

Abstract
Thymidylate kinase (TMK) is a potential chemotherapeutic target because it is directly involved in the synthesis of an essential component, thymidine triphosphate, in DNA replication. All reported TMK inhibitors are thymidine analogs, which might retard their development as potent therapeutics due to cell permeability and off-target activity against human TMK. A small molecule hit (1, IC50 = 58 μM), which has reasonable inhibition potency against Pseudomonas aeruginosa TMK (PaTMK), was identified by the analysis of the binding mode of thymidine or TP5A in a PaTMK homology model. This hit (1) was co-crystallized with PaTMK, and several potent PaTMK inhibitors (leads, 46, 47, 48, and 56, IC50 = 100–200 nM) were synthesized using computer aided design approaches including virtual synthesis/screening, which was used to guide the design of inhibitors. The binding mode of the optimized leads in PaTMK overlaps with that of other bacterial TMKs, but not with human TMK which shares few common features with the bacterial enzymes. Therefore, the optimized TMK inhibitors described here should be useful for the development of antibacterial agents targeting TMK without undesired off-target effects. In addition, an inhibition mechanism associated with the LID loop, which mimics the process of phosphate transfer from ATP to dTMP, was proposed based on X-ray co-crystal structures, homology models, and SAR results.
INTRODUCTION
Most classes of antibacterial agents currently employed in clinical use were discovered over 45 years ago, with the exception of the oxazolidinone class (Linezolid), introduced in 2000 to treat Gram positive infections. New antibacterial therapeutics which utilize new mechanisms of action are urgently needed to combat growing resistance to existing antibacterial agents for both Gram positive and Gram negative infections. Although discovery of new antibacterial classes is extraordinarily difficult,1 the need is especially high for Gram negative organisms prevalent in the hospital and in particular for infections caused by Pseudomonas aeruginosa (Pa), for which treatment options are often limited.2–4
Thymidylate kinase (TMK) has emerged as an attractive therapeutic target because inhibiting TMK functions blocks DNA synthesis in replicating organisms.5 TMK phosphorylates thymidine monophosphate (dTMP) to thymidine diphosphate (dTDP), using ATP as a phosphoryl donor.6 In addition, TMK is the last specific enzyme in the pathways for the synthesis of thymidine triphosphate (dTTP), which is an essential component in DNA synthesis.7 Therefore, targeting bacterial TMK has been the subject of recent investigation with inhibitors of Mycobacterium tuberculosis,8–12 Staphylococcus aureus,13 and Bacillus anthracis14 TMKs reported in conjunction with their cognate protein crystal structures.
However, most of the reported TMK inhibitors are thymidine derived (Figure 1).15–21 Although inhibitor design has been enhanced by utilizing protein structures,21 computer aided design,20 and QSAR methods,19 the thymine head group of the inhibitors always remains (Figure 1). The thymine head group was viewed as a hindrance for designing inhibitors that penetrate the complex cell membrane and avoid efflux pumps prevalent in Pa. Therefore, small molecule TMK inhibitors, or non-substrate analogs, are necessary to develop antibacterial therapeutics. Herein, we report a non-thymidine inhibitor (1) targeting PaTMK, the co-crystal structures of 1 and the evolved inhibitor analog 17 with PaTMK, and structure guided development of TMK inhibitors. To our knowledge, this is the first report of TMK inhibitors dispossessing the thymidine moiety and of its co-crystal structure with PaTMK. In addition, we describe the use of computer aided design, including virtual synthesis and screening, with the co-crystal structures to expedite rational design and synthesis of more potent PaTMK inhibitors.
Figure 1.

Known TMK inhibitors
RESULTS AND DISCUSSION
Hit generation
Structural analysis of a PaTMK homology model built based on co-crystal structure (PDB ID: 4TMK) of E.coli TMK22 with the thymidine based dual substrate inhibitor TP5A led us to intimately understand the interactions required for thymidine binding to its binding site of PaTMK (Figure 2(a)). Furthermore, we found that a commercial compound, 1-methyl-6-phenyl imidazopyridinone (1), has similar chemical properties to thymidine, although their two-dimensional structures have low similarity. Flexible alignment of 1 to thymidine structure was performed to recognize their geometrical similarity using Molecular Operating Environment program (MOE).23 The result showed that 1 and thymidine are well overlapped with identical pharmacophore (Figure 2(b)). In the enzyme assay, compound 1 proved to be an inhibitor of PaTMK with approximately three-fold less potency than the known TMK inhibitor, dFTM (IC50 = 58 μM vs. 20 μM in Table 1).
Figure 2.

Key interactions of dTMP and 1 in PaTMK. (a) Thymidine interactions in the active site of PaTMK at the homology model are represented in two-dimensional view; (b) The result of flexible alignment of 1 to dTMP shows clear pharmacophore match between 1 and dTMP. The structure of dTMP is represented as red line, and that of 1 is as stick with atom-type color (gray for carbon, blue for nitrogen, red for oxygen, and orange for phosphorous); (c) The overall X-ray co-crystal structure of PaTMK and 1 shows 1 binds where dTMP bound (left panel). The structure of TP5A was merged from the reported X-ray structure (PDB ID: 4TMK) to compare similarity of binding site of 1 and TP5A. The binding mode of 1 in PaTMK at the X-ray co-crystal structure shows the binding pose of 1 at PaTMK is identical to that of dTMP. α-Helix is red, β-sheet is yellow, and loop is green (left panel). The water molecule is represented with a red sphere, 1 and amino acids associated with the binding of 1 are represented as sticks with atom-type color, and H-bond interactions are represented with black lines. π-Cation stacking between the phenyl ring of 1 and Arg96 is not marked. Hydrogen atoms are omitted for clarity. Figures are generated with Pymol program.
Table 1.
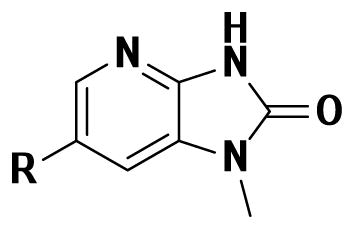 | ||
|---|---|---|
| Compound | R | IC50 (μM) |
| 1 |
|
58 ± 11 |
| dFTM | N/A | 20 ± 4 |
| 9 |
|
>200 |
| 10 |
|
66 ± 2 |
| 11 |
|
>200 |
| 12 |
|
11 ± 2 |
| 13 |
|
63 |
| 14 |
|
>200 |
| 15 |
|
>200 |
| 16 |
|
37 ± 2 |
| 17 |
|
0.44 ± 0.13 |
| 18 |
|
>200 |
| 19 |
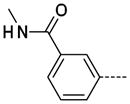
|
2.2 ± 0.4 |
| 20 |
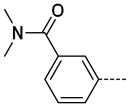
|
104 |
| 21 |
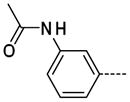
|
3.4 ± 0.7 |
| 22 |
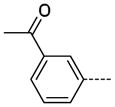
|
13 |
| 23 |
|
21 |
| 24 |
|
152 |
| 25 |
|
4.9 |
| 26 |
|
>200 |
Each reported C50 was determined at least in duplicate and is an average of the individual values determined. Each individual IC50 value in a single determination was the result of a 20 point IC50 curve with each point determined in duplicate.
X-ray structure analysis
To elucidate the binding mode of 1 to facilitate the structure based development of TMK inhibitors, 1 was co-crystallized with PaTMK, as described in the supplementary material, yielding co-crystals that diffracted to 1.91 Å. Gratifyingly, many of the interactions predicted in our homology model and flexible alignment are identical to those in the X-ray co-crystal structure (Figure 2(c)). Especially important for molecular recognition is the ability of the cyclic imidazopyridinone functionality to satisfy the hydrogen bond donors and acceptors of the typical thymidine substrate: specifically accepting hydrogen bonds from Arg74, Thr101, while both donating and accepting hydrogen bonds with Gln105 (Figure 2(c)). Other important interactions with the distal phenyl ring are the box-like face to face and edge to face aromatic interactions24 with Phe155 and Tyr104 which are complemented by the π-cation interaction with Arg96.25–27 Also apparent is a structural water molecule that bridges between the phenolic –OH group of Tyr104 and the amide backbone (-NH) of Glu12. Although this bridging water does not offer a favorable binding interaction with the inhibitor, it is located only 3.8 Å from the meta-position of the distal phenyl ring. Thus, the bridging structural water suggests a design opportunity for increasing potency via either displacement or additional interaction through hydrogen bonding.28
Synthesis of 1 and its analogs
Synthesis of analogs of 1 was accomplished using 6 as a key intermediate (Scheme 1). Briefly, 5-bromo 2,3-diaminopyridine 2 was treated with benzyl chloroformate to give benzyl carbamate 3.29 The urethane was reduced to a methyl group with lithium aluminum hydride and the diamine was cyclized to the urea 5, utilizing triphosgene.30 Palladium mediated coupling reactions of 5 were accomplished efficiently when the acidic urea NH was protected with a trityl group as in 6. The coupling reaction of 6 with phenyl boronic acid provided 7 in good yield when performed under microwave conditions.31–34 Removal of the trityl group with trifluoroacetic acid provided the final inhibitor, 1. Various aryl boronic acids proposed by computational studies were coupled to the intermediate 6 to produce the first inhibitor library.
Scheme 1a.

aReagents and conditions: (a) ClCO2Bn, Pyridine, 0 °C to rt 5 h, 48%; (b) LiAlH4, Et2O, 0 °C to rt 6 h, 99%; (c) triphosgene, THF, rt to reflux, 4 h, 83%; (d) Ph3CCl, Et3N, CH2Cl2, rt, 1 h, 84%; (e) aryl boronic acid, 1 mol% Pd2(dba)3, 2 mol% PCy3, K3PO4, dioxane, 100 °C (microwave), 1 h; (f) TFA, CH2Cl2, rt, 30 min., 50 ~ 99% (over 2 steps).
Virtual synthesis/screening of first round PaTMK inhibitors
As described in the analysis of the co-crystal structure, the phenyl ring of 1 can be further modified to increase inhibition potency by replacing it with electron rich aromatic rings and/or by incorporating functional groups replacing the water molecule near Tyr104. In terms of synthesis, aryl boronic acids can be coupled with intermediate 6 followed by trityl deprotection to produce 1 and its analogs (Scheme 1). Therefore, to increase the diversity of inhibitors being designed, virtual synthesis of inhibitors followed by docking and scoring was applied as outlined in Scheme 2. First, 5000 commercially available boronic acids were computationally attached to the 6-position of imidazopyridine-2-one 5 with the combi-gen program in MOE. The virtually synthesized 5000 compounds were docked into the active site of PaTMK with the water molecule removed using Glide 5.5.35 Hits were ranked and sorted with docking score function ranging from −11.5 to −2.3 kcal/mol. In this virtual screening the lead inhibitor 1 ranked 1933th with a score of −8.7 kcal/mol. The virtual compounds with scores lower than 1 were visually inspected using the maestro9.0 program36; most aryl appendages targeted nearby amino acid residues such as the hydroxyl group of Tyr104, the carboxyl groups of Asp153 and Glu156, and the guanidine group of Arg50. About 20 compounds were selected for synthesis, each of which had small aryl groups which offered favorable interactions with the protein, along with polar functionality to generally improve physical properties while preserving the hydrogen bonding network of the imidazopyridinone. This structure guided design and synthesis resulted in the discovery of 17 (IC50 = 0.44 μM) and 19 (IC50 = 2.2 μM) which have 130-fold and 30-fold improved inhibition over 1, respectively.
Scheme 2.

Analysis of first round PaTMK inhibitors and X-ray co-crystal structure of 17 with PaTMK
The tactic of displacing the water molecule near Tyr104 with simple H-bond donors and acceptors was explored using the docking result as a guide. The assay results showed good consistency with the rationale proposed with the X-ray structure and the modeling technique with minor differences. The phenol substitution in 12 provided about a four-fold increase in potency, while the cyano-group in 14 resulted in loss of activity. Not only was the cyano-compound 14 inactive, it also lacked the H-bond donating ability of the phenol in 12; therefore follow-up focused on incorporating hydrogen bond donating groups that were larger than the cyano-group. The meta-aminomethyl group in 15 was chosen as there are several acidic protein residues in the vicinity of the inhibitor binding site such as Glu12, Glu156 and Asp153. The lack of activity observed for this analog contrasts with the good potency of para-aminomethyl analog 25, indicating that the para-amino methyl group must extend towards the complementary acidic residues providing a favorable protein-ligand interaction. The similar in size but uncharged meta-hydroxymethyl group in 16 provided a 37 μM inhibitor, a two-fold increase in potency over 1, suggesting the positive charge in 15 was problematic.
In an opposite tactic to displacing the bridging structural water molecule, interactions through this water molecule was probed by incorporation of pyridine, pyrimidine and fluorine groups as potential hydrogen bond acceptors as in 9–11. Only the pyridine containing inhibitor 10 retained the meaningful activity of the parent compound 1 (Table 1). As described, the phenyl ring of 1 forms a π-cation interaction with Arg96.25–27 Therefore, not surprisingly, compounds with electron withdrawing groups such as 9, 11, 14, 18, and 26 significantly lost electronic potential at the phenyl ring followed by inhibition potency (IC50 >200). However, interestingly, 17 has dramatically increased inhibition potency (130-fold over initial lead 1 and a 60-fold over a standard inhibitor dFTM), although it possesses an electron-poor aryl ring; this is also the case with 19, 21, and 22, which however are less potent than 17. These results can be rationalized in the context of the solved X-ray co-crystal structure of 17 and PaTMK (Figure 3).
Figure 3.

X-ray co-crystal structure of 17 and PaTMK. Key interactions of 17 in the active site of PaTMK are described as 2–D (up) and 3–D (down) views. For clarity, interactions associated with imidazopyridinone moiety and hydrogen atoms are omitted. Furthermore, water molecules, which are not involved in binding of 17, are omitted. The water molecule is represented as a red sphere, 17 and amino acids are represented as sticks with atom-type color, and H-bond interactions are represented with black lines.
Inhibitor 17 was co-crystallized with PaTMK and provided a 1.7 Å structure. The binding pose of 17 in virtual synthesis/screening procedure is very consistent with that of the X-ray structure. Indeed, the structural water targeted in the design was clearly displaced with the carboxamide of 17 and a direct hydrogen bond interaction between the carbonyl oxygen of 17 and the phenolic oxygen of Tyr104 is apparent (Figure 3). Also present was a water molecule mediated H-bond network that further hydrates the carboxamide of 17 and could surround the entrance of the inhibitor (or dTMP) binding site to make the release of 17 slow. As a combined effect of H-bond interactions with Tyr104 and the H-bond network, PaTMK inhibition by 17 was significantly increased compared to 1: ca. 1000-fold increase over 26 (p-carboxamide), which possesses an electron withdrawing group but without the aforementioned cooperative hydrogen bonding interactions. The effect of the H-bond interactions mediated by a network of water molecules appears to be much more important than H-bond interactions with Tyr104 because of decreased potency of 19 and 20. The activity of inhibitor 19 (N-methylamide), which presumably does not support the extensive H-bond network seen in 17, was decreased 5-fold compared with 17. The importance of the hydrogen bonds with the structural water molecules is supported by the significant, 240-fold decreased inhibition observed for 20 (N,N-dimethylamide), which likely would lose the H-bond network while retaining a H-bond interaction with Tyr104. Consequently, as rationalized in the computer guided design approach, the π-cation interaction of an electron-rich phenyl ring with Arg96 and H-bond interactions of the carbonyl oxygen of the carboxamide with Tyr104 are important for inhibition of PaTMK. In addition, the X-ray co-crystal structure of 17 provided critical clues, specifically the water mediated H-bond network, that provides dramatically increased potency of 17, though possessing an electron-withdrawing group. Since the secondary amide 19 retained reasonable potency, and intermediate 28 (Scheme 3) was conducive to rapid analog production, this vector was explored with the aim of increasing potency.
