

Abstract
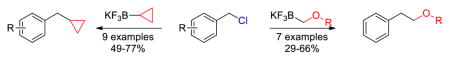
Efficient Csp3-Csp3 Suzuki couplings have been developed with both potassium cyclopropyl- and alkoxymethyltrifluoroborates. Moderate to good yields have been achieved in the cross-coupling of potassium cyclopropyltrifluoroborate with benzyl chlorides possessing electron-donating or electron-withdrawing substituents. Benzyl chloride was also successfully cross-coupled to potassium alkoxymethyltrifluoroborates derived from primary, secondary, and tertiary alcohols.
Cyclopropyl groups are of great interest because of their occurrence in many natural products and synthesized drug molecules.1,2 Phenylethoxy moieties are also important in bioactive compounds because they can enhance molecular properties, in particular the solubility, of drug molecules.3,4,5 Both cyclopropyl and phenylethoxy subunits also present the advantage of preventing the metabolic breakdown of active pharmaceutical ingredients.
Cross-coupling approaches to introduce these subunits into aliphatic core structures can present difficulties because alkyl-alkyl Suzuki-Miyaura coupling reactions remain a challenge.6,7 Indeed, alkyl halides are less reactive toward oxidative addition than their unsaturated analogues.8 In the past fifteen years, diverse boronic acids and alkylboronates have been tested in their reactions toward numerous alkyl electrophiles.9,10 Most of these studies were carried out on alkyl bromides,11,12,13 but Csp3-Csp3 bond formations have been also reported with iodoalkanes14 and alkyl tosylates.15 Fu and co-workers developed a method to couple a range of alkyl chlorides with B-alkyl-9-BBN reagents, utilizing Pd2(dba)3 in conjunction with tricyclohexylphosphine as a ligand.16
Benzyl chlorides are activated alkyls and therefore have been described as good electrophiles in Suzuki-Miyaura cross-couplings with aryl and (hetero)arylboronic acids,17 but they have rarely been used with alkylboron species. Pertinent to the current work, Deng and co-workers have reported the coupling of benzyl bromides with substituted cyclopropylboronic acids, in which the use of expensive Ag2O as a base was required to enhance the rate of the reaction.18 Only a few additional literature accounts describe the use of cyclopropylboron species in Suzuki coupling reactions with sp3-hybridized electrophiles,19,20 and even cyclopropyl Kumada21 and Negishi22 couplings with benzylic halides are exceedingly rare.
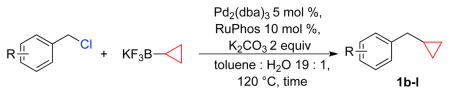
Similarly, although Suzuki-Miyaura couplings between potassium alkoxymethyltrifluoroborates and aryl chlorides have been reported,23 their coupling to sp3-hybridized halides remains unexplored. Having recently demonstrated that good yields are obtained for the Suzuki-Miyaura cross-coupling between substituted benzyl halides with potassium aryltrifluoroborates,24 we sought to extend this success to two select alkyltrifluoroborate systems. Herein, we describe alkyl-alkyl Suzuki-Miyaura cross-coupling reactions of benzyl chlorides with potassium cyclopropyltrifluoroborate and alkoxymethyltrifluoroborates, neither class of which has ever been cross-coupled to sp3-hybrized electrophiles.
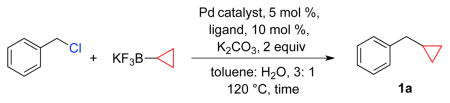
During initial optimization studies of the cyclopropyl system, we investigated several palladium catalysts in combination with a variety of ligands.25,26 For cyclopropyltrifluoroborate, Pd(OAc)2/RuPhos gave the best conversions when compared to the other biarylphosphines (e.g., SPhos and XantPhos, Figure 1) and BINAP, resulting in 57% yield of product 1a (Table 1, entry 1). Interestingly, changing the palladium source from Pd(OAc)2 to Pd2(dba)3 27,28 resulted in a higher conversion to alkylated product 1a (entry 5). Other palladium catalysts such as tetrakis(triphenylphosphine)palladium(0) (entry 6) and bis(triphenylphosphine)palladium(II) chloride (entry 7) gave moderate conversions to the desired product.
Figure 1.

Structures of ligands and PEPPSI precatalyst
Table 1.
