Bcl-xL were studied. β-actin was used as a loading control. Similar results were obtained in three independent experiments.
Synergistic effects detected in combination therapies
First, Western blot was applied to detect changes of caspase 3 (a key apoptosis executioner) and PARP (a main target of caspase 3). We showed that although any intervention could induce cleavages of caspase 3 (subunits p19 and p17) and PARP (p89), their levels increased significantly further by combined treatments (Figure 6). In Table 1, ANOVA and LSD showed significant enhancements in combined therapies than in mono-therapies (P<0.05). Then, to further confirm the effects on apoptosis, double staining flow cytometry was performed. Results showed that while any therapy could induce apoptosis, combined treatments increased apoptosis synergistically due to significant enhancements of Annexin V positively stained cells (Figure 7 and Table 1).
Figure 6. Effect of different treatments on apoptotic cascade.
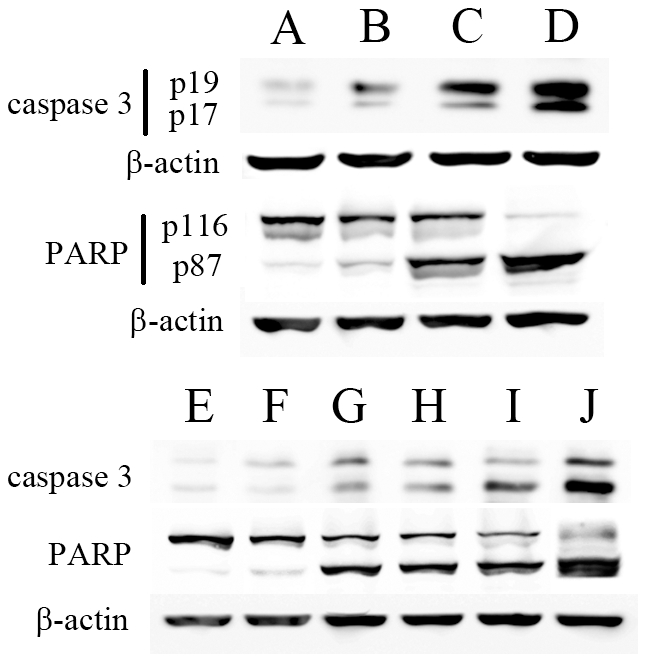
KTC-1 cells were left untreated as controls (A), or treated with 20 MBq/ml 131I (B), 5 µmol/L Bay 11-7082 (C) or combination (D) for 24 hours. Then whole cell lysates were examined by Western blot for caspase 3 and PARP. In the second set of experiments, KTC-1 cells were treated with no oligonucleotide control (E), scrambled oligonucleotides (F) or p65 siRNA transfection (G) first. Then these three groups of cells were exposed to 131I (20 MBq/ml) for 24 hours (H–J). Caspase 3 and PARP protein levels were examined by Western blot. β-actin was used as a loading control. Similar results were obtained in three independent experiments.
Table 1. Synergy confirmed by Western blot and flow cytometry.
| Western blot with semi-quantification* | Flow cytometry | ||||
| Caspase 3 p19 | Caspase 3 p17 | PARP p89 | PARP p116 | Annexin V positive cells | |
| Control | 4.72±0.69 | 4.46±0.55 | 4.65±0.70 | 93.02±13.44 | 0.36±0.18 |
| 131I(1) | 25.68±6.35 | 19.64±3.89 | 32.70±7.93 | 64.12±10.14 | 9.96±2.01 |
| Bay 11-7082(2) | 59.53±8.55 | 41.29±8.51 | 74.89±11.90 | 45.68±8.33 | 19.43±4.29 |
| Combined therapy(3) | 103.74±12.41 | 92.41±10.97 | 101.53±15.78 | 3.84±0.55 | 49.95±6.95 |
| F (P)** | 83.79 (<0.01) | 85.06 (<0.01) | 49.22 (<0.01) | 47.43 (<0.01) | 78.15 (<0.01) |
| P (1) ∶ (3) *** | <0.01 | <0.01 | <0.01 | <0.01 | <0.01 |
| P (2) ∶ (3) *** | <0.01 | <0.01 | <0.05 | <0.01 | <0.01 |
calculated by protein/β-actin with Quantity One Software;
analyzed by one-way analysis of variance;
analyzed by least significant difference test.
Figure 7. Apoptotic changes measured by flow cytometry.