Scheme 3a.

aReagents and conditions: (a) 3-carboxyphenylboronic acid, 1 mol% Pd2(dba)3, 2 mol% PCy3, K3PO4, dioxane, 100 °C (microwave), 1 h, then aq. HCl, 99%; (b) pentafluorophenyl trifluoroacetate, N(i-Pr)2Et, CH2Cl2, 0 °C to rt, 1 h, 81%; (c) primary amines, N(i-Pr)2Et, CH2Cl2, rt, 1 h; (d) TFA, CH2Cl2, rt, 30 min., 100 ~ 56% (over 2 steps).
Virtual synthesis/screening of second round PaTMK inhibitors
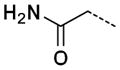
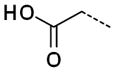
A second round of virtual synthesis designed to efficiently expand diversity on lead 19. Virtual synthesis was performed with 1188 commercially available primary amines to obtain various alkyl tethered benzamide derivatives that were then virtually screened by docking into the active site of PaTMK using Glide 5.5. About 900 hits were ranked and sorted with docking scores ranging from −14.6 to −0.9 kcal/mol. Among them, inhibitor 19 ranked 390th with a score of −11.4 kcal/mol. With a focus on improving potency, twenty compounds were selected for synthesis by visually inspecting the top scoring compounds followed by ranking for size and enhanced interaction with the protein while maintaining the integrity of the imidazopyridinone hydrogen bonding interactions (Table 2).
Table 2.
 | ||
|---|---|---|
| Compound | R | IC50 (μM) |
| 19 |
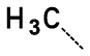
|
2.2 ± 0.4 |
| 30 |
|
0.88 ± 0.31 |
| 31 |
|
0.88 ± 0.08 |
| 32 |
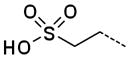
|
1.3 ± 0.22 |
| 33 |
|
2.1 ± 0.3 |
| 34 |
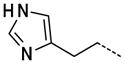
|
2.3 ± 0.1 |
| 35 |
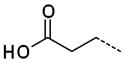
|
2.6 ± 0.5 |
| 36 |
|
2.6 ± 0.2 |
| 37 |
|
3.2 ± 0.7 |
| 38 |
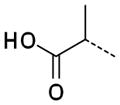
|
5.0 |
| 39 |
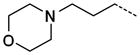
|
5.8 |
| 40 |
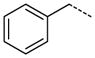
|
1.0 ± 0.4 |
| 41 |
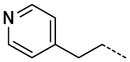
|
2.0 ± 1.0 |
| 42 |
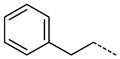
|
2.9 ± 1.6 |
| 43 |
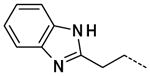
|
3.0 ± 1.3 |
| 44 |
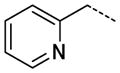
|
6.2 ± 1.6 |
| 45 |
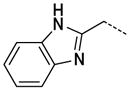
|
68 ± 28 |
| 46 |
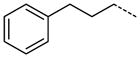
|
0.12 ± 0.08 |
| 47 |
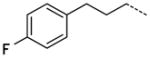
|
0.12 ± 0.05 |
| 48 |
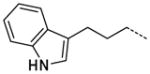
|
0.20 ± 0.08 |
| 49 |
|
0.51 ± 0.1 |
| 50 |
|
0.99 ± 0.14 |
| 51 |
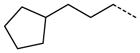
|
0.81 ± 0.23 |
| 52 |
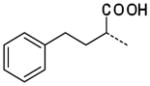
|
12 ± 2 |
| 53 |
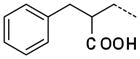
|
0.25 ± 0.13 |
| 57 |
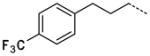
|
0.16 ± 0.05 |
| 58 |
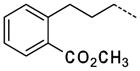
|
0.37 ± 0.15 |
| 59 |
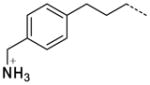
|
3.2 |
| 60 |
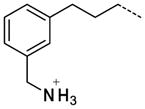
|
2.2 |
| 61 |
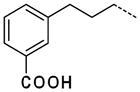
|
2.4 |
| 62 |
|
2.7 |
| 63 |
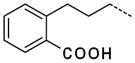
|
2.0 ± 0.7 |
Each reported IC50 was determined at least in duplicate and is an average of the individual values determined. Each individual IC50 value in a single determination was the result of a 20 point IC50 curve with each point determined in duplicate.
Synthesis of 19 and its analogs
Synthesis of analogs of 19 was achieved using 28 as a key intermediate (Scheme 3). Intermediate 6 in Scheme 1 was coupled with 3-carboxylphenylboronic acid to provide 27 in near quantitative yield. The acid in 27 was activated as the pentafluorophenyl ester 28, which was stable to storage (0.1 M solution of CH2Cl2). About 0.2 mmol of 28 (2 mL solution of CH2Cl2) was treated with the various primary amines and diisopropylethylamine to provide the desired amides. Removal of the trityl group with TFA provided the final inhibitors. In addition, for modification of the terminal phenyl ring of 46, intermediate 28 was treated with propargylamine to produce 54 (Scheme 4). The terminal alkyne of 54 was coupled with various aryl bromides mediated by Pd(0) in the presence of a catalytic amount of copper(I) iodide to give 55. Hydrogenation of 55 provided arylpropyl amides 56 from which the trityl group was removed with TFA to give the desired inhibitors 57–63.
Scheme 4a.

aReagents and conditions: (a) mono-propagylamine hydrochloride, N(i-Pr)2Et, CH2Cl2, rt to 50 °C, 1 h, 98%; (b) Aryl bromide, 10 mol% Pd(PPh3)4, NEt3, 5 mol% CuI, DMF, 80 °C (microwave), 1 h, 73%; (c) H2, Pd/C, MeOH, 12 h, 63%; (d) TFA, CH2Cl2, rt, 30 min., 46%; (e) 10% KOH(aq), MeOH, 60 °C, 12 h, then TFA, CH2Cl2, rt, 30 min., 22% (over 4 steps).
Assay results of second round PaTMK inhibitors
A number of small polar secondary amides were synthesized to explore functionality accommodated by the binding pocket 30 – 38 (Table 2). The glycinamide and glycine analogs 30 and 31 proved to be the most potent polar analogs synthesized. Each has submicromolar activity and improved potency about 2.5-fold over secondary amide 19, though they were both less potent than the parent amide analog 17. Both negatively and positively charged groups as in 31, 32, 35, and 37 also were accommodated without large changes in potency when compared with 19. The X-ray co-crystal structure of 17 indicated that the area immediately adjacent to the m-carboxamide might also have hydrophobic character (Figure 4) so small hydrophobic extensions of the amide were investigated as in compounds 40–47 (Table 2). Inhibitors 40 and 41 both retained the potency of the parent methylamide 19 showing that larger groups could be accommodated in this area. Since potency did not increase in a manner commensurate with the increase in size, 40 and 41 were deemed inefficient binding ligands. More polar heterocycles as in 43 – 45 lost potency compared with 19 which were in contrast to the glycinamide 30 where a polar group increased potency. Exploring extension of the hydrophobic group in the series 40 to 42 to 46/47/57 indicated that a propyl spacer between the amide was optimal with inhibitor 46 having 0.12 μM inhibitory activity, an 18-fold improvement over the methyl amide 19. Inhibitors 47 and 48 both retain a high level of potency indicating that electronic characteristics and steric bulk can be varied. Addition of polar groups in 59–63 to the phenyl ring resulted in a 4 to 20-fold loss of inhibition compared with 46. Introduction of the carboxyl group into the propyl side chain, making hybrids of 31 and 46 provided racemic compound 52, lost significant potency. However, the hybrid of 35 and 46 giving racemic inhibitor 53 with a carboxyl group proved to be a useful combination where a good level of potency was retained.
Figure 4.

Homology model of PaTMK and 46 based on the X-ray structure of PaTMK and 17. The blue loop is the generated LID loop in homology modeling. In the close view (right panel), the terminal phenyl ring of 46 occupies the hydrophobic pocket composed of Pro11 (facing to the plane of the phenyl ring), Phe163, Val139, Leu137, Ile141, and Leu143 (surround the terminal phenyl ring of 46). Other residues in the loop are oriented toward solvent accessible area such as Pro138, Glu140, and Gly142, which were omitted for clarity. In addition, interactions associated with imidazopyridinone are same as those in the previous X-ray co-crystal structure complexed with 1 or 17 as in Figure 2(c), thus these interactions and hydrogen atoms are omitted for clear view.
Analysis of second round PaTMK inhibitors
The trend of the second round structure activity relationship is that compared to 19, ca. 18-fold increased potency of inhibitors 46, 47, 48, 53, and 57, which possessing hydrophobic aromatic rings linked with aliphatic propyl chain. However, substitution with small functional groups in 30–38 and an aromatic ring linked with a short aliphatic chain in 40–45 did not provide significantly increased inhibition of PaTMK. For rationalization of these results, homology modeling has been performed to fill out the missing amino acids (Val139 to Arg151, a part of LID loop22, 37) in the X-ray co-crystal structure of PaTMK and 17, because the terminal phenyl ring of 46 is oriented toward the region where the LID loop is involved (Figure 4). Interestingly, the results of the homology modeling showed the terminal phenyl ring of 46 occupies a hydrophobic pocket composed of residues such as Pro11, Leu137, Val139, Ile141, Leu143, and Phe163. Therefore, the favorable hydrophobic contact between the terminal phenyl ring of 46 and hydrophobic pocket may be the main reason for the increased potency of 46. Furthermore, 47, 57, and 58 with p-fluoro, p-trifluoromethyl, and o-methoxycarbonyl groups, respectively, have very similar potency to 46. However, a terminal aryl ring with charged functional groups as in 59–63 lost about 10-fold inhibition potency. The LID loop is not clearly solved in most X-ray structures of TMK because it is highly flexible. However, it is known that this highly adaptable loop undergoes substantial conformational changes to close the ATP and dTMP binding sites when the phosphoryl donor, ATP, binds to the enzyme for phosphoryl transfer. In addition, TP5A, which is a phosphoryl transfer mimetic, has high binding affinity due to LID loop closure (Kd = 20 nM).22 Therefore, the LID loop is directly involved in the mechanism of phosphoryl transfer by covering the active site when both ATP and dTMP are bound. Based on our structural model and SAR results, it can be proposed that the terminal phenyl ring of 46 forces shielding of the active site by the LID loop via forming direct hydrophobic contacts between the inhibitor and the hydrophobic pocket of PaTMK, resulting in increased binding affinity of 46.
Biological evaluation of inhibitors
Each compound in Table 1 and 2 was tested against Pseudomonas aeruginosa PA01 and PAO280 (MexABoprM, MexXY, MexZ efflux pump knock out) and were found to have MICs >256 μg/mL. Inhibitors 46 (and also 47/48/57) have nanomolar enzyme inhibition, and have comparable enzyme inhibition to the cellularly active TMKmt inhibitor (thiourea-α-TM in Figure 1: Ki = 1.0 μM (against TMKmt) and MIC (M. bovis) = 25 μg/ml). Therefore, lacking cellular activity in an efflux pump knock out strain suggests penetration of these inhibitors into the cytosol is probably poor due to the complex cell membrane of Gram negative bacteria.38
Sequence similarity and selectivity with human TMK
Structure based multiple sequence alignment of PaTMK with other species including human has been performed to elucidate selectivity of inhibitors over human TMK (21% sequence identity between P. aeruginosa and human TMKs). As indicated in Figure 5, the important amino acids directly involved in the binding of 1, 17, and 46 at PaTMK are significantly different from those of hTMK39: Arg74, Arg96, Thr101, Tyr104, Gln105, and Phe163 of PaTMK are equivalent to Arg76, Arg97, Gly102, Phe105, Thr106, and Gln157 respectively. We envisage Arg76 and Arg97 of hTMK, having a weak H-bond interaction with imidazopyridinone moiety and π-cation interaction with the central phenyl ring of 46. Importantly, the hTMK interactions with 1 is missing key H-bond interactions of imidazopyridinone moiety with Thr101 and Gln105 of PaTMK. In addition, Phe105 of hTMK is corresponds to Tyr104 of PaTMK which eliminates the phenolic oxygen which is involved in the potency boosting H-bond interaction with carboxamide oxygen of 17 and 46. Furthermore, Gln157 of hTMK is matching to Phe163 of PaTMK which is associated with LID closing and hydrophobic contacts with 46. Collectively, the inhibitors presented here would be predicted to have reduced activity toward human TMK.
Figure 5.

Comparison of the active sites of PaTMK (left) and hTMK (right). Amino acids associated with the interaction of 46 in PaTMK and their corresponding amino acids in hTMK are represented as sticks with atom type color. The structure of 46 and PaTMK was obtained from the docking study with the X-ray co-crystal structure of PaTMK complexed with 17. The structure of 46 and hTMK was obtained through structure based multiple sequence alignment followed by merging the coordinate of 46 to hTMK structure (PDB ID: 1E2F) from PaTMK structure. H-bond interactions are highlighted with dashed black lines. Edge to face hydrophobic interactions and π-cation stacking are annotated with black arrows.
As a verification of this prediction, five compounds 1, 17, 46, 47, and 57 were assayed toward human TMK. As shown in Table 3, 1 and 17 are inactive toward hTMK even at high concentration (% inhibition at 200 μM = 17% and 12%, respectively). In addition, 46, 47, and 57 possess meaningful inhibition only at very high concentrations (IC50 = >200, 123, and 124 μM, and % inhibition of 43%, 58%, and 55%, respectively, at 200 μM). Consequently, these inhibitors have over 1000-fold selectivity for PaTMK over hTMK. That 46, 47, and 57 exhibit very weak inhibition of hTMK at high concentration is consistent with the fact that the hydrophobic pocket formed by LID closing is highly conserved between PaTMK and hTMK (the hydrophobic residues of Val14, Leu135, Leu137, Ala140, and Phe146 of hTMK (PDB ID: 1E2F) are oriented toward the area where the terminal phenyl ring of 46, 47, or 57 occupies, and these amino acids are at the equivalent positions of Pro11, Leu137, Val139, Ile141, and Leu143 of PaTMK in Figure 4). Therefore, due to the hydrophobic contacts of the terminal phenyl ring of 46, 47, or 57, these inhibitors are able to inhibit hTMK, but only at high concentration. In addition, these results indirectly support the proposed inhibition mechanism via hydrophobic contacts assisted by LID loop closing.
Table 3.
Activity of Selected Inhibitors toward hTMK
| Compound | IC50 (μM) | % inhibition at 200 μM |
|---|---|---|
| DTBN | 1.53 | - |
| 1 | >200 | 17 |
| 17 | >200 | 12 |
| 46 | >200 | 43 |
| 47 | 123 | 58 |
| 57 | 124 | 55 |
CONCLUSION
Visual inspection of the binding of the thymidine mimetic TP5A in a homology model of PaTMK derived from E. coli led to the identification of a novel thymidine mimetic, 1, that had an IC50 of 58 μM. Inhibitor 1 was co-crystallized with PaTMK to give the first Pseudomonas aeruginosa TMK co-crystal structure. The binding orientation of 1 is identical to what we expected by the modeling approaches. Affinity optimization of 1 using both structure-based design and its directed library build-up provided inhibitors with nanomolar potency (IC50 = ca. 100 – 200 nM). The SAR results are fully rationalized with the X-ray co-crystal structures and modeling structures. In particular, the water molecule mediated H-bond network in 17, which was not recognized in the modeling approach, plays a significant role for high potency toward PaTMK. In addition, LID loop closing mediated by hydrophobic contacts between the terminal phenyl ring of 46 and the hydrophobic pocket of the PaTMK could be the main reason for high inhibition of 46 toward PaTMK.