Optimization
 | |||
|---|---|---|---|
| entry | catalyst | ligand | yield (%) a |
| 1 | Pd(OAc)2 | RuPhos | 57 |
| 2 | Pd(OAc)2 | SPhos | 41 |
| 3 | Pd(OAc)2 | XantPhos | 23 |
| 4 | Pd(OAc)2 | BINAP | 55 |
| 5 | Pd2(dba)3 | RuPhos | 62 |
| 6 | Pd(PPh3)4 | - | 29 |
| 7 | Pd(PPh3)2Cl2 | PPh3 | 44 |
| 8 | PEPPSI | - | 89 |
Benzyl chloride (0.2 mmol), potassium cyclopropyltrifluoroborate (0.35 mmol), Pd catalyst (0.01 mmol), ligand (0.02 mmol), K2CO3 (0.4 mmol), toluene/H2O 3:1 (0.1 M), 120 °C.
GC/MS yield determined using dodecane as the internal standard.
N-Heterocyclic carbene ligands (NHC), discovered by Öfele,29 represent a second class of ligands that are commonly used in C-C bond couplings. These compounds are neutral, electron rich, excellent σ-donors, and have a poor capacity to accept π back donation from the metal center.30 Organ and co-workers recently employed an NHC precatalyst in the formation of Csp3-Csp3 bonds with alkylboronates.31 Further to this, they reported Csp2-Csp3 Suzuki couplings between potassium organotrifluoroborates and alkyl halides using PEPPSI (Pyridine Enhanced Precatalyst Preparation Stabilization and Initiation) as a catalyst.32 In the present system, the PEPPSI precatalyst also appeared to be efficient, resulting in a conversion of 89% of 1a (entry 8).
Optimization also involved screening various conventional inorganic bases; in this regard potassium carbonate gave the best conversion. Additionally, it was important to limit the concentration of the reaction to 0.1 M to suppress dimer formation. Incorporating these parameters, we obtained the highest conversions using two different catalytic systems; Pd2(dba)3/RuPhos and the PEPPSI precatalyst (entries 5 and 8). Subsequently, we also noticed that the mixture of toluene : water in a ratio of 19:1 was important to avoid the formation of benzylic alcohols derived from hydrolysis of solvolytically reactive benzyl chlorides.
Utilizing this reaction protocol, the reactivity of various benzyl chlorides toward potassium cyclopropyltrifluoroborate was evaluated. We initially studied the PEPPSI precatalyst, but its practical application was mostly limited to electron deficient benzyl chlorides (Table 2). The scope of the reaction proved to be much broader using Pd2(dba)3 and RuPhos as the catalytic system, as both electron rich and electron poor substrates were successfully cross-coupled with moderate to good yields.
Table 2.
Optimization on Substituted Benzyl Chlorides.
| entry | benzyl chloride | catalytic system | GC/MS yield (%) a |
|---|---|---|---|
| 1 |
|
Pd2(dba)3 / RuPhos | 93 |
| 2 | PEPPSI | 75 | |
| 3 |
|
Pd2(dba)3 / RuPhos | 92 |
| 4 | PEPPSI | 96 |
Benzyl chloride (0.2 mmol), potassium cyclopropyltrifluoroborate (0.35 mmol), Pd catalyst (0.01 mmol), ligand (0.02 mmol), K2CO3 (0.4 mmol), toluene/H2O 19:1 (0.1 M), 120 °C.
GC/MS yield determined using dodecane as the internal standard.
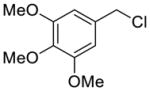
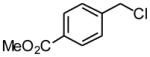
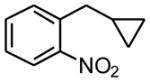
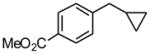
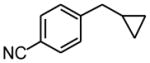
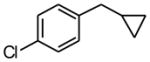
Using the RuPhos system, benzyl chlorides decorated with electron donating groups in the ortho, meta and para positions proved to be suitable substrates for the reactions, resulting in yields of the desired products as high as 80% (Table 3). The coupling between 3,4,5-trimethoxybenzyl chloride and potassium cyclopropyltrifluoroborate could be performed on one gram scale, demonstrating the scalability of these couplings. The desired compound 1f was isolated in a yield of 72% (entry 5). Electron-deficient substituents such as carbonyl-, cyano-, and nitro-groups were tolerated when they were placed para (entries 6, 9, 10) or meta to the chloromethyl group (entry 7). However, no coupling was observed when 2-nitrobenzyl chloride was tested (entry 8). Using optimized conditions, we obtained modest chemoselectivity in the cross-coupling of 4-chlorobenzyl chloride (entry 11), giving the desired Csp3-alkylated compound 1l in 34% yield, with 1-chloro-4-methylbenzene as well as dialkylated materials being observed among the byproducts.