The first set of experiments included four groups: KTC-1 cells received no treatment (A), 20 MBq/ml 131I (B), 5 µmol/L Bay 11-7082 (C) or combination (D) for 24 hours. Then cells were harvested by trypsinization, double stained by FITC-conjugated annexin V and propidium iodide, and then subjected for flow cytometry. In the second set of experiments, KTC-1 cells were first treated with no oligonucleotide control (E), scrambled oligonucleotides (F) or p65 siRNA transfection (G). Then these three groups of cells were exposed to 131I (20 MBq/ml) for 24 hours (H–J). And flow cytometry was conducted. Similar results were obtained in three independent experiments.
Iodide uptake in DTC cells under NF-κB blocking
To determine whether NF-κB pathway inhibition influences iodine uptake, we tested radioactive iodine uptake changes with or without NF-κB inhibition by Bay 11-7082 or p65 siRNA transfection. Figure 8 demonstrated that there were only slight changes after therapeutic interventions, no significant differences were observed (P>0.05), which indicated that NF-κB did not control iodine uptake in KTC-1 cells.
Figure 8. Iodide uptake assay.

(A) KTC-1 cells were left untreated or treated with Bay 11-7082 (5 µmol/L) for 24 hours. Then cells were cultured in the present of Na125I (37 KBq/mL) for 1 hour. Afterwards, the cells were washed and cell number was counted. Then radioactivity was measured. (B) KTC-1 cells were treated with no oligonucleotide control, scrambled oligonucleotides or p65 siRNA transfection first. Then cells were exposed to Na125I, and radioactivity was measured. Similar results were obtained in three independent experiments.
Discussion
The most effective post-surgical treatment of DTC lesions is 131I therapy, due to the inherent ability of thyroid cancer cells to concentrate iodine. 131I emits short pathlength (0.5–2.2 mm) β-radiation that is cytotoxic to the cells, which is intrinsically very similar to external radiotherapy. However, this methodology is ineffective in patients whose tumors no longer concentrate 131I efficiently [4], [19], [20]. We tested our hypothesis in this study, that NF-κB could be an important factor regulating the anti-apoptotic process during 131I therapy. NF-κB has been shown to play an important role in many types of cancer [14] including thyroid cancer [7], [8], [13], [21], [22], which heralded searches for drugs capable of suppressing NF-κB activity [7], [18]. Till now, a number of NF-κB inhibitors have been demonstrated to induce thyroid cancer cell apoptosis, for instance, SN50, DHMEQ, bortezomib and Bay 11-7082. And when they were used in combination with chemotherapy or radiotherapy, synergism could be achieved [9], [10], [11], [12]. Another approach of p65 siRNA transfection has been tried in many types of cancers [15], [23], [24], [25], and enhanced chemosensitivity was demonstrated as well [15], [23]. Yet, combined therapy including p65 siRNA transfection has not been tested in thyroid cancer so far.
In the current study, the effects of 131I on NF-κB function and expression in DTC cells were analyzed first. DNA-binding assay showed that NF-κB activation was dramatically increased at 6-hour time point, reaching peaks at 24-hour time point for KTC-1 and WRO cells. Increased NF-κB reporter activation was also proved by luciferase assay. However, these activations could be significantly suppressed by a NF-κB inhibitor or by p65 siRNA transfection. We then used Western blot data to show that 131I could increase nuclear protein levels of NF-κB, while Bay 11-7082 or p65 siRNA transfection could dramatically inhibit NF-κB protein levels.
NF-κB mediated anti-apoptosis are essentially depended on its ability to enhance transcriptions of cell death suppressive genes [7], [18], [26]. The most representative NF-κB controlled anti-apoptotic factors are XIAP, cIAP1 and Bcl-xL, which can prevent tumor cell killing effects [7], [18]. XIAP can inhibit caspase 3 and 7, and it is also involved in the suppression of the pro-apoptotic JNK activity. cIAP1 is another potent inhibitor of apoptosis, which can bind and inhibit caspase 3 and 8. And Bcl-xL is known to inhibit cytochrome C release from mitochondria. In this investigation, we found that XIAP, cIAP1 and Bcl-xL were significantly up-regulated after exposure to 131I. Inhibition of NF-κB by Bay 11-7082 or by p65 siRNA transfection during 131I exposure could change the balance toward significant down-regulation of these factors.
Then, in order to confirm the possibility of synergistic effects induced by combined therapies, we performed Western blot on key apoptotic proteins and conducted flow cytometry to assess apoptosis. Our results demonstrated significant synergy if NF-κB was inhibited during 131I therapy. Our study also showed that NF-κB pathway inhibition did not control iodine uptake, which indicates its pro-apoptotic effects is the main mechanism for the synergistic results.