There have been many reports of TMK inhibitors using thymidine analogs targeting M. tuberculosis, S. aureus, and B. anthracis. Therefore, we anticipate that the lead compounds 1, 17, and 46 that we developed and the knowledge from structure-based SAR will be useful for the development of inhibitors targeting the TMK’s of these other species because of high sequence identity within the bacterial TMK’s. In particular, the amino acids directly involved in interaction of 1, 17 or 46 are very similar within the bacterial species, but not in human TMK. Therefore, due to the low similarity of the amino acid residues in the active sites of PaTMK and hTMK, particularly the residues associated with inhibitor binding, the inhibitors described here should enable off-target effects to be avoided. Therefore, the present set of inhibitors has great potential as leads for the development of novel anti-bacterial agents.
Unfortunately, all of the TMK inhibitors described here were inactive against Pseudomonas aeruginosa bacterial strains. The complex cell membrane of Pseudomonas aeruginosa is an effective barrier for preventing cellular activity of these new TMK inhibitors because growth inhibition did not occur even in an efflux pump knockout strain. It has been proposed that quinolones penetrate the outer membrane of Gram negative bacteria by translocation through the OmpF channel (in case of P. aeruginosa, OprD40) due to their unique zwitterionic nature41; the inner membrane is crossed and the cytosol accessed due to the substantial amount of uncharged properties in the zwitterions.42 Therefore, incorporating zwitterions into the inhibitors is a plausible next strategy for penetrating the complex cell membrane of Gram negative bacteria and to inhibit P. aeruginosa bacteria cell growth and division.
Experimental Details
Cloning and expression of Pseudomonas aeruginosa TMK
Pa TMK was subcloned in the pET15b vector (EMD Biosciences) and expressed in E. coli BL21 DE3Cells were grown in 30L batches using fermentation at 37 °C in LB broth supplemented with 150 mg/L amplicillin. Cells were grown to late log phase, pelleted by centrifugation and stored at − 80 °C.
Purification
Cell pellets were resuspended in lysis buffer (50 mL 50 mM NaH2PO4, 2 mM TCEP, 5 mM imidazole, pH 7.5, and 300 mM NaCl per 10 g cells). 1 Roche EDTA-free protease inhibitor tablets was added per 50 mL lysate. The solution was brought up to 0.002% Benzonase, 10 mM MgCl2 and lysed by sonication on ice. Lysate was clarified by centrifugation at 20200xg for 1 h. Clarified lysate was bound to a HisTrap HP (GE Life Sciences) column that had been equilibrated in 50 mM NaH2PO4, 2 mM TCEP, 5 mM imidazole, pH 7.5, 300 mM NaCl. The bound material was washed with 5 column volumes 50 mM NaH2PO4, 2 mM TCEP, 5 mM imidazole, pH 7.5, 300 mM NaCl followed by 5 column volumes 50 mM NaH2PO4, 2 mM TCEP, 30 mM imidazole, pH 7.5, 300 mM NaCl. The bound protein was eluted with a gradient of 5%–50% 50 mM NaH2PO4, 2 mM TCEP, 500 mM imidazole, pH 7.5, 300 mM NaCl for 20 column volumes followed by 100% 50 mM NaH2PO4, 2 mM TCEP, 500 mM imidazole, pH 7.5, 300 mM NaCl for 10 column volumes. Fractions containing Pa TMK were pooled and dialyzed against 8L 25 mM HEPES, pH 7.5, 2 mM TCEP, and 300 mM NaCl overnight at 4C. The dialyzed material was concentrated for loading onto a Superdex 200 column (GE Life Sciences) that had been equilibrated with 25 mM HEPES, pH 7.5, 2 mM TCEP, 300 mM NaCl. Fractions containing Pa TMK were pooled and concentrated to 10mg/ml for crystallization.
Crystallization
Pa TMK at 10 mg/ml was mixed with 2 mM inhibitor (from a 100 mM stock in DMSO) and incubated on ice for 30 minutes. Precipitated material was removed by centrifugation at 12000xg for 5 minutes. The 1, dFTM, and 17 complexes were crystallized by hanging drop vapor diffusion. The protein was mixed 1:1 with a reservoir solution containing 30% PEG 4000, 0.2M MgCl2, and 0.1 M Tris pH 7.5 – 8.5 and incubated at 22 °C. Crystals were prepared for data collection by cryoprotection in a mother liquor solution containing an additional 20% ethylene glycol and flash freezing in liquid nitrogen.
Data collection
Diffraction data were collected at 100K by the rotation method (360 frames, 0.5° oscillation per frame) at beam line 17-ID (λ = 1.0 Å) at the Advance Photon Source. The data were processed with HKL2000.43
Structure solution and refinement
The structure of Pa TMK in complex with 1 was solved by molecular replacement at 3 Å resolution using the structure of S.pneumo TMK as a search model in Molrep.44 Calculation of the Matthew’s coefficient indicated that this crystal form contained two molecules in the asymmetric unit. The correlation coefficient and R-factor from the molecular replacement solutions indicated that the correct space group was P21212. Rigid body and restrained refinement were performed in REFMAC45 at 3.0 Å and 1.9 Å, respectively. Five percent of randomly selected reflections were designated as test reflections for use in the Free-R cross-validation method46 and used throughout the refinement. During the refinement, residues which differed in identity between the Pa and S. Pneumo enzymes were mutated to Ala and then built into SigmaA-weighted47 |Fo|-|Fc| electron density maps contoured at 2σ using the visualization and model building program Coot.48 1 was modeled into the SigmaA-weighted |Fo|-|Fc| electron density maps contoured at 2σ. One hundred water molecules and one magnesium atom were added. The model was refined to a final Rcryst/Rfree of 19.5/24.2% in REFMAC (Supplemental Table I).
The structure of Pa TMK in complex with dFTM was solved by molecular replacement at 3 Å resolution using the structure of Pa DHFR from the 1 complex as a search model in PHASER.49 Calculation of the Matthew’s coefficient indicated that this crystal form contained 4 molecules in the asymmetric unit. The correlation coefficient and R-factor from the molecular replacement solutions indicated that the correct space group was P21. Rigid body and restrained refinement were performed in REFMAC45 at 3.0 Å and 1.9 Å, respectively. Five percent of randomly selected reflections were designated as test reflections for use in the Free-R cross-validation method46 and used throughout the refinement. dFTM was modeled into the SigmaA-weighted47 |Fo|-|Fc| electron density maps contoured at 2σ. Three hundred seventy seven water molecules and four Magnesium atoms were added. The model was refined to a final Rcryst/Rfree of 17.5/23.6% in REFMAC (Supplemental Table I).
The structure of Pa TMK in complex with 17 was solved by molecular replacement at 3 Å resolution using the structure of Pa DHFR from the 1 complex as a search model in Molrep.44 Calculation of the Matthew’s coefficient indicated that this crystal form contained 2 molecules in the asymmetric unit. The correlation coefficient and R-factor from the molecular replacement solutions indicated that the correct space group was P2. Rigid body and restrained refinement were performed in BUSTER50 at 3.0 Å and 1.7 Å, respectively. Five percent of randomly selected reflections were designated as test reflections for use in the Free-R cross-validation method46 and used throughout the refinement. 17 was modeled into the SigmaA-weighted47 |Fo|-|Fc| electron density maps contoured at 2σ. Two hundred water molecules were added. The model was refined to a final Rcryst/Rfree of 19.2/21.2% in BUSTER50 (Supplemental Table I).
Coordinates and structure factors of PaTMK complexed with 1, 17, and dFTM are available from the Protein Data Bank with accession codes, 3UWK, 3UWO, and 3UXM, respectively.
Thymidylate Kinase Kinetic Assay Materials and Methods.51
Thymidylate monophosphate kinase (TMK) catalyzes the phosphorylation of thymidine-5′-monophosphate (dTMP) to form thymidine-5′-diphosphate (dTDP) in both de novo and salvage pathways of dTTP (thymidine-5′-triphosphate) synthesis and is the last unique enzyme in the pathway specific for dTMP. A 384-well enzyme assay was performed in which dTDP production is enzymatically coupled to NADH oxidation. This permits the rate of TMK turnover to be monitored by the decrease in absorbance at 340 nm. The amounts of each coupling enzyme were optimized to ensure that TMK turnover is rate limiting. The assay utilized 40 data points (absorbance readings) over a 20 minute time period to calculate the initial rates produced in the presence of compound relative to that of the inhibited (enzyme in the presence of EDTA) and uninhibited controls (enzyme alone).
The assay components consisted of 2 mM adenosine 5′-triphosphate (ATP), 2 mM phosphoenolpyruvate (PEP), 5 U/ml pyruvate kinase (PK), 5 U/ml lactate dehydrogenase (LDH), 1.2 U/ml nucleotide diphosphate kinase (NDP Kinase), 0.22 mM (Nicotinamide adenine di-nucleotide, reduced form (NADH), 54 μM thymidine-5′-monophosphate (TMP) in 40 mM HEPES pH 8.0, 80 mM KCl, and 1.6 mM MgCl2. Pseudomonas aeruginosa thymidylate monophosphate kinase (TMK) enzyme was cloned, expressed and purified using standard molecular biology techniques and diluted in enzyme diluent buffer containing 25 mM Tris, pH 7.8, 250 mM NaCl, 5% glycerol, 1 mM DTT, and 0.0025% Triton X-100. Test compounds were solvated in 100% DMSO at 30 mM concentration and twenty 1.67-fold serial dilutions starting at a concentration of 200 μM or 100 μM (final concentration) were performed in 96 well polypropylene plates. The Biomek FX was used to dilute and spot 5 μL=solution containing compound in duplicate into Corning #3702 384-well clear flat bottom polystyrene microplates. The 50 μL reaction was initiated by the addition of 40 nM enzyme and the oxidation of NADH was monitored for 20 minutes at 25 °C at absorbance 340 nm using a Molecular Devices Spectramax Plus plate reader. The initial rates from the kinetic time course absorbance data for duplicates of each compound concentration were exported into an XLfit4 Excel-based plug-in spreadsheet which allowed curve fitting and statistical analysis to determine IC50 values for each compound tested. Compounds that resulted in single-digit μM potency were re-tested to confirm activity.
Human Thymidylate Kinase Assay
The Human Thymidylate Kinase Assay Kit Plus (Catalog No. HTMK500KE at Profoldin) was used for measurement of the inhibition IC50s. The kinase assay is based on detection of ADP generated by the kinase reaction in the presence of the kinase substrate dTMP. The total volume of each assay reaction mixture was 50 μl. In a black 96-well plate, 1 μl of inhibitor, 27 μl of H2O, 5 μl of 10 x assay buffer, 2.5 μl of 1 mM ATP, 2 μl of 10 mM dTMP and 2.5 μl of 1 μM human thymidylate kinase were mixed. The reaction mixture was incubated at room temperature for 2 min. Then 5 μl of 10 x MUK Reagent A and 5 μl of 10 x MUK Reagent B were added. The reaction was incubated at room temperature for 30 min. Finally 50 μl of the fluorescence dye was added and the fluorescence at 535 nm with excitation at 485 nm was measured. The final concentrations were 50 mM Tris-HCl, pH 8.0, 3 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 50 mM NaCl, 0.003% Brij-35, 50 μM ATP, 400 μM dTMP, 50 nM human thymidylate kinase, 0.39 to 200 μM inhibitor. Negative controls (no enzyme control): The assay reactions without the kinase were used as negative controls to observe the fluorescence of the compounds at each concentration. The fluorescence reading without the kinase reaction without compound was used as the 100% inhibition value. Assay controls: The assay reactions with the product of the kinase (5 μM ADP) were used as assay controls to observe the assay interference of the compounds at each concentration. The data from the assay controls were used for correction of the assay values. The percentage inhibition values were calculated from the corrected assay values and used for IC50 curve fitting.
Virtual synthesis/screening
The in-house co-crystal structure of PaTMK and 1 was initially applied for docking studies. Protein Preparation Wizard in maestro9.0 was employed to refine the structure for Glide docking by deleting unnecessary metals and water molecules followed by optimizing hydrogen-bonding network. After removing three water molecules near 1, a grid was generated using default values except constraints such as aforementioned three H-bond interactions of 1 with Arg74 and Gln105. The structures of commercially available aryl boronic acids (ca. 5000) were obtained from ZINC database. Virtual inhibitors (5000 analogs of 1) possessing various aryl groups at the 6-position of 5 in Scheme 1 were generated using ‘Combigen’ in MOE program package. These were docked into the active site of PaTMK using Glide 5.0 with XP mode and three predefined constraints. The docking results were scored and sorted based on docking scores. The virtual screening results were visually inspected in maestro9.0, and 20 inhibitors were selected for synthesis and screening.
Similarly, the co-crystal structure of PaTMK and 17 was refined using Protein Preparation Wizard in maestro9.0, and a grid was generated for Glide docking. The structures of primary alkyl and aryl amines were obtained from Sigma-Aldrich, Acros and Maybridge websites. ‘Combigen’ in MOE was applied to generate 1188 virtual inhibitors (analogs of 19 in Scheme 3), which were docked into the active site of PaTMK using Glide 5.0. The docking results were scored and visually inspected to select amine reagents for synthesis and screening.
Loop searching by homology modeling
The co-crystal structure of PaTMK with 17 was applied to fill in the missing loop region (Glu140 ~ Gly150). The complete sequence of P. aeruginosa TMK was obtained from Uniprot database (ID: Q9HZN8) and aligned to that of the co-crystal structure using blosum62 algorithm and the tree-based build-up method. The final model was obtained by generating 100 intermediates and averaging these intermediates followed by minimization using Amber99 forcefield with 0.1 RMS gradient. The coordination of 46 was docked into the final model by superimposing the model and the result of Glide docking.
Synthesis
General Methods
All reaction solvents were purified before use. Dichloromethane, tetrahydrofuran, dimethylform amide and toluene were purified by passing through a solvent column composed of activated A-1 alumina. All other reagents purchased from commercial suppliers were used as received. All reactions sensitive to moisture or oxygen were conducted under an argon atmosphere using flame-dried (under vacuum) or oven-dried glassware (overnight). Removal of solvents was accomplished on a rotary evaporator under reduced pressure in the water bath below 35 °C followed by using high vacuum pump. Microwave assisted reactions were performed using a Biotage® Initiator microwave reactor.
Proton nuclear magnetic resonance (1H NMR) spectra and carbon-13 (13C) NMR spectra were recorded on a commercial 400 MHz Bruker NMR spectrometer. Chemical shifts are reported in ppm (δ) relative to tetramethylsilane as an internal standard. Coupling constants (J) are reported in hertz (Hz)
Analytical thin layer chromatography (TLC) was performed on glass plates precoated with a 0.25-mm thickness of silica gel. The TLC plates were visualized with UV light. Column chromatography was performed using a Biotage® Isolera flash purification system using Biotage® SNAP HP-SIL cartridge (30 μm silica, 10 g to 100 g size) and SNAP C18 cartridges. Unless noted otherwise, all compounds isolated by chromatography were sufficiently pure by 1H NMR analysis for use in subsequent reactions.
All final compounds were further introduced to high performance liquid chromatography (HPLC, Varian 1100 series) on a reverse phase ZORBAX Eclipse XDB-C18 column (4.6 × 150 mm, 5 μm). A linear gradient elution was performed ranging from 2% to 98% CH3CN and H2O (containing 0.1% TFA and 1% CH3CN) at 1.5 mL/min. The purity of all final compounds (typically ≥96%) was assayed at 254 nm wavelength.