Table 3.
Scope of Substituted Benzyl Chlorides.
 | ||||
|---|---|---|---|---|
| entry | product | time | yield (%) | |
| 1 |
 1b |
5 h | 58 | |
| 2 |
 1c |
3 h | 77 | |
| 3 |
 1d |
3 h | 76 | |
| 4 |
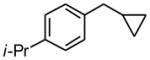 1e |
7 h | 62 | |
| 5 |
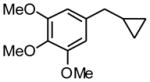 1f |
5 h | 56 (72)a | |
| 6 |
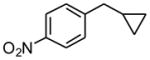 1g |
3 h | 49 | |
| 7 |
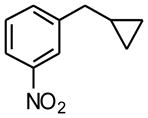 1h |
7 h | 80 | |
| 8 |
 1i |
6 h | 0 | |
| 9 |
 1j |
4 h | 73 | |
| 10 |
 1k |
2.5 h | 59 | |
| 11 |
 1l |
24 h | 34 | |
Benzyl chloride (2.0 mmol), potassium cyclopropyltrifluoroborate (3.5 mmol), Pd2(dba)3 (0.1 mmol), RuPhos (0.2 mmol), K2CO3 (4.0 mmol), toluene/H2O 19:1 (0.1 M), 120 °C.
Reaction performed on 1 g scale.
Our previously optimized conditions using PEPPSI precatalyst proved best for the coupling of potassium alkoxymethyltrifluoroborates with benzyl chloride. The desired coupling products were isolated in yields between 29% and 66% (Table 4). The cross coupling was adversely affected by steric hindrance. Potassium alkoxymethyltrifluoroborates derived from primary alcohols gave moderate to good yields (entries 1–4). Potassium alkoxymethyltrifluoroborates bearing secondary substituents afforded products 2e-f with yields up to 42% yield (entries 5–6). Finally, the cross-coupling of potassium (tert-butoxymethyl)trifluoroborate led to the alkylated compound 2g with only 29% yield (entry 7).
Table 4.
Scope of Potassium Alkoxymethyltrifluoroborates.
 | |||
|---|---|---|---|
| entry | product | time | yield (%) |
| 1 |
 2a
2a
|
24 h | 66 |
| 2 |
|
16 h | 62 |
| 3 |
|
3 h | 45 |
| 4 |
 2d
2d
|
5 d | 45 |
| 5 |
 2e
2e
|
3 d | 40 |
| 6 |
|
4 d | 42 |
| 7 |
 2g
2g
|
20 h | 29 |
Benzyl chloride (2.0 mmol), potassium alkoxymethyltrifluoroborate (3.5 mmol), PEPPSI (0.1 mmol), K2CO3 (4.0 mmol), toluene/H2O 19:1 (0.1 M), 120 °C.
In summary, we have developed Suzuki-Miyaura conditions that allow the formation of Csp3-Csp3 bonds between potassium cyclopropyl- or alkoxymethyltrifluoroborates and benzyl chlorides with yields up to 77%. Electron-rich and electron-poor substituents on the benzyl chlorides are allowed, and potassium alkoxymethyltrifluoroborates derived from primary, secondary, and tertiary alcohol precursors are all suitable reagents for the process. This method broadens the application of potassium organotrifluoroborates in Csp3-Csp3 bond formation.