Apoptosis is an essential process of eliminating destined cells during development or after damage from chemotherapy or radiotherapy. It is known that 131I-induced cell death can be both apoptosis and necrosis, the nature of which is dose-dependent [27]. Marx et al. [27] demonstrated that, in B-CPAP cells (another DTC cell line), apoptosis was detectable after incubation with 1 MBq/ml of 131I for 2 days. At medium 131I activity concentration (1–10 MBq/ml) apoptosis was predominant, while at much higher 131I activity concentration (especially higher than 100 MBq/ml) necrosis became predominant. In our investigation, we used 131I activity concentration of 20 MBq/ml, which could produce enough apoptosis for different experiments. The slight dosage difference could be from different cell lines and different laboratory conditions.
Destruction of pro-apoptotic or anti-apoptotic balance is proved to cause carcinogenesis. In cancer treatment settings, aberrant apoptotic signaling could induce resistance to chemotherapy or radiotherapy. Therefore, restoration of functional apoptosis in cancer cells could be a useful approach to enhance therapeutic efficacies. NF-κB pathway hypothesis has been tested true in thyroid cancer already [9], [10]. Increased NF-κB activity reinforces the intrinsic therapy-resistance phenotype of thyroid cancer cells, and its inhibition has been showing promising results in combination with chemotherapy or radiotherapy [9], [10]. The present data adds to the hypothesis, that 131I could induce NF-κB activation which attenuates 131I efficacy in thyroid cancer cells. NF-κB inhibition is effective in suppressing 131I related NF-κB induction and anti-apoptotic changes. In combined regimen apoptosis can be achieved synergistically.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This investigation was supported by China National Natural Science Foundation grant 30900376, Key Project of Tianjin Science and Technology Committee Foundation grant 10JCZDJC19000, Tianjin Medical University Scientific Research grant 2008KY20, and Tianjin Medical University New Century Excellent Talent Program (awarded to ZM). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA. 2006;295:2164–2167. doi: 10.1001/jama.295.18.2164. [DOI] [PubMed] [Google Scholar]
- 2.Eustatia-Rutten CF, Corssmit EP, Biermasz NR, Pereira AM, Romijn JA, et al. Survival and death causes in differentiated thyroid carcinoma. J Clin Endocrinol Metab. 2006;91:313–319. doi: 10.1210/jc.2005-1322. [DOI] [PubMed] [Google Scholar]
- 3.Durante C, Haddy N, Baudin E, Leboulleux S, Hartl D, et al. Long-term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J Clin Endocrinol Metab. 2006;91:2892–2899. doi: 10.1210/jc.2005-2838. [DOI] [PubMed] [Google Scholar]
- 4.Pfister DG, Fagin JA. Refractory thyroid cancer: a paradigm shift in treatment is not far off. J Clin Oncol. 2008;26:4701–4704. doi: 10.1200/JCO.2008.17.3682. [DOI] [PubMed] [Google Scholar]
- 5.Tan J, Zhang G, Xu W, Meng Z, Dong F, et al. Thyrotoxicosis due to functioning metastatic follicular thyroid carcinoma after twelve I-131 therapies. Clin Nucl Med. 2009;34:615–619. doi: 10.1097/RLU.0b013e3181b06b2d. [DOI] [PubMed] [Google Scholar]
- 6.O'Neill CJ, Oucharek J, Learoyd D, Sidhu SB. Standard and emerging therapies for metastatic differentiated thyroid cancer. Oncologist. 2010;15:146–156. doi: 10.1634/theoncologist.2009-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pacifico F, Leonardi A. Role of NF-kappaB in thyroid cancer. Mol Cell Endocrinol. 2010;321:29–35. doi: 10.1016/j.mce.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Namba H, Saenko V, Yamashita S. Nuclear factor-kB in thyroid carcinogenesis and progression: a novel therapeutic target for advanced thyroid cancer. Arq Bras Endocrinol Metabol. 2007;51:843–851. doi: 10.1590/s0004-27302007000500023. [DOI] [PubMed] [Google Scholar]
- 9.Meng Z, Mitsutake N, Nakashima M, Starenki D, Matsuse M, et al. Dehydroxymethylepoxyquinomicin, a novel nuclear Factor-kappaB inhibitor, enhances antitumor activity of taxanes in anaplastic thyroid cancer cells. Endocrinology. 2008;149:5357–5365. doi: 10.1210/en.2008-0279. [DOI] [PubMed] [Google Scholar]