Benzyl (2-amino-5-bromopyridin-3-yl)carbamate, 3
To a solution of 5-bromopyridine-2,3-diamine (8.0 g, 42 mmol) and pyridine (12 mL) in dry THF (200 mL) was added benzyl chloroformate (11 mL) using a syringe pump (rate 2 mL/h) at 0 °C. After the addition of benzyl chloroformate was complete, the reaction mixture was stirred at room temperature for 12 h. Excess of ethyl acetate was added to the crude mixture, which was washed with saturated aq. NaHCO3 and brine. The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. Purification of the crude product by flash chromatography eluting with a linear gradient ranging from 12% to 100% EtOAc-hexane provided 6.6 g (48%) of 3 as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.00 (s, 1H), 7.90 (s, 1H), 7.80 (d, J = 2.0 Hz, 1H), 7.45–7.32 (m, 5H), 6.11 (s, 2H), 5.17 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 154.03, 151.04, 142.76, 136.32, 128.45, 128.16, 128.11, 120.16, 104.65, 66.29; MS (ESI) 322, 324 m/z [M+H]+.
5-Bromo-N3-methylpyridine-2,3-diamine, 4
To a solution of LiAlH4 (3.2 g) in dry ether was added 3 (6.6 g, 20 mmol) at 0 °C. The reaction mixture was stirred for 15 min, then was warmed to ambient temperature and stirred for 18 h. The mixture was cooled to 0 °C, then 3.2 mL of water, 3.2 mL of 10% NaOH (aq), and 3.2 mL of water were sequentially added. The precipitated white solid was filtered off, and the filtrate was concentrated with a rotary evaporator. The crude product was purified by flash chromatography eluting with a linear gradient ranging from 12% to 100% EtOAc-hexane yielded 4.1 g (99%) of 4 as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 7.28 (d, J = 2.4 Hz, 1H), 6.56 (d, J = 2.0 Hz, 1H), 5.64 (s, 2H), 5.22 (d, J = 4.8 Hz, 1H), 2.69 (d, J = 4.8 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 147.35, 133.47, 133.01, 114.06, 107.14, 29.49; MS (ESI) 202, 204 m/z [M+H]+.
6-Bromo-1-methyl-1H-imidazo[4,5-b]pyridin-2(3H)-one, 5
To a solution of triphosgene (27 g) in dry toluene (200 mL) was added a solution of 4 (10 g, 50 mmol) in dry THF (20 mL) dropwise at room temperature. The reaction mixture was stirred at 100 °C for 5 h. The solution was cooled to ambient, then the solvent was removed under reduced pressure. Ethyl acetate and 10% NaOH (aq) were added to the product mixture. The aqueous layer was collected and treated with 1N HCl (aq) to neutralize, and a yellow solid was formed. The product was obtained by filtering, and further purification by flash chromatography eluting with a linear gradient ranging from 16% to 100% EtOAc-hexane yielded 5 (9.2 g, 80%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 11.71 (s, 1H), 7.96 (d, J = 2.0 Hz, 1H), 7.70 (d, J = 2.0 Hz, 1H), 3.27 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 153.79, 142.53, 139.57, 126.42, 116.18, 111.32, 26.50; MS (ESI) 228, 230 m/z [M+H]+; HRMS (ESI) calcd for C7H7BrN3O [M+H]+ 227.9772, found 227.9773.
6-Bromo-1-methyl-3-trityl-1H-imidazo[4,5-b]pyridin-2(3H)-one, 6
To a solution of 5 (9.2 g, 40 mmol) in dry CH2Cl2 (150 mL) were added trityl chloride (13 g, 46 mmol) and NEt3 (10 mL) at room temperature. The reaction mixture was stirred overnight (ca. 16 h), and the solvent was removed under reduced pressure. The product mixture was diluted with ethyl acetate and washed with saturated NaHCO3 (aq) and brine. The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. Purification of the crude product by flash chromatography eluting with a linear gradient ranging from 6% to 50% EtOAc-hexane provided 17 g (88%) of 6 as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 2.0 Hz, 1H), 7.63 (d, J = 7.6 Hz, 6H), 7.32 (t, J = 7.4 Hz, 6H), 7.23 (t, J = 7.4 Hz, 3H), 7.17 (d, J = 2.0 Hz, 1H), 3.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.68, 142.52, 139.80, 129.03, 127.38, 126.62, 125.94, 115.20, 112.72, 74.84, 26.88; MS (ESI) 243 m/z [decomposition of trityl group, M+H]+, HRMS (ESI) calcd for C26H21BrN3O [M+H]+ 470.0868, found : 470.0876.
General Procedure A for Synthesis of Aryl-Substituted Analogues (1, 9 – 26) in Scheme 1
1-Methyl-6-phenyl-3-trityl-1H-imidazo[4,5-b]pyridin-2(3H)-one, 7
A reaction mixture of 6 (0.11 g, 0.23 mmol), phenyl boronic acid (36 mg, 0.30 mmol), Pd2(dba)3 (5 mg, 2 mol%), PCy3 (5 mg, 5 mol%), and K3PO4 (1M, 1 mL) in dioxane (4 mL) was stirred under microwave heating (100 °C) for 1 h. The palladium catalyst was removed by filtration. The product mixture was diluted with ethyl acetate (30 mL) and washed with water (10 mL) and brine (10 mL). The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. The resulting product was purified by flash chromatography eluting with a linear gradient ranging from 6% to 50% EtOAc-hexane to afford 7 (0.12 g) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 2.0 Hz, 1H), 7.46–7.06 (m, 22H), 3.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 154.37, 143.50, 142.99, 138.58, 138.41, 130.72, 129.35, 129.13, 127.62, 127.48, 127.12, 126.69, 125.26, 111.29, 74.91, 27.04; MS (ESI) 243 m/z [decomposition of trityl group, M+H]+; HRMS (ESI) calcd for C32H26N3O [M+H]+ 468.2076, found 468.2090.
1-Methyl-6-phenyl-1H-imidazo[4,5-b]pyridin-2(3H)-one, 1
To a solution of 7 (0.12 g) in CH2Cl2 (5 mL) was added trifluoroacetic acid (1 mL) at room temperature. The reaction mixture was stirred for 30 min. The solvent and trifluoroacetic acid were removed in vacuo. The product 8 was purified by flash chromatography eluting with a linear gradient ranging from 12% to 100% EtOAc-hexane as a white solid (49 mg, 93% over 2 steps). 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1H), 8.21 (d, J = 2.0 Hz, 1H), 7.74 (d, J = 2.0 Hz, 1H), 7.72–7.69 (m, 2H), 7.37 (tt, J = 7.4, 1.2 Hz, 1H), 3.36 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.09, 143.09, 138.05, 129.49, 128.95, 127.27, 126.65, 125.40, 112.21, 26.39; MS (ESI) 226 m/z [M+H]+; HRMS (ESI) calcd for C13H12N3O [M+H]+ 226.0980, found 226.0981; Purity (≥98%, tr = 6.58 min).
6-(3-Fluorophenyl)-1-methyl-1H-imidazo[4,5-b]pyridin-2(3H)-one, 9
The general procedure A was followed using 6 (185 mg, 393 μmol) and 3-fluorophenylboronic acid to give 9 as a white solid (76 mg, 314 μmol, 80%). 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 8.26 (d, J = 2.0 Hz, 1H), 7.79 (d, J = 2.0 Hz, 1H), 7.59–7.55 (m, 2H), 7.52–7.46 (m, 1H), 7.18 (tdd, J = 8.4, 2.4, 1.2 Hz, 1H), 3.35 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 163.93, 161.51, 154.10, 143.52, 140.57, 140.49, 138.26, 130.90, 125.43, 122.60, 122.57, 114.03, 113.82, 113.40, 113.18, 112.23, 26.44; MS (ESI) 244.2 m/z [M+H]+; Purity (96%, tr = 6.85 min).
1-Methyl-6-(pyridin-3-yl)-1H-imidazo[4,5-b]pyridin-2(3H)-one, 10
The general procedure was followed using 6 (194 mg, 412 μmol) and pyridine-3-boronic acid to provide 13 as a white solid (97 mg, 431 μmol, 100%). 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 8.98 (d, J = 1.6 Hz, 1H), 8.61 (d, J = 3.6 Hz, 1H), 8.29 (d, J = 2.0 Hz, 1H), 8.21 (dt, J = 4.0, 2.4 Hz, 1H), 7.86 (d, J = 2.0 Hz, 1H), 7.57 (dd, J = 8.0, 4.8 Hz, 1H), 3.36 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.06, 147.41, 146.64, 143.73, 138.34, 134.98, 134.03, 125.97, 125.54, 124.23, 112.20, 26.47; MS (ESI) 227.3 m/z [M+H]+; Purity (≥98%, tr = 3.74 min).
1-Methyl-6-(pyrimidin-5-yl)-1H-imidazo[4,5-b]pyridin-2(3H)-one, 11
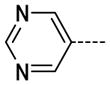
The general procedure A was followed using 6 (202 mg, 431 μmol) and pyrimidine-5-boronic acid to provide 11 as a yellow solid (78 mg, 346 μmol, 80%). 1H NMR (400 MHz, DMSO-d6) δ 11.75 (s, 1H), 9.19 (s, 1H), 8.36 (d, J = 2.0 Hz, 1H), 7.94 (d, J = 2.0 Hz, 1H), 3.35 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 157.07, 154.47, 154.02, 144.11, 138.37, 131.49, 125.61, 123.03, 111.99, 26.52; MS (ESI) 228.4 m/z [M+H]+; Purity (≥98%, tr = 4.48 min).
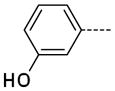
6-(3-Hydroxyphenyl)-1-methyl-1H-imidazo[4,5-b]pyridin-2(3H)-one, 12
The general procedure A was followed using 6 (192 mg, 408 μmol) and 3-hydroxyphenylboronic acid to provide 12 as a dime yellow solid (66 mg, 247 μmol, 67%). 1H NMR (400 MHz, DMSO-d6) δ 11.58 (s, 1H), 9.68 (br, 1H), 8.13 (d, J = 2.0 Hz, 1H), 7.66 (d, J = 1.6 Hz, 1H), 7.25 (t, J = 7.8 Hz, 1H), 7.09–7.06 (m, 2H), 6.78 (ddd, J = 8.0, 2.0, 0.8 Hz, 1H), 3.34 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 158.02, 154.19, 143.13, 139.49, 137.98, 129.99, 129.79, 125.42, 117.45, 114.39, 113.63, 112.25, 105.86, 26.43; MS (ESI) 242.2 m/z [M+H]+; Purity (96%, tr = 5.48 min).
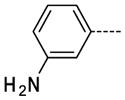
6-(3-Aminophenyl)-1-methyl-1H-imidazo[4,5-b]pyridin-2(3H)-one, 13
The general procedure A was followed using 6 (121 mg, 255 μmol) and 3-aminobenzeneboronic acid to provide 13 as a yellow solid (61 mg, 254 μmol, 100%). 1H NMR (400 MHz, DMSO-d6) δ 11.71 (s, 1H), 10. 46 (br, 2H), 8.19 (d, J = 2.0 Hz, 1H), 7.75–7.72 (m, 2H), 7.68 (d, J = 1.6 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.38 (dd, J = 8.0, 1.2 Hz, 1H), 3.36 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.08, 143.55, 139.56, 137.98, 133.16, 130.38, 128.24, 126.16, 125.56, 121.96, 121.09, 112.15, 26.45; MS (ESI) 241.2 m/z [M+H]+; Purity (≥98%, tr = 4.25 min).
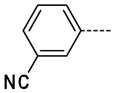
3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzonitrile, 14
The general procedure A was followed using 6 (480 mg, 1.02 μmol) and 3-cyanophenylboronic acid to provide 14 as a white solid (265 mg, 537 μmol, 53%). 1H NMR (400 MHz, DMSO-d6) δ 11.68 (s, 1H), 8.31 (d, J = 2.0 Hz, 1H), 8.22 (t, J = 1.6 Hz, 1H), 8.08 (ddd, J = 8.0, 2.0, 1.2 Hz, 1H), 7.88 (d, J = 2.0 Hz, 1H), 7.82 (dt, J = 8.0, 1.2 Hz, 1H), 7.67 (t, J = 8.0 Hz, 1H), 3.36 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.10, 143.75, 139.23, 138.41, 131.24, 130.79, 130.15, 130.07, 127.28, 125.51, 118.79, 112.27, 112.14, 26.47; MS (ESI) 251.1 m/z [M+H]+; Purity (≥98%, tr = 6.32 min).
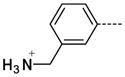
6-(3-(Aminomethyl)phenyl)-1-methyl-1H-imidazo[4,5-b]pyridin-2(3H)-one hydrochloride, 15
The general procedure A was followed using 6 (790 mg, 1.66 mmol) and 3-N-Boc-aminomethylphenylboronic acid (0.88 g, 3.09 mmol) to provide 15 as a white solid (0.23 mg, 0.791 mmol, 48%). 1H NMR (400 MHz, D2O) δ 7.85 (d, J = 2.0 Hz, 1H), 7.39–7.26 (m, 4H), 7.15 (d, J = 1.6 Hz, 1H), 4.11 (s, 2H), 3.06 (s, 3H); 13C NMR (100 MHz, D2O) δ 154.92, 141.02, 137.70, 136.97, 133.13, 129.65, 129.13, 127.74, 126.43, 125.99, 124.79, 112.94, 42.92, 26.09; MS (ESI) 255 m/z [M+H]+; Purity (≥98%, tr = 4.50 min).
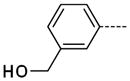
6-(3-(Hydroxymethyl)phenyl)-1-methyl-1H-imidazo[4,5-b]pyridin-2(3H)-one, 16
The general procedure A was followed using 6 (127 mg, 270 μmol) and 3-(hydroxymethyl)phenylboronic acid to provide 16 as a white solid (44 mg, 170 μmol, 63%). 1H NMR (400 MHz, CDCl3) δ 11.25 (br, 1H), 8.28 (d, J = 1.2 Hz, 1H), 7.57–7.33 (m, 5H), 5.41 (s, 2H), 3.46 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 154.84, 143.42, 139.06, 139.06, 134.38, 130.63, 129.88, 128.26, 128.05, 127.62, 126.01, 112.71, 69.57, 26.98; MS (ESI) 256.4 m/z [M+H]+; Purity (95%, tr = 5.35 min).
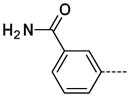
3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide
The general procedure A was followed using 6 (208 mg, 443 μmol) and 3-carbamoylphenylboronic acid to provide 17 as a white solid (51 mg, 190 μmol, 43%). 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.18 (t, J = 1.6 Hz, 1H), 8.09 (br, 1H), 7.87 (t, J = 1.6 Hz, 1H), 7.85 (t, J = 2.0 Hz, 1H), 7.81 (d, J = 2.0 Hz, 1H), 7.55 (t, J = 2.0 Hz, 1H), 7.45 (br, 1H), 3.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.72, 154.09, 143.30, 138.26, 138.01, 135.01, 129.31, 128.95, 128.90, 126.42, 125.51, 125.45, 112.21, 26.42; MS (ESI) 269.1 m/z [M+H]+; HRMS (ESI) calcd for C14H13N4O2 [M+H]+ 269.1039, found 269.1041; Purity (≥98%, tr = 5.00 min).
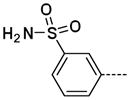
3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzenesulfonamide, 18
The general procedure A was followed using 6 (233 mg, 490 μmol) and benzenesulfonamide-3-boronic acid pinacol ester to provide 18 as a white solid (18 mg, 59 μmol, 12%). 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 8.26 (d, J = 2.0 Hz, 1H), 8.15 (t, J = 1.6 Hz, 1H), 7.94 (ddd, J = 7.6, 1.6, 0.8 Hz, 1H), 7.82 (ddd, J = 8.0, 1.6, 1.2 Hz, 1H), 7.77 (d, J = 2.0 Hz, 1H), 7.67 (t, J = 7.6 Hz, 1H), 7.41 (br, 2H), 3.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.11, 144.91, 143.65, 138.84, 138.25, 129.92, 129.69, 128.18, 125.56, 124.24, 123.59, 112.08, 26.47; MS (ESI) 305.1 m/z [M+H]+; Purity (≥98%, tr = 5.07 min).