Experimental Section
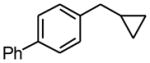
Procedure A: 1-Cyclopropylmethyl-4-phenylbenzene (1b)
A Biotage microwave vial was charged with 4-phenylbenzyl chloride (413.6 mg, 2.0 mmol), potassium cyclopropyltrifluoroborate (443.9 mg, 3.0 mmol), Pd2(dba)3 (91.6 mg, 0.1 mmol), RuPhos (98.2 mg, 0.2 mmol) and K2CO3 (552.8 mg, 4.0 mmol). The tube was sealed and purged with nitrogen. A degassed mixture of toluene : water, 19 mL : 1 mL was added under a nitrogen atmosphere. The reaction was stirred at 120 °C for 7 h. After cooling to room temperature, the reaction mixture was filtered through Celite and MgSO4. The solvent was removed in vacuo, and the residue was purified by preparative plate chromatography (silica gel, heptanes : EtOAc 95 : 5) to obtain 1b as a colorless oil (240.8 mg, 58%). 1H NMR (360 MHz, CDCl3): δ 7.62-7.59 (m, 2H), 7.57-7.53 (m, 2H), 7.47-7.42 (m, 2H), 7.37-7.32 (m, 3H), 2.61 (d, J = 7.0 Hz, 2H), 1.10-0.99 (m, 1H), 0.58-0.58 (m, 2H), 0.26-0.22 (m, 2H); 13C NMR (90 MHz, CDCl3): δ 141.4, 141.3, 138.9, 128.9 (2C), 128.8 (2C), 127.1 (3C), 127.0 (2C), 40.1, 12.0, 4.9 (2C); HRMS (EI) calcd for C16H16 [(M+.)] 208.1252; found: 208.1247; IR (neat) ν= 3001, 2913, 1487, 1016, 825, 697 cm-1.
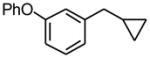
1-Cyclopropylmethyl-3-phenoxybenzene (1c)
Following standard procedure A, the reaction was performed starting from 3-phenoxybenzyl chloride (446.3 mg, 2.0 mmol). After 3 h, the resulting crude was purified by preparative plate chromatography (heptane : EtOAc 98 : 2), 1c was obtained as a colorless oil (343.5 mg, 77%). 1H NMR (360 MHz, CDCl3): δ 7.38 (td, J = 8.1, 1.8 Hz, 2H), 7.30 (td, J = 3.7, 1.8 Hz, 1H), 7.14 (t, J = 7.0 Hz, 1H), 7.08-7.04 (m, 3H), 7.00 (br s, 1H), 6.88 (dd, J = 7.9, 2.4 Hz, 1H), 2.57 (d, J = 7.0 Hz, 2H), 1.07-0.96 (m, 1H), 0.59-0.54 (m, 2H), 0.25-0.21 (m, 2H); 13C NMR (90 MHz, CDCl3): δ 157.5, 157.2, 144.4, 129.8 (2C), 129.5, 123.4, 123.2, 119.1, 118.9 (2C), 116.4, 40.3, 11.8, 4.8 (2C); HRMS (EI) calcd for C16H16O [(M+.)] 224.1201; found: 224.1185; IR (neat) ν= 3075, 3000, 1582, 1485, 1250 cm-1.
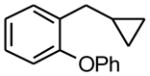
1-Cyclopropylmethyl-2-phenoxybenzene (1d)
Following standard procedure A, the reaction was performed starting from 2-phenoxybenzyl chloride (437.4 mg, 2.0 mmol). After, 3 h, the resulting crude was purified by preparative plate chromatography (heptane : EtOAc 98 : 2), 1d was obtained as a colorless oil (341.9 mg, 76%). 1H NMR (360 MHz, CDCl3): δ 7.40-7.29 (m, 3H), 7.08-7.05 (m, 5H), 6.88 (dd J = 7.9, 2.0 Hz, 1H), 2.57 (d, J = 7.0 Hz, 2H), 1.02-0.91 (m, 1H), 0.58-0.45 (m, 2H), 0.25-0.21 (m, 2H); 13C NMR (90 MHz, CDCl3): δ 157.5, 157.2, 144.3, 129.8 (2C), 129.5, 123.4, 123.1, 116.1, 118.8 (2C), 116.4, 40.2, 11.8, 4.8 (2C); HRMS (EI) calcd for C16H16O [(M+.)] 224.1201; found: 224.1223; IR (neat) ν= 3074, 3001, 1582, 1486, 1250 cm−1.
1-Cyclopropylmethyl-4-(propan-2-yl)benzene (1e)
Following standard procedure A, the reaction was performed starting from 4-iso-propylbenzyl chloride (347.8 mg, 2.0 mmol). After 7 h, the resulting crude was purified by preparative plate chromatography (heptane : EtOAc 85 : 15), 1e was obtained as a colorless oil (217.1 mg, 62%). 1H NMR (