- 10.Starenki D, Namba H, Saenko V, Ohtsuru A, Yamashita S. Inhibition of nuclear factor-kappaB cascade potentiates the effect of a combination treatment of anaplastic thyroid cancer cells. J Clin Endocrinol Metab. 2004;89:410–418. doi: 10.1210/jc.2003-031216. [DOI] [PubMed] [Google Scholar]
- 11.Starenki DV, Namba H, Saenko VA, Ohtsuru A, Maeda S, et al. Induction of thyroid cancer cell apoptosis by a novel nuclear factor kappaB inhibitor, dehydroxymethylepoxyquinomicin. Clin Cancer Res. 2004;10:6821–6829. doi: 10.1158/1078-0432.CCR-04-0463. [DOI] [PubMed] [Google Scholar]
- 12.Mitsiades CS, McMillin D, Kotoula V, Poulaki V, McMullan C, et al. Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. J Clin Endocrinol Metab. 2006;91:4013–4021. doi: 10.1210/jc.2005-2472. [DOI] [PubMed] [Google Scholar]
- 13.Palona I, Namba H, Mitsutake N, Starenki D, Podtcheko A, et al. BRAFV600E promotes invasiveness of thyroid cancer cells through nuclear factor kappaB activation. Endocrinology. 2006;147:5699–5707. doi: 10.1210/en.2006-0400. [DOI] [PubMed] [Google Scholar]
- 14.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 15.Duan J, Friedman J, Nottingham L, Chen Z, Ara G, et al. Nuclear factor-kappaB p65 small interfering RNA or proteasome inhibitor bortezomib sensitizes head and neck squamous cell carcinomas to classic histone deacetylase inhibitors and novel histone deacetylase inhibitor PXD101. Mol Cancer Ther. 2007;6:37–50. doi: 10.1158/1535-7163.MCT-05-0285. [DOI] [PubMed] [Google Scholar]
- 16.Wu X, Zhu Y, Yan H, Liu B, Li Y, et al. Isothiocyanates induce oxidative stress and suppress the metastasis potential of human non-small cell lung cancer cells. BMC Cancer. 2010;10:269. doi: 10.1186/1471-2407-10-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss SJ, Philp NJ, Grollman EF. Iodide transport in a continuous line of cultured cells from rat thyroid. Endocrinology. 1984;114:1090–1098. doi: 10.1210/endo-114-4-1090. [DOI] [PubMed] [Google Scholar]
- 18.Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woyach JA, Shah MH. New therapeutic advances in the management of progressive thyroid cancer. Endocr Relat Cancer. 2009;16:715–731. doi: 10.1677/ERC-08-0335. [DOI] [PubMed] [Google Scholar]
- 20.Ganti AK, Cohen EE. Iodine-refractory thyroid carcinoma. Rev Recent Clin Trials. 2006;1:133–141. doi: 10.2174/157488706776876526. [DOI] [PubMed] [Google Scholar]
- 21.Pacifico F, Mauro C, Barone C, Crescenzi E, Mellone S, et al. Oncogenic and anti-apoptotic activity of NF-kappa B in human thyroid carcinomas. J Biol Chem. 2004;279:54610–54619. doi: 10.1074/jbc.M403492200. [DOI] [PubMed] [Google Scholar]
- 22.Visconti R, Cerutti J, Battista S, Fedele M, Trapasso F, et al. Expression of the neoplastic phenotype by human thyroid carcinoma cell lines requires NFkappaB p65 protein expression. Oncogene. 1997;15:1987–1994. doi: 10.1038/sj.onc.1201373. [DOI] [PubMed] [Google Scholar]
- 23.Guo J, Verma UN, Gaynor RB, Frenkel EP, Becerra CR. Enhanced chemosensitivity to irinotecan by RNA interference-mediated down-regulation of the nuclear factor-kappaB p65 subunit. Clin Cancer Res. 2004;10:3333–3341. doi: 10.1158/1078-0432.CCR-03-0366. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Sheng G, Lu J, Xie L, Bai S, et al. Effect of RNAi-induced down regulation of nuclear factor kappa-B p65 on acute monocytic leukemia THP-1 cells in vitro and vivo. Mol Cell Biochem. 2012;359:125–133. doi: 10.1007/s11010-011-1006-z. [DOI] [PubMed] [Google Scholar]
- 25.Wu W, Yao D, Wang Y, Qiu L, Sai W, et al. Suppression of human hepatoma (HepG2) cell growth by nuclear factor-kappaB/p65 specific siRNA. Tumour Biol. 2010;31:605–611. doi: 10.1007/s13277-010-0076-y. [DOI] [PubMed] [Google Scholar]
- 26.Kucharczak J, Simmons MJ, Fan Y, Gelinas C. To be, or not to be: NF-kappaB is the answer–role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 2003;22:8961–8982. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- 27.Marx K, Moka D, Schomacker K, Fischer T, Gabruk-Szostak B, et al. Cell death induced by 131 I in a differentiated thyroid carcinoma cell line in vitro: necrosis or apoptosis? Nucl Med Commun. 2006;27:353–358. doi: 10.1097/01.mnm.0000199475.08407.e2. [DOI] [PubMed] [Google Scholar]