N-(3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)phenyl)acetamide, 21
The general procedure A was followed using 6 (487 mg, 1.04 mmol) and 3-acetamidophenylboronic acid to provide 21 as a yellow solid (105 mg, 373 μmol, 36%). 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 10.04 (s, 1H), 8.13 (d, J = 2.0 Hz, 1H), 7.86 (s, 1H), 7.65 (d, J = 1.6 Hz, 1H), 7.59 (d, J = 7.6 Hz, 1H), 7.41–7.33 (m, 2H), 3.35 (s, 3H), 2.06 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 168.43, 154.10, 143.16, 139.90, 138.58, 137.89, 129.64, 129.35, 125.44, 121.56, 118.00, 117.29, 112.16, 26.39, 24.04; MS (ESI) 283.2 m/z [M+H]+; Purity (98%, tr = 5.44 min).
6-(3-Acetylphenyl)-1-methyl-1H-imidazo[4,5-b]pyridin-2(3H)-one, 22
The general procedure A was followed using 6 (82 mg, 161 μmol) and 3-acetyphenylboronic acid to provide 22 as a white solid (29 mg, 108 μmol, 67%). 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.29 (d, J = 2.0 Hz, 1H), 8.22 (t, J = 1.6 Hz, 1H), 7.98–7.93 (m, 2H), 7.83 (d, J = 2.0 Hz, 1H), 7.63 (t, J = 8.0 Hz, 1H), 3.37 (s, 3H), 2.70 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 198.00, 154.09, 143.41, 138.54, 138.32, 137.56, 131.29, 129.38, 128.66, 126.91, 126.31, 125.46, 112.32, 26.95, 26.47; MS (ESI) 288.2 m/z [M+H]+; Purity (≥98%, tr = 6.19 min).
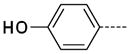
6-(4-Hydroxyphenyl)-1-methyl-1H-imidazo[4,5-b]pyridin-2(3H)-one, 23
The general procedure A was followed using 6 (205 mg, 430 μmol) and 4-hydroxyphenylboronic acid to provide 23 as a dark yellow solid (12 mg, 52 μmol, 12%). 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 9.53 (s, 1H), 8.11 (d, J = 2.0 Hz, 1H), 7.63 (d, J = 2.0 Hz, 1H), 7.50 (dt, J = 8.8, 2.0 Hz, 1H), 6.85 (dt, J = 8.4, 2.0 Hz, 1H), 3.36 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 156.98, 154.08, 142.34, 137.36, 129.70, 128.77, 127.75, 125.31, 115.75, 111.73, 26.33; MS (ESI) 242.2 m/z [M+H]+; Purity (≥98%, tr = 5.17 min).
1-Methyl-6-(pyridin-4-yl)-1H-imidazo[4,5-b]pyridin-2(3H)-one, 24
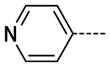
The general procedure A was followed using 6 (149 mg, 317 μmol) and pyridine-4-boronic acid to provide 24 as a light yellow solid (56 mg, 246 μmol, 78%). 1H NMR (400 MHz, DMSO-d6) δ 11.81 (s, 1H), 8.71 (s, 2H), 8.47 (d, J = 2.0 Hz, 1H), 7.97 (d, J = 2.0 Hz, 1H), 7.95 (d, J = 6.0 Hz, 2H), 3.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.06, 148.23, 147.26, 144.84, 139.10, 125.65, 125.58, 121.39, 112.00, 26.53; MS (ESI) 227.2 m/z [M+H]+; Purity (≥98%, tr = 3.80 min).
6-(4-(Aminomethyl)phenyl)-1-methyl-1H-imidazo[4,5-b]pyridin-2(3H)-one hydrochloride, 25
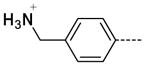
The general procedure A was followed using 6 (613 mg, 1.29 mmol) and 4-(N-Boc-aminomethyl)phenylboronic acid to provide 25 as a white solid (71 mg, 279 μmol, 22%), which was purified by flash chromatography using reverse phase C13 column eluting with a linear gradient ranging from 0% to 100% Acetonitrile/Water. 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.52 (d, J = 15.6 Hz, 2H), 8.25 (d, J = 2.0 Hz, 1H), 7.79–7.76 (m, 3H), 7.60 (d, J = 8.0 Hz, 2H), 5.41 (br, 2H), 3.2 (t, J = 3.2 Hz, 2H), 3.36 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 154.08, 143.24, 138.03, 133.08, 129.59, 128.76, 126.63, 125.49, 112.11, 99.51, 41.80, 26.45; MS (ESI) 255 m/z [M+H]+; Purity (≥98%, tr = 4.32 min).
4-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 26
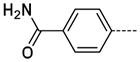
The general procedure A was followed using 6 (200 mg, 421 mmol) and 4-carbamoylphenylboronic acid to provide 26 as a white solid (54 mg, 200 μmol, 48%). 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.04 (br, 1H), 7.98 (d, J = 8.4 Hz, 2H), 7.83 (d, J = 2.0 Hz, 1H), 7.81 (d, J = 8.8 Hz, 2H), 7.38 (br, 1H), 3.64 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.47, 154.10, 143.51, 140.68, 138.36, 132.85, 128.48, 128.17, 126.25, 125.48, 112.20, 26.44; MS (ESI) 269.1 m/z [M+H]+; Purity (≥98%, tr = 4.80 min).
3-(1-Methyl-2-oxo-3-trityl-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzoic acid, 27
A reaction mixture of 6 (6.22 g, 13.1 mmol), phenyl boronic acid (2.81 g, 16.9 mmol), Pd2(dba)3 (99 mg, 1 mol%), PCy3 (108 mg, 2 mol%), and K3PO4 (2 M, 12 mL) in dioxane (28 mL) was stirred under microwave heating (100 °C) for 1 h. The palladium catalyst was removed by filtration. The filtrate was diluted with ethyl acetate (100 mL) and treated with 1 N HCl (aq.) to acidify. The aqueous layer was removed, and then the organic layer was washed with water (20 mL) and brine (20 mL). The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. Purification of the crude product by flash chromatography eluting with a linear gradient ranging from 25% to 100% EtOAc-hexane provided 6.76 g (99%) of 27 as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 13.07 (s, 1H), 8.18 (t, J = 1.2 Hz, 1H), 8.06 (d, J = 2.0 Hz, 1H), 7.92–7.90 (m, 2H), 7.80 (d, J = 2.0 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.51 (d, J = 7.6 Hz, 6H), 7.23 (t, J = 7.6 Hz, 6H), 7.15 (d, J = 7.2 Hz, 3H), 3.34 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.17, 153.32, 142.96, 137.95, 136.81, 131.59, 130.98, 129.27, 128.82, 128.53, 128.21, 127.34, 127.27, 126.23, 125.40, 112.06, 73.78, 26.84; MS (ESI) 510.4 m/z [M-H]−; HRMS (ESI) calcd for C33H26N3O3 [M+H]+ 512.1974, found 512.1978.
Pentafluorophenyl 3-(1-Methyl-2-oxo-3-trityl-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzoate, 28
To a solution of 27 (0.502 g, 0.96 mmol) and diisopropylethyl amine (0.35 mL) in dry CH2Cl2 (10 mL) was added pentafluorophenyl trifluoroacetate (0.25 mL, 1.45 mmol) dropwise at 0 °C. The mixture was stirred for 10 min, then was warmed to the ambient temperature and stirred for 1 h. The solvent was removed under reduced pressure, then purification of the crude product by flash chromatography eluting with a linear gradient ranging from 6% to 50% EtOAc-hexane yielded 0.528 g (81%) of 28 as viscous yellow oil. A solution of 28 (0.1 M) in dry CH2Cl2 was prepared for the following amide formation reaction and stored at room temperature. 1H NMR (400 MHz, CDCl3) δ 8.39 (t, J = 1.6 Hz, 1H), 8.22 (dt, J = 7.6, 1.2 Hz, 1H), 8.11 (d, J = 2.0 Hz, 1H), 7.87 (ddd, J = 8.0, 1.6, 0.8 Hz, 1H), 7.66–7.60 (m, 7H), 7.34 (d, J = 2.0 Hz, 1H), 7.31–7.28 (m, 6H), 7.25–7.21 (m, 3H), 3.42 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.57, 154.30, 144.09, 142.85, 139.63, 138.39, 133.18, 129.79, 129.67, 129.30, 129.21, 129.06, 127.85, 127.50, 126.74, 125.45, 111.10, 75.03, 27.12; MS (ESI) 243 m/z [decomposition of trityl group, M+H]+, HRMS (ESI) calcd for C39H25F5N3O3 [M+H]+ 678.1816, found 678.1814.
General Procedure B for Synthesis of the Substituted Benzamide Analogues (19, 20, 30 – 53) in Scheme 3
To a solution of 28 (0.1 M) and diisopropylethyl amine (ca. 3 eq.) was added the appropriate primary amine (1.2 eq) at 0 °C. The reaction mixture was warmed to the ambient temperature and stirred for 1 h. The solvent was removed by reduced pressure. The crude product was diluted with ethyl acetate (30 mL) and washed with saturated aq. NaHCO3 (10 mL) and brine (10 mL). The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. Purification of the crude product by flash chromatography eluting with a linear gradient ranging from 25% to 100% EtOAc-hexane provided trityl protected benzamide analogues 29. To a solution of 29 in CH2Cl2 (5 mL) was added trifluoroacetic acid (1 mL) at room temperature. The reaction mixture was stirred for 30 min. After the solvent and trifluoroacetic acid were removed in vacuo, the desired product (19, 20, 30–53) was purified by flash chromatography eluting with a linear gradient ranging from 0% to 20% MeOH/EtOAc.
N-Methyl-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 19
The general procedure B was followed using 28 (216 mg, 318 μmol) and methylamine to provide 19 as a white solid (86 mg, 305 μmol, 96%). 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.59 (d, J = 4.4 Hz, 1H), 8.29 (d, J = 2.0 Hz, 1H), 8.14 (t, J = 1.6 Hz, 1H), 7.86–7.81 (m, 3H), 7.55 (t, J = 8.0 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 3.37 (s, 3H), 2.82 (d, J = 4.8 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 166.49, 154.11, 143.33, 138.27, 138.07, 135.29, 129.14, 129.01, 128.94, 126.10, 125.46, 125.12, 112.23, 26.43, 26.23; MS (ESI) 283.0 m/z [M+H]+; HRMS (ESI) calcd for C15H15N4O2 [M+H]+ 283.1195, found 283.1194; Purity (≥98%, tr = 5.28 min).
N,N-Dimethyl-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 20
The general procedure B was followed using 28 (123 mg, 182 μmol) and dimethylamine to provide 20 as a white solid (26 mg, 89 μmol, 50%). 1H NMR (400 MHz, CDCl3) δ 11.05 (s, 1H), 8.27 (d, J = 2.0 Hz, 1H), 7.63 (t, J = 1.6 Hz, 1H), 7.59 (dt, J = 8.0, 1.6 Hz, 1H), 7.46 (t, J = 7.6 Hz, 1H), 7.38 (dt, J = 7.6, 1.6 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 3.43 (s, 3H), 3.13 (s, 3H), 3.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.48, 154.77, 143.34, 139.08, 138.89, 137.40, 130.59, 129.21, 128.33, 126.24, 126.08, 125.95, 112.71, 39.85, 35.62, 26.96; MS (ESI) 297.3 m/z [M+H]+; Purity (≥98%, tr = 5.58 min).
N-(2-Amino-2-oxoethyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 30
The general procedure B was followed using a 0.1 M solution of 28 (5 mL, 500 μmol) and glycine benzyl ester and then followed by heating with 2.0 M NH3 in MeOH (5 mL) at 100 °C for 5 h to provide 30 as a white solid (87 mg, 268 μmol, 54%). 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.81 (t, J = 5.6 Hz, 1H), 8.32 (d, J = 2.0 Hz, 1H), 8.21 (t, J = 1.6 Hz, 1H), 7.87 (t, J = 8.0 Hz, 1H), 7.81 (d, J = 2.0 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.40 (s, 1H), 7.06 (s, 1H), 3.86 (d, J = 6.0 Hz, 2H), 3.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 170.99, 166.29, 154.09, 143.34, 138.31, 138.00, 134.85, 129.37, 128.99, 128.89, 126.38, 125.46, 125.41, 112.17, 42.44, 26.44; MS (ESI) 326.3 m/z [M+H]+; Purity (66%, tr = 4.78 min).
2-(3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamido)acetic acid, 31
The general procedure B was followed using a 0.1 M solution of 28 (3 mL, 300 μmol) and glycine benzyl ester and followed by de-benzylation using H2 and Pd/C in MeOH (10 mL) to provide 31 as a pale yellow solid (98 mg, 300 μmol, 100%). 1H NMR (400 MHz, DMSO-d6) δ 8.37 (t, J = 5.6 Hz, 1H), 8.28 (d, J = 2.0 Hz, 1H), 8.14 (t, J = 1.6 Hz, 1H), 7.84–7.80 (m, 3H), 3.70 (d, J = 5.2 Hz, 2H), 3.36 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 171.63, 165.41, 154.13, 143.33, 138.26, 138.07, 135.53, 129.10, 129.00, 128.97, 126.03, 125.45, 125.15, 112.25, 43.57, 26.53; MS (ESI) 327.2 m/z [M+H]+; Purity (64%, tr = 5.52 min).
2-(3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamido)ethanesulfonic acid, 32
The general procedure B was followed using a 0.1 M solution of 28 (3 mL, 300 μmol) and taurine to provide 32 as a pale yellow solid (83 mg, 221 μmol, 74%). 1H NMR (400 MHz, DMSO-d6) δ 11.68 (s, 1H), 8.63 (t, J = 5.6 Hz, 1H), 8.28 (d, J = 1.6 Hz, 2H), 8.10 (s, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.81 (d, J = 2.0 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H), 3.57 (q, J = 7.6 Hz, 2H), 3.37 (s, 3H), 2.74 (t, J = 7.6 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 165.62, 154.08, 143.22, 138.10, 137.94, 135.36, 129.29, 129.09, 128.95, 125.91, 125.59, 125.13, 112.40, 50.28, 36.22, 26.48; MS (ESI) 332.3 m/z [M+H]+; Purity (96%, tr = 4.42 min).
N-(Cyanomethyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 33
The general procedure B was followed using a 0.1 M solution of 28 (229 mg, 526 μmol) and aminoacetonitrile to provide 33 as a white solid (29 mg, 95 μmol, 18%). 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 9.31 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 1.6 Hz, 1H), 8.18 (t, J = 1.6 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.82 (d, J = 2.0 Hz, 1H), 7.61 (t, J = 8.0 Hz, 1H), 4.37 (d, J = 5.6 Hz, 2H), 3.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 166.52, 154.09, 143.42, 138.35, 138.32, 133.57, 130.09, 129.29, 128.68, 126.34, 125.47, 125.40, 117.64, 112.21, 27.75, 26.45; MS (ESI) 308.2 m/z [M+H]+; Purity (93%, tr = 5.46 min).
N-(2-(1H-Imidazol-5-yl)ethyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 34
The general procedure B was followed using a 0.1 M solution of 28 (3 mL, 300 μmol) and histamine to provide 34 as a white solid (113 mg, 300 μmol, 100%). 1H NMR (400 MHz, DMSO-d6) δ 14.39 (br, 1H), 11.65 (s, 1H), 8.99 (d, J =1.2 Hz, 1H), 8.75 (t, J = 5.6 Hz, 1H), 8.28 (d, J = 2.0 Hz, 1H), 8.10 (s, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.80–7.78 (m, 2H), 7.56 (t, J = 8.0 Hz, 1H), 7.48 (s, 1H), 3.60 (q, J = 6.8 Hz, 2H), 3.37 (s, 3H), 2.95 (t, J = 6.8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.29, 154.11, 143.36, 138.29, 138.13, 135.07, 133.84, 131.11, 129.42, 129.03, 128.90, 126.19, 125.46, 125.21, 116.24, 112.19, 38.18, 26.45, 24.50; MS (ESI) 363 m/z [M+H]+; Purity (≥98%, tr = 4.84 min).
3-(3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamido)propanoic acid, 35
The general procedure B was followed using a 0.1 M solution of 28 (3 mL, 300 μmol) and β-alanine benzylester hydrochloride, and then benzyl ester deprotection was performed with Pd/C and H2 in MeOH (10 mL) to provide 35 as a white solid (33 mg, 97 μmol, 32%). 1H NMR (400 MHz, DMSO-d6) δ 12.52 (s, 1H), 11.64 (s, 1H), 8.67 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 1.6 Hz, 1H), 8.14 (s, 1H), 7.87–7.80 (m, 3H), 7.56 (t, J = 8.0 Hz, 1H), 3.50 (q, J = 6.8 Hz, 2H), 3.37 (s, 3H), 2.55 (t, J = 6.8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 172.92, 166.11, 154.10, 143.34, 138.32, 138.09, 135.12, 129.33, 129.02, 128.92, 126.20, 125.46, 125.21, 112.23, 35.65, 33.79, 26.46; MS (ESI) 339 m/z [M-H] −; Purity (72%, tr = 5.75 min).
N-(2-hydroxyethyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 36
The general procedure B was followed using 28 (114 mg, 261 μmol) and ethanolamine to provide 36 as a white solid (22 mg, 69 μmol, 26%). 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 8.56 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.16 (t, J = 1.6 Hz, 1H), 7.86–7.83 (m, 2H), 7.81 (d, J = 2.0 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 4.75 (br, 1H), 3.54 (t, J = 5.6 Hz, 2H), 3.39–3.35 (m, 5H); 13C NMR (100 MHz, DMSO-d6) δ 166.18, 154.09, 143.32, 138.32, 138.02, 135.30, 129.21, 128.96, 128.94, 126.24, 125.44, 125.23, 112.21, 59.76, 42.23, 26.45; MS (ESI) 313 m/z [M+H]+; Purity (≥98%, tr = 4.95 min).
N-(2-Aminoethyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide hydrochloride, 37
The general procedure B was followed using a 0.1 M solution of 37 (3 mL, 300 μmol) and N-Boc-ethylenediamine. After adding 1N HCl (aq), purification with reverse phase flash chromatography eluting with a linear gradient ranging from 5 to 100% acetonitrile/water provided 37 as a white solid (30 mg, 96 μmol, 28%). 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 9.00 (t, J = 5.6 Hz, 1H), 8.33 (d, J = 2.0 Hz, 1H), 8.31 (s, 1H), 8.13 (br, 2H), 7.96 (d, J = 2.0 Hz, 1H), 7.91–7.88 (m, 2H), 7.58 (t, J = 8.0 Hz, 1H), 3.57 (q, J = 6.0 Hz, 2H), 3.38 (s, 3H), 3.03 (q, J = 6.0 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.59, 154.09, 143.29, 138.13, 137.96, 134.74, 129.35, 129.01, 128.79, 126.49, 125.52, 125.43, 112.44, 38.62, 37.17, 26.55; MS (ESI) 312 m/z [M+H]+; Purity (≥98%, tr = 4.63 min).
2-(3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamido)propanoic acid, 38
The general procedure B was followed using 28 (213 mg, 314 μmol) and D/L-alanine benzylester and followed by de-benzylation under H2 and Pd/C in MeOH (10 mL) to provide 38 as a white solid (51 mg, 151 μmol, 48%). 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 8.80 (d, J = 7.2 Hz, 1H), 8.58 (br, 1H), 8.32 (d, J = 2.0 Hz, 1H), 8.20 (s, 1H), 7.89–7.86 (m, 2H), 7.81 (d, J = 2.0 Hz, 1H), 7.58 (d, J = 7.6 Hz, 1H), 4.47 (p, J = 7.2 Hz, 1H), 3.37 (s, 3H), 1.43 (d, J = 7.6 Hz, 3H); 13C NMR (100 MHz, DMSO-d6) δ 174.20, 165.96, 154.12, 143.37, 138.36, 138.11, 134.63, 129.57, 129.02, 128.92, 126.50, 125.47, 125.42, 112.24, 48.23, 26.47, 12.33; MS (ESI) 341 m/z [M+H]+; Purity (34%/66%, tr = 5.32 and 5.90 min).
3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)-N-(3-morpholinopropyl)benzamide, 39
The general procedure B was followed using a 0.1 M solution of 28 (1 mL, 100 μmol) and N-(3-aminopropryl)morpholine to provide 39 as a pale yellow solid (34 mg, 86 μmol, 86%). 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 8.75 (br, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.15 (s, 1H), 7.89–7.83 (m, 2H), 7.81 (d, J = 2.0 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 3.76–3.56 (m, 4H), 3.41–3.33 (m, 5H), 3.15–3.10 (m, 4H), 1.92–1.91 (br, 2H), 1.25 (q, J = 6.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.33, 154.10, 143.36, 138.28, 138.12, 135.05, 129.41, 129.05, 128.87, 126.23, 125.46, 125.20, 112.20, 74.12, 63.78, 53.53, 26.46, 18.04, 16.71; MS (ESI) 396 m/z [M+H]+; Purity (≥98%, tr = 4.93 min).
N-Benzyl-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 40
The general procedure B was followed using 28 (88 mg, 203 μmol) and benzylamine to provide 40 as a white solid (106 mg, 295 μmol, 99%). 1H NMR (400 MHz, DMSO-d6) δ 11.62 (s, 1H), 9.15 (t, J = 6.0 Hz, 1H), 8.31 (d, J = 2.0 Hz, 1H), 8.21 (t, J = 1.2 Hz, 1H), 7.90–7.86 (m, 2H), 7.81 (d, J = 2.0 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.36–7.31 (m, 4H), 7.27–7.23 (m, 1H), 4.53 (d, J = 6.0 Hz, 2H), 3.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 166.03, 154.08, 143.34, 139.60, 138.33, 138.15, 135.03, 129.41, 129.07, 128.90, 128.28, 127.24, 126.75, 126.30, 125.44, 125.26, 112.21, 42.66, 26.44; MS (ESI) 359 m/z [M+H]+; Purity (≥98%, tr = 6.90 min).
3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)-N-(2-(pyridin-4-yl)ethyl)benzamide, 41
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 200 μmol) and 4-(2-aminoethyl)pyridine to provide 41 as a white solid (64 mg, 171 μmol, 86%). 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 8.71 (t, J = 5.6 Hz, 1H), 8.66 (br, 2H), 8.27 (d, J = 2.0 Hz, 1H), 8.08 (t, J = 1.6 Hz, 1H), 7.85 (dd, J = 7.6, 1.2 Hz, 1H), 7.79–7.77 (m, 2H), 7.64 (d, J = 5.6 Hz, 2H), 7.56 (t, J = 7.6 Hz, 1H), 3.63 (q, J = 6.8 Hz, 2H), 3.37 (s, 3H), 3.05 (t, J = 6.8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.20, 154.36, 154.09, 145.52, 143.35, 138.27, 138.12, 135.08, 129.38, 129.05, 128.88, 126.12, 125.81, 125.45, 125.16, 112.18, 34.67, 26.44; MS (ESI) 374 m/z [M+H]+; Purity (≥98%, tr = 4.98 min).
3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)-N-phenethylbenzamide, 42
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 200 μmol) and phenethylamine to provide 42 as a white solid (63 mg, 170 μmol, 85%). 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.68 (t, J = 5.6 Hz, 1H), 8.29 (d, J = 2.0 Hz, 1H), 8.12 (t, J = 1.6 Hz, 1H), 7.87–7.79 (m, 3H), 7.56 (t, J = 7.6 Hz, 1H), 7.33–7.18 (m, 5H), 3.52 (q, J = 6.8 Hz, 2H), 3.37 (s, 3H), 2.88 (t, J = 7.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.01, 154.09, 143.33, 139.51, 138.28, 138.06, 135.35, 129.22, 129.00, 128.92, 128.66, 128.33, 126.13, 126.09, 125.45, 125.17, 112.19, 40.94, 35.12, 26.44; MS (ESI) 373 m/z [M+H]+; Purity (98%, tr = 7.15 min).
N-(2-(1H-Benzo[d]imidazol-2-yl)ethyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 43
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 200 μmol) and 2-(1H-benzimidazol-2-yl)ethanamine hydrochloride to provide 43 as a white solid (24 mg, 57 μmol, 29%). 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 8.89 (t, J = 5.6 Hz, 1H), 8.28 (d, J = 2.0 Hz, 1H), 8.14 (s, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.82–7.80 (m, 2H), 7.58–7.54 (m, 3H), 7.23 (dd, J = 6.0, 3.2 Hz, 2H), 3.79 (q, J = 6.8 Hz, 2H), 3.36 (s, 3H), 3.21 (t, J = 6.8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.33, 158.19, 157.89, 154.10, 152.75, 143.34, 138.28, 138.07, 135.12, 129.34, 129.01, 128.88, 126.24, 125.45, 125.28, 122.41, 114.27, 112.21, 37.85, 28.37, 26.46; MS (ESI) 413 m/z [M+H]+; Purity (≥98%, tr = 5.42 min).
3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)-N-(pyridin-2-ylmethyl)benzamide, 44
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 200 μmol) and 2-(aminomethyl)pyridine to provide 44 as a white solid (53 mg, 148 μmol, 74%). 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 9.26 (t, J = 6.0 Hz, 1H), 8.54 (d, J = 4.8 Hz, 1H), 8.32 (d, J = 5.0 Hz, 1H), 8.25 (s, 1H), 7.92–7.88 (m, 2H), 7.82–7.77 (m, 2H), 7.59 (t, J = 7.6 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.30 (dd, J = 7.2, 4.8 Hz, 1H), 4.63 (d, J = 5.6 Hz, 2H), 3.36 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 166.25, 158.59, 154.09, 148.57, 143.36, 138.34, 138.17, 137.10, 134.87, 129.50, 129.11, 128.88, 126.34, 125.46, 125.32, 122.23, 121.19, 112.21, 44.63, 26.45; MS (ESI) 360 m/z [M+H]+; Purity (≥98%, tr = 4.94 min).
N-((1H-Benzo[d]imidazol-2-yl)methyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 45
The general procedure B was followed using a 0.1 M solution of 28 (2.5 mL, 250 μmol) and (1H-benzimidazol-2-ylmethyl)amine hydrochloride to provide 45 as a white solid (65 mg, 164 μmol, 66%). 1H NMR (400 MHz, DMSO-d6) δ 11.66 (s, 1H), 9.49 (t, J = 5.2 Hz, 1H), 8.33 (d, J = 2.0 Hz, 1H), 8.29 (s, 1H), 7.93 (dd, J = 7.6, 1.6 Hz, 1H), 7.81 (d, J = 2.0 Hz, 1H), 7.68–7.60 (m, 3H), 7.36 (dd, J = 6.0, 3.2 Hz, 2H), 4.88 (d, J = 5.6 Hz, 2H), 3.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 166.76, 154.09, 152.37, 143.41, 138.30, 138.15, 134.16, 129.81, 129.16, 128.76, 126.54, 125.54, 125.48, 123.66, 114.35, 112.11, 36.98, 26.45; MS (ESI) 399 m/z [M+H]+; Purity (88%, tr = 5.35 min).
3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)-N-(3-phenylpropyl)benzamide, 46
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 200 μmol) and 3-phenylpropylamine to provide 46 as a white solid (43 mg, 111 μmol, 56%). 1H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1H), 8.60 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.14 (t, J = 1.6 Hz, 1H), 7.86–7.82 (m, 2H), 7.80 (d, J = 2.0 Hz, 1H), 7.56 (t, J = 8.0 Hz, 1H), 7.30–7.15 (m, 5H), 3.37–3.29 (m, 5H), 2.65 (t, J = 7.6 Hz, 2H), 1.86 (p, J = 7.6 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.04, 154.10, 143.32, 141.75, 138.32, 138.07, 135.42, 129.21, 128.99, 128.97, 128.31, 128.28, 126.20, 125.73, 125.45, 125.21, 112.24, 38.96, 32.67, 30.89, 26.46; MS (ESI) 387.3 m/z [M+H]+; HRMS (ESI) calcd for C23H23N4O2 [M+H]+ 387.1821, found 387.1818; Purity (≥98%, tr = 7.58 min).
N-(3-(4-Fluorophenyl)propyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 47
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 200 μmol) and 3-(4-fluorophenyl)propan-1-amine to provide 47 as a white solid (56 mg, 137 μmol, 69%). 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 8.58 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.13 (t, J = 1.6 Hz, 1H), 7.86–7.80 (m, 2H), 7.80 (d, J = 2.0 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.30–7.25 (m, 2H), 7.13–7.07 (m, 2H), 3.37 (s, 3H), 3.31 (q, J = 7.2 Hz, 2H), 2.64 (t, J = 7.6 Hz, 2H), 1.84 (p, J = 7.6, 7.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.04, 154.09, 143.32, 138.31, 138.07, 137.85, 137.82, 135.40, 130.06, 129.98, 129.20, 128.97, 128.96, 126.18, 125.44, 125.20, 114.99, 114.79, 112.22, 38.81, 31.74, 30.93, 26.45; MS (ESI) 405.2 m/z [M+H]+; HRMS (ESI) calcd for C23H22FN4O2 [M+H]+ 405.1727, found 405.1727; Purity (≥98%, tr = 7.66 min).
N-(3-(1H-Indol-3-yl)propyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 48
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 200 μmol) and 3-(1H-indol-3-yl)propan-1-amine hydrochloride to provide 48 as a white solid (51 mg, 119 μmol, 59%). 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 10.76 (s, 1H), 8.61 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.15 (t, J = 1.6 Hz, 1H), 7.86–7.82 (m, 2H), 7.80 (d, J = 2.0 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.33 (d, J = 8.0 Hz, 1H), 7.17 (d, J = 2.0 Hz, 1H), 7.06 (td, J = 7.2, 1.2 Hz, 1H), 6.96 (td, J = 8.0, 0.8 Hz, 1H), 3.40–3.33 (m, 5H), 2.76 (t, J = 7.2 Hz, 2H), 1.94 (p, J = 7.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.02, 154.09, 143.31, 138.31, 138.07, 136.32, 135.49, 129.17, 128.97, 127.13, 126.19, 125.44, 125.20, 122.27, 120.81, 118.26, 118.07, 114.00, 112.23, 111.31, 29.83, 26.45, 22.23; MS (ESI) 426.2 m/z [M+H]+; HRMS (ESI) calcd for C25H24N5O2 [M+H]+ 426.1930, found 426.1928; Purity (≥98%, tr = 7.38 min).
3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)-N-(3-(pyridin-3-yl)propyl)benzamide, 49
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 200 μmol) and 3-(pyridin-3-yl)propan-1-amine hydrochloride to provide 49 as a white solid (55 mg, 141 μmol, 71%). 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.67–8.63 (m, 2H), 8.54 (d, J = 4.4 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.14 (s, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.86–7.82 (m, 2H), 7.80 (d, J = 2.0 Hz, 1H), 7.58–7.54 (m, 2H), 3.37–3.31 (m, 5H), 2.75 (t, J = 7.2 Hz, 2H), 1.90 (p, J = 7.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.12, 154.10, 146.76, 144.51, 143.33, 139.35, 138.70, 138.31, 138.08, 135.34, 129.24, 128.98, 128.95, 126.20, 125.45, 125.22, 124.58, 112.23, 38.61, 30.26, 29.48, 26.45; MS (ESI) 388.2 m/z [M+H]+; Purity (≥98%, tr = 5.12 min).
N-(3-(1H-Imidazol-4-yl)propyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 50
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 200 μmol) and 3-(1H-imidazol-4-yl)propan-1-amine hydrochloride to provide 50 as a white solid (43 mg, 113 μmol, 57%). 1H NMR (400 MHz, DMSO-d6) δ 14.24 (br, 1H), 11.65 (s, 1H), 8.96 (d, J = 1.2 Hz, 1H), 8.70 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.15 (s, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 7.6 Hz, 1H), 7.81 (d, J = 1.6 Hz, 1H), 7.57 (t, J = 7.6 Hz, 1H), 7.47 (s, 1H), 3.37–3.32 (m, 5H), 2.72 (t, J = 7.2 Hz, 2H), 1.90 (p, J = 7.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.19, 154.10, 143.35, 138.29, 138.11, 135.22, 133.71, 133.06, 129.32, 129.03, 128.91, 126.20, 125.46, 125.18, 115.58, 112.21, 38.37, 28.03, 26.46, 21.55; MS (ESI) 377.3 m/z [M+H]+; Purity (96%, tr = 5.02 min).
N-(3-Cyclopentylpropyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide, 51
The general procedure B was followed using a 0.1 M solution of 28 (2.0 mL, 200 μmol) and 3-cyclopentylpropan-1-amine to provide 51 as a white solid (32 mg, 84 mmol, 42%). 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 8.55 (d, J = 5.6 Hz, 1H), 8.29 (d, J = 2.0 Hz, 1H), 8.13 (d, J = 1.6 Hz, 1H), 7.86–7.81 (m, 2H), 7.80 (d, J = 2.0 Hz, 1H), 7.55 (t, J = 7.6 Hz, 1H), 3.37 (s, 3H), 3.28 (q, J = 6.8 Hz, 2H), 1.81–1.69 (m, 3H), 1.60–1.44 (m, 6H), 1.36–1.30 (m, 2H), 1.10–1.04 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 165.86, 154.09, 143.32, 138.31, 138.06, 135.44, 129.16, 128.97, 126.17, 125.45, 125.17, 112.23, 39.37, 33.02, 32.22, 28.41, 26.45, 24.70; MS (ESI) 379.4 m/z [M+H]+; Purity (≥98%, tr = 8.39 min).
2-(3-(1-Nethyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamido)-4-phenylbutanoic acid, 52
The general procedure was followed using a 0.1 M solution of 28 (3 mL, 300 μmol) and benzyl 2-amino-4-phenylbutanoate followed by debenzylation using Pd/C, H2, MeOH (10 mL) for 12 h at room temperature to provide 52 as a white solid (47 mg, 110 μmol, 37% over 3 steps). 1H NMR (400 MHz, DMSO-d6) δ 12.66 (s, 1H), 11.64 (s, 1H), 8.81 (d, J = 7.6 Hz, 1H), 8.33 (d, J = 2.0 Hz, 1H), 8.21 (s, 1H), 7.90 (dt, J = 7.6, 2.0 Hz, 2H), 7.82 (d, J = 2.0 Hz, 1H), 7.60 (t, J = 7.6 Hz, 1H), 7.31–7.17 (m, 5H), 4.42–4.37 (m, 1H), 3.38 (s, 3H), 2.80–2.65 (m, 2H), 2.16–2.08 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 173.70, 166.46, 154.10, 143.36, 141.06, 138.36, 138.14, 134.71, 129.61, 129.02, 128.92, 128.40, 128.33, 126.51, 125.91, 125.52, 125.47, 112.25, 52.14, 32.43, 31.84, 26.46; MS (ESI) 429.4 m/z [M-H]−; Purity (53%/47%, tr = 7.00 and 7.71 min).
2-Benzyl-3-(3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamido)propanoic acid, 53
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 200 μmol) and methyl 3-amino-2-benzylpropanoate hydrochloride followed by hydrolysis using 10% KOH (aq) (10 mL) and MeOH (5 mL) at 60 °C for 12 h to provide 53 as a white solid (69 mg, 160 μmol, 80%). 1H NMR (400 MHz, DMSO-d6) δ 12.30 (s, 1H), 11.65 (s, 1H), 8.73 (t, J = 5.6 Hz, 1H), 8.31 (d, J = 2.0 Hz, 1H), 8.14 (s, 1H), 7.87 (d, J = 7.6 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 1.6 Hz, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.31–7.18 (m, 5H), 3.55–3.42 (m, 2H), 3.38 (s, 3H), 3.00 (p, J = 7.6 Hz, 1H), 2.91–2.81 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ 174.87, 166.28, 154.10, 143.35, 139.17, 138.32, 138.07, 135.14, 129.34, 129.01, 128.91, 128.77, 128.25, 126.24, 126.17, 125.46, 125.26, 112.21, 46.74, 41.26, 35.52, 26.46; MS (ESI) 429.4 m/z [M-H] −; HRMS (ESI) calcd for C24H23N4O4 [M+H]+ 431.1719, found 431.1724; Purity (60%/24%, tr = 6.66 and 7.43 min).
General Procedure C for Synthesis of the Substituted Benzamide Analogues (57 – 63) in Scheme 4
3-(1-methyl-2-oxo-3-trityl-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)-N-(prop-2-yn-1-yl)benzamide
To a solution of 28 (0.1 M CH2Cl2, 5 mL, 5.0 mmol) were added mono-propagylamine hydrochloride (79 mg) and diisopropylethylamine (1.0 mL) at room temperature. The reaction mixture was stirred at 50 °C for 1 h. The mixture was cooled to ambient, then solvent was removed by evaporation. The crude mixture was diluted with ethyl acetate (40 mL) and was washed with saturated aq. NaHCO3 (10 mL) and brine (10 mL). The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. Purification of the crude product by flash chromatography eluting with a linear gradient ranging from 12% to 100% EtOAc-hexane provided 0.27 g (98%) of 54 as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 8.99 (t, J = 5.6 Hz, 1H), 8.17 (d, J = 2.0 Hz, 1H), 8.14 (t, J = 1.2 Hz, 1H), 7.87–7.82 (m, 3H), 7.56–7.50 (m, 7H), 7.26–7.22 (m, 6H), 7.17–7.13 (m, 3H), 4.09 (dd, J = 5.6, 2.4, 2H), 3.33 (s, 3H), 3.15 (t, J = 2.4, 1H); 13C NMR (100 MHz, DMSO-d6) δ 165.64, 153.31, 142.95, 142.92, 137.54, 137.02, 134.41, 129.46, 129.12, 128.85, 128.53, 127.26, 126.55, 126.23, 125.36, 125.01, 111.81, 81.17, 73.78, 72.99, 28.51, 26.84; MS (ESI) 243 m/z [decomposition of trityl group, M+H]+;HRMS (ESI) calcd for C36H29N4O2 [M+H]+ 549.2291, found 549.2297.
3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)-N-(3-(4-(trifluoromethyl)phenyl)propyl)benzamide, 57
A mixture of 54 (0.17 g, 0.310 mmol), 1-bromo-4-(trifluoromethyl) benzene (0.110 g, 0.431 mmol), Pd(PPh3)4 (40 mg, 10 mol%), NEt3 (0.5 mL), and CuI (5 mg) in DMF (5 mL) was stirred under heating (80 °C) by microwave for 1 h. The product mixture was diluted with ethyl acetate (30 mL) and washed with water (10 mL) and brine (10 mL). The combined organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. The resulting product was purified by flash chromatography eluting with a linear gradient ranging from 25 to 100% EtOAc-hexane to afford 55a as a pale yellow solid (0.15 g, 0.215 mmol), which was directly used for the next step (MS (ESI) 450 m/z [M+H]+, trityl decomposition). To a solution of 55a in MeOH (10 mL) was added 10% Pd/C (ca. 30 mg) at room temperature. After removing air from the flask using a vacuum pump, hydrogen gas was introduced using a balloon. The reaction mixture was stirred for 1 h, the flask was evacuated under vacuum and refilled with hydrogen gas. This procedure was repeated three times and the reaction mixture was stirred overnight at ambient temperature. Palladium on carbon was removed by filtration through a Celite pad. The filtrate was collected and evaporated to give the crude product 56a, from which the trityl group was removed upon treatment with TFA (1 mL) in CH2Cl2 (10 mL) for 1 h. The TFA and solvent were then removed under reduced pressure. The resulting product was purified by flash chromatography eluting with a linear gradient ranging from 0 to 20% MeOH/EtOAc to afford 57 (67 mg, 0.148 mmol, 34% over 3 steps) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.61 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.13 (s, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.80 (d, J = 1.6 Hz, 1H), 7.65 (d, J = 8.0 Hz, 2H), 7.57 (d, J = 7.6 Hz, 1H), 7.49 (d, J = 8.0 Hz, 2H), 3.38–3.31 (m, 5H), 2.76 (t, J = 7.6 Hz, 2H), 1.90 (p, J = 7.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.07, 154.08, 146.80, 143.32, 138.30, 138.07, 135.36, 129.21, 129.15, 129.15, 128.96, 128.94, 126.17, 125.44, 125.19, 125.10, 125.06, 112.21, 32.39, 30.46, 26.44; MS (ESI) 455 m/z [M+H]+; HRMS (ESI) calcd for C24H22F3N4O2 [M+H]+ 455.1695, found 455.1694; Purity (≥98%, tr = 8.32 min).
Methyl 2-(3-(3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamido)propyl)benzoate, 58
The general procedure B was followed using a 0.1 M solution of 28 (2 mL, 0.2 mmol) and methyl 2-(3-aminopropyl)benzoate hydrochloride to provide 58 as a white solid (47 mg, 0.105 mmol, 52%). 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 8.65 (t, J = 5.6 Hz, 1H), 8.29 (d, J = 2.0 Hz, 1H), 8.15 (t, J = 2.0 Hz, 1H), 7.86–7.83 (m, 2H), 7.79 (d, J = 2.0 Hz, 1H), 7.65 (dd, J = 7.6, 1.6 Hz, 1H), 7.58–7.50 (m, 2H), 7.16 (d, J = 7.6 Hz, 1H), 7.01 (td, J = 7.6, 0.8 Hz, 1H), 4.13 (t, J = 6.0 Hz, 2H), 3.79 (s, 3H), 3.51 (q, J = 6.8 Hz, 2H), 3.36 (s, 3H), 2.02 (p, J = 6.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.25, 166.20, 157.66, 154.09, 143.31, 138.30, 138.06, 135.37, 133.51, 130.69, 129.22, 128.96, 126.22, 125.44, 125.26, 120.16, 120.09, 113.58, 112.22, 66.24, 51.75, 36.47, 28.88, 26.44; MS (ESI) 444.3 m/z [M+H]+; Purity (≥98%, tr = 7.23 min).
N-(3-(4-(Aminomethyl)phenyl)propyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide hydrochloride, 59
The general procedure C was followed using 54 (0.19 g, 0.349 mmol), tert-butyl 4-bromobenzyl carbamate (0.27, 0.94 mmol), Pd(PPh3)4 (40 mg, 10 mol%), NEt3 (0.5 mL), and CuI (5 mg) in DMF (5 mL) to provide 55b (0.17 g, 0.254 mmol, 73%) which was hydrogenated to give 56b (0.12 g, 0.159 mmol, 63%). To a solution of 56b (0.12 g, 0.159 mmol) in CH2Cl2 (5 mL) was added trifluoroacetic acid (ca. 1 mL). The reaction mixture was stirred for 1 h at room temperature. After evaporation of the solvent and trifluoroacetic acid, 1N aq. HCl was added to the product mixture. The resulting product was purified by reverse phase flash chromatography eluting with a linear gradient ranging from 5% to 100% acetonitrile-water to provide 59 (33 mg, 0.074 mmol, 46%) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.68 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.28 (br, 3H), 8.17 (t, J = 1.6 Hz, 1H), 7.87–7.83 (m, 3H), 7.56 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 8.0 Hz, 2H), 3.98 (s, 2H), 3.37 (s, 3H), 3.3–3.29 (m, 2H), 2.67 (t, J = 7.6 Hz, 2H), 1.86 (p, J = 7.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.04, 154.10, 143.32, 142.19, 138.29, 138.06, 135.37, 131.42, 129.20, 128.98, 128.94, 128.92, 128.57, 126.23, 125.45, 125.22, 112.28, 41.98, 32.32, 30.82, 26.48; MS (ESI) 416 m/z [M+H]+; Purity (≥98%, tr = 5.57 min).
N-(3-(3-(Aminomethyl)phenyl)propyl)-3-(1-methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamide hydrochloride, 60
The procedure for the synthesis of 59 was followed using 54 (144 mg, 262 μmol) and tert-butyl 3-bromobenzyl carbamate to provide 60 as a white solid (35 mg, 78 μmol, 30% over 4 steps). 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 8.76 (t, J = 5.6 Hz, 1H), 8.35 (br, 2H), 8.31 (d, J = 2.0 Hz, 1H), 8.20 (s, 1H), 7.87–7.84 (m, 3H), 7.56 (t, J = 7.6 Hz, 1H), 7.38 (s, 1H), 7.36–7.25 (m, 3H), 3.99 (d, J = 5.2 Hz, 2H), 3.38–3.31 (m, 5H), 2.67 (t, J = 7.6 Hz, 2H), 1.89 (p, J = 7.6, 7.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.03, 154.10, 143.31, 142.15, 138.28, 138.05, 135.34, 133.97, 129.17, 128.99, 128.92, 128.88, 128.56, 128.43, 126.28, 126.24, 125.46, 125.21, 112.32, 42.20, 32.55, 30.72, 26.50; MS (ESI) 416 m/z [M+H]+; Purity (≥98%, tr = 5.64 min).
3-(3-(3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamido)propyl)benzoic acid, 61
The general procedure C were followed using 54 (153 mg, 278 μmol) and methyl 3-bromobenzoate to provide 56d. A solution of 56d in MeOH (5 mL) and 10% KOH (aq) (10 mL) was stirred at 60 °C for 24 h. The reaction mixture was cooled to ambient, then 1 N HCl was added to acidify the product mixture, and a white solid precipitated. The mixture was diluted with ethyl acetate (30 mL) and washed with water (10 mL) and brine (10 mL). The organic layer was dried over magnesium sulfate, filtered, and concentrated in vacuo. Purification of the crude product by flash chromatography eluting with a linear gradient ranging from 0 to 30% MeOH/EtOAc provided 61 (26 mg, 61 μmol, 22%, 4 steps). 1H NMR (400 MHz, DMSO-d6) δ 12.90 (br, 1H), 11.63 (s, 1H), 8.60 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 1.6 Hz, 1H), 8.14 (s, 1H), 7.86–7.82 (m, 3H), 7.80 (d, J = 2.0 Hz, 1H), 7.77 (d, J = 7.6 Hz, 1H), 7.56 (t, J = 7.6 Hz, 1H), 7.48 (d, J = 7.6 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 3.37–3.30 (m, 5H), 2.72 (t, J = 7.6 Hz, 2H), 1.88 (p, J = 7.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 166.08, 154.09, 143.31, 142.07, 138.31, 138.07, 135.41, 129.19, 129.09, 128.96, 128.40, 128.37, 126.81, 125.44, 125.22, 112.23, 32.40, 30.77, 26.45; MS (ESI) 429 m/z [M-H]−; Purity (95%, tr = 6.58 min).
4-(3-(3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamido)propyl)benzoic acid, 62
The procedure for the synthesis of 61 was followed using 54 (0.16 g, 0.293 mmol) and methyl 4-bromobenzoate to provide 62 as a white solid (26 mg, 60.8 μmol, 21% over 4 steps); 1H NMR (400 MHz, DMSO-d6) δ 12.77 (br, 1H), 11.63 (s, 1H), 8.62 (t, J = 5.6 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.14 (s, 1H), 7.88–7.80 (m, 5H), 7.56 (t, J = 7.6 Hz, 1H), 7.41–7.36 (m, 2H), 3.37–3.30 (m, 5H), 2.73 (t, J = 7.6 Hz, 2H), 1.89 (p, J = 7.6 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 167.24, 166.05, 154.08, 147.17, 143.31, 138.30, 138.06, 135.37, 129.37, 129.19, 128.94, 128.51, 128.38, 126.18, 125.43, 125.20, 112.23, 32.60, 30.47, 26.44; MS (ESI) 429 m/z [M-H] −; Purity (≥98%, tr = 6.44 min).
2-(3-(3-(1-Methyl-2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-6-yl)benzamido)propyl)benzoic acid, 63
The procedure for the synthesis of 61 was followed using 58 (0.16 g, 0.360 mmol) to provide 63 as a white solid (0.12 g, 0.279 mmol, 78%). 1H NMR (400 MHz, DMSO-d6) δ 12.55 (br, 1H), 11.63 (s, 1H), 8.66 (t, J = 5.6 Hz, 1H), 8.29 (d, J = 2.0 Hz, 1H), 8.14 (s, 1H), 7.86–7.79 (m, 2H), 7.79 (d, J = 2.0 Hz, 1H), 7.64 (dd, J = 7.6, 2.0 Hz, 1H), 7.55 (d, J = 7.6 Hz, 1H), 7.48 (td, J = 8.4, 2.0 Hz, 1H), 7.13 (d, J = 8.4 Hz, 1H), 6.99 (td, J = 7.6, 0.8 Hz, 1H), 4.13 (d, J = 6.4 Hz, 2H), 3.50 (d, J = 6.4 Hz, 2H), 3.36 (s, 3H), 2.03 (p, J = 6.4 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 167.38, 166.22, 157.41, 154.09, 143.32, 138.31, 138.07, 135.33, 132.94, 130.63, 129.23, 128.96, 126.22, 125.44, 125.25, 121.63, 120.08, 113.53, 112.23, 66.20, 36.43, 30.66, 28.87, 26.44; MS (ESI) 429 m/z [M-H] −; Purity (≥98%, tr = 6.50 min).
Supplementary Material
Acknowledgments
Support by the Pfizer-Scripps Florida postdoctoral research fellow program to J.Y.C. is gratefully acknowledged. Additional financial support for J.Y.C. and W.R.R. at Scripps Florida was provided by NIH grant U54MH084512, Hugh Rosen, Principal Investigator. The authors also thank Lisa Mullins and Joe Penzien of Pfizer for performing preliminary anti-bacterial assays.
Abbreviations used
- TP5A
P1-(5′-adenosyl)-P5-(5′-thymidyl)pentaphosphate
- dFTM
deoxy-fluorothymidine
- dFTM
5′-fluorodeoxythymidine
- AZTMP
3′-azidodeoxythymidine monophosphate
- acyTM
acyclic thymidine
- thiourea-α-TM
5′-deoxy-5′-N-arylthiourea α-thymidine derivatives
- DTBN
5, 5′-dithio-bis (2-nitrobenzoic acid)
- MIC
minimum inhibitory concentration
- IC50
half-maximal inhibitory concentration
Footnotes
Accession Codes: Coordinates and structure factors of PaTMK complexed with 1, 17, and dFTM are available from the Protein Data Bank with accession codes, 3UWK, 3UWO, and 3UXM, respectively.
Supporting Information Available: Data collection and refinement statistics for the X-ray co-crystallization of PaTMK with 1, 17, and dFTM. Illustration of the four reactions which comprise the assay system. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov. 2007;6(1):29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 2.Obritsch MD, Fish DN, MacLaren R, Jung R. Nosocomial infections due to multidrug-resistant Pseudomonas aeruginosa: epidemiology and treatment options. Pharmacotherapy. 2005;25(10):1353–1364. doi: 10.1592/phco.2005.25.10.1353. [DOI] [PubMed] [Google Scholar]
- 3.Rice LB. The clinical consequences of antimicrobial resistance. Curr Opin Microbiol. 2009;12(5):476–481. doi: 10.1016/j.mib.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Rice LB. Emerging issues in the management of infections caused by multidrug-resistant gram-negative bacteria. Cleve Clin J Med. 2007;74(Suppl 4):S12–S20. doi: 10.3949/ccjm.74.suppl_4.s12. [DOI] [PubMed] [Google Scholar]
- 5.Kandeel M, Kato A, Kitamura Y, Kitade Y. Thymidylate kinase: the lost chemotherapeutic target. Nucleic Acids Symp Ser (Oxf) 2009;(53):283–284. doi: 10.1093/nass/nrp142. [DOI] [PubMed] [Google Scholar]
- 6.Anderson E. In the enzymes. 3. Vol. 8. Academic Press; New York: 1973. Nucleoside and nucleotide kinases; pp. 49–96. [Google Scholar]
- 7.Van Calenbergh S. Structure-aided design of inhibitors of Mycobacterium tuberculosis thymidylate kinase. Verh K Acad Geneeskd Belg. 2006;68(4):223–248. [PubMed] [Google Scholar]
- 8.Fioravanti E, Adam V, Munier-Lehmann H, Bourgeois D. The crystal structure of Mycobacterium tuberculosis thymidylate kinase in complex with 3′-azidodeoxythymidine monophosphate suggests a mechanism for competitive inhibition. Biochemistry. 2005;44(1):130–137. doi: 10.1021/bi0484163. [DOI] [PubMed] [Google Scholar]
- 9.Gasse C, Douguet D, Huteau V, Marchal G, Munier-Lehmann H, Pochet S. Substituted benzyl-pyrimidines targeting thymidine monophosphate kinase of Mycobacterium tuberculosis: synthesis and in vitro anti-mycobacterial activity. Bioorg Med Chem. 2008;16(11):6075–6085. doi: 10.1016/j.bmc.2008.04.045. [DOI] [PubMed] [Google Scholar]
- 10.Gasse C, Huteau V, Douguet D, Munier-Lehmann H, Pochet S. A new family of inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase. Nucleosides Nucleotides Nucleic Acids. 2007;26(8–9):1057–1061. doi: 10.1080/15257770701513349. [DOI] [PubMed] [Google Scholar]
- 11.Vanheusden V, Munier-Lehmann H, Froeyen M, Busson R, Rozenski J, Herdewijn P, Van Calenbergh S. Discovery of bicyclic thymidine analogues as selective and high-affinity inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase. J Med Chem. 2004;47(25):6187–6194. doi: 10.1021/jm040847w. [DOI] [PubMed] [Google Scholar]
- 12.Vanheusden V, Van Rompaey P, Munier-Lehmann H, Pochet S, Herdewijn P, Van Calenbergh S. Thymidine and thymidine-5′-O-monophosphate analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. Bioorg Med Chem Lett. 2003;13(18):3045–3048. doi: 10.1016/s0960-894x(03)00643-7. [DOI] [PubMed] [Google Scholar]
- 13.Kotaka M, Dhaliwal B, Ren J, Nichols CE, Angell R, Lockyer M, Hawkins AR, Stammers DK. Structures of S. aureus thymidylate kinase reveal an atypical active site configuration and an intermediate conformational state upon substrate binding. Protein Sci. 2006;15(4):774–784. doi: 10.1110/ps.052002406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byun Y, Vogel SR, Phipps AJ, Carnrot C, Eriksson S, Tiwari R, Tjarks W. Synthesis and biological evaluation of inhibitors of thymidine monophosphate kinase from Bacillus anthracis. Nucleosides Nucleotides Nucleic Acids. 2008;27(3):244–260. doi: 10.1080/15257770701845238. [DOI] [PubMed] [Google Scholar]
- 15.Andrade CH, Pasqualoto KF, Ferreira EI, Hopfinger AJ. 3D-Pharmacophore mapping of thymidine-based inhibitors of TMPK as potential antituberculosis agents. J Comput Aided Mol Des. 2010;24(2):157–172. doi: 10.1007/s10822-010-9323-y. [DOI] [PubMed] [Google Scholar]
- 16.Cui H, Ruiz-Perez LM, Gonzalez-Pacanowska D, Gilbert IH. Potential application of thymidylate kinase in nucleoside analogue activation in Plasmodium falciparum. Bioorg Med Chem. 2010;18(20):7302–7309. doi: 10.1016/j.bmc.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Familiar O, Munier-Lehmann H, Ainsa JA, Camarasa MJ, Perez-Perez MJ. Design, synthesis and inhibitory activity against Mycobacterium tuberculosis thymidine monophosphate kinase of acyclic nucleoside analogues with a distal imidazoquinolinone. Eur J Med Chem. 2010;45(12):5910–5918. doi: 10.1016/j.ejmech.2010.09.056. [DOI] [PubMed] [Google Scholar]
- 18.Frecer V, Seneci P, Miertus S. Computer-assisted combinatorial design of bicyclic thymidine analogs as inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase. J Comput Aided Mol Des. 2011;25(1):31–49. doi: 10.1007/s10822-010-9399-4. [DOI] [PubMed] [Google Scholar]
- 19.Andrade CH, Pasqualoto KF, Ferreira EI, Hopfinger AJ. Rational design and 3D-pharmacophore mapping of 5′-thiourea-substituted alpha-thymidine analogues as mycobacterial TMPK inhibitors. J Chem Inf Model. 2009;49(4):1070–1078. doi: 10.1021/ci8004622. [DOI] [PubMed] [Google Scholar]
- 20.Douguet D, Munier-Lehmann H, Labesse G, Pochet S. LEA3D: a computer-aided ligand design for structure-based drug design. J Med Chem. 2005;48(7):2457–2468. doi: 10.1021/jm0492296. [DOI] [PubMed] [Google Scholar]
- 21.Van Daele I, Munier-Lehmann H, Froeyen M, Balzarini J, Van Calenbergh S. Rational design of 5′-thiourea-substituted alpha-thymidine analogues as thymidine monophosphate kinase inhibitors capable of inhibiting mycobacterial growth. J Med Chem. 2007;50(22):5281–5292. doi: 10.1021/jm0706158. [DOI] [PubMed] [Google Scholar]
- 22.Lavie A, Ostermann N, Brundiers R, Goody RS, Reinstein J, Konrad M, Schlichting I. Structural basis for efficient phosphorylation of 3′-azidothymidine monophosphate by Escherichia coli thymidylate kinase. Proc Natl Acad Sci U S A. 1998;95(24):14045–14050. doi: 10.1073/pnas.95.24.14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MOE. Chemical Computing Group Inc; Montreal: 2009. p. 10. [Google Scholar]
- 24.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53(14):5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma JC, Dougherty DA. The Cation-pi Interaction. Chem Rev. 1997;97(5):1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 26.Mecozzi S, West AP, Jr, Dougherty DA. Cation-pi interactions in aromatics of biological and medicinal interest: electrostatic potential surfaces as a useful qualitative guide. Proc Natl Acad Sci U S A. 1996;93(20):10566–10571. doi: 10.1073/pnas.93.20.10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dougherty DA. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271(5246):163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 28.Michel J, Tirado-Rives J, Jorgensen WL. Energetics of displacing water molecules from protein binding sites: consequences for ligand optimization. J Am Chem Soc. 2009;131(42):15403–15411. doi: 10.1021/ja906058w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grivas S, Lindstrom S. Palladium(0)-catalyzed phenylation of imidazo[4, 5-b]pyridines. J Heterocyclic Chem. 1995;32:467–471. [Google Scholar]
- 30.Savelli F, Boido A, Vazzana I, Sparatore F. Tetrahydrocyclopenta[e]pyrido[3, 2-b][1, 4]diazepin and -cyclopenta[e]pyrido[2, 3-b][1, 4]diazepine derivatives. J Heterocyclic Chem. 1987;24:1709–1716. [Google Scholar]
- 31.Kudo N, Perseghini M, Fu GC. A versatile method for Suzuki cross-coupling reactions of nitrogen heterocycles. Angew Chem Int Ed Engl. 2006;45(8):1282–1284. doi: 10.1002/anie.200503479. [DOI] [PubMed] [Google Scholar]
- 32.Barder TE, Walker SD, Martinelli JR, Buchwald SL. Catalysts for Suzuki-Miyaura coupling processes: scope and studies of the effect of ligand structure. J Am Chem Soc. 2005;127(13):4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 33.Arvela RK, Leadbeater NE. Suzuki coupling of aryl chlorides with phenylboronic acid in water, using microwave heating with simultaneous cooling. Org Lett. 2005;7(11):2101–2104. doi: 10.1021/ol0503384. [DOI] [PubMed] [Google Scholar]
- 34.Leadbeater NE. Fast, easy, clean chemistry by using water as a solvent and microwave heating: the Suzuki coupling as an illustration. Chem Commun (Camb) 2005;(23):2881–2902. doi: 10.1039/b500952a. [DOI] [PubMed] [Google Scholar]
- 35.Glide 55. Schrodinger Inc; New York: [Google Scholar]
- 36.Maestro90. Schrodinger Inc; New York: [Google Scholar]
- 37.Lavie A, Konrad M, Brundiers R, Goody RS, Schlichting I, Reinstein J. Crystal structure of yeast thymidylate kinase complexed with the bisubstrate inhibitor P1-(5′-adenosyl) P5-(5′-thymidyl) pentaphosphate (TP5A) at 2.0 A resolution: implications for catalysis and AZT activation. Biochemistry. 1998;37(11):3677–3686. doi: 10.1021/bi9720787. [DOI] [PubMed] [Google Scholar]
- 38.Cabeen MT, Jacobs-Wagner C. Bacterial cell shape. Nat Rev Microbiol. 2005;3(8):601–610. doi: 10.1038/nrmicro1205. [DOI] [PubMed] [Google Scholar]
- 39.Ostermann N, Schlichting I, Brundiers R, Konrad M, Reinstein J, Veit T, Goody RS, Lavie A. Insights into the phosphoryltransfer mechanism of human thymidylate kinase gained from crystal structures of enzyme complexes along the reaction coordinate. Structure. 2000;8(6):629–642. doi: 10.1016/s0969-2126(00)00149-0. [DOI] [PubMed] [Google Scholar]
- 40.Biswas S, Mohammad MM, Patel DR, Movileanu L, van den Berg B. Structural insight into OprD substrate specificity. Nat Struct Mol Biol. 2007;14(11):1108–1109. doi: 10.1038/nsmb1304. [DOI] [PubMed] [Google Scholar]
- 41.Mach T, Neves P, Spiga E, Weingart H, Winterhalter M, Ruggerone P, Ceccarelli M, Gameiro P. Facilitated permeation of antibiotics across membrane channels--interaction of the quinolone moxifloxacin with the OmpF channel. J Am Chem Soc. 2008;130(40):13301–13309. doi: 10.1021/ja803188c. [DOI] [PubMed] [Google Scholar]
- 42.Nikaido H, Thanassi DG. Penetration of lipophilic agents with multiple protonation sites into bacterial cells: tetracyclines and fluoroquinolones as examples. Antimicrob Agents Chemother. 1993;37(7):1393–1399. doi: 10.1128/aac.37.7.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carter JCW, Sweet RM. Macromolecular Crystallography, part A. Vol. 2. Academic Press; New York: 1997. pp. 307–326. [Google Scholar]
- 44.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 45.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 46.Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355(6359):472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 47.Read RJ. Improved fourier coefficients for maps using phases from partial structures with errors. Acta Crystallographica Section A. 1986;42:140–149. [Google Scholar]
- 48.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Read RJ. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt 10):1373–1382. doi: 10.1107/s0907444901012471. [DOI] [PubMed] [Google Scholar]
- 50.Bricogne G, Blanc E, Brandl ME, Flensburg C, Keller P, Paciorek W, Roversi P, Smart O, Vonrhein C, Womack T. BUSTER, version 280. Cambridge: United Kingdom:Global Phasing Ltd; 2009. [Google Scholar]
- 51.Petit CM, Koretke KK. Characterization of Streptococcus pneumoniae thymidylate kinase: steady-state kinetics of the forward reaction and isothermal titration calorimetry. Biochem J. 2002;363(Pt 3):825–s831. doi: 10.1042/0264-6021:3630825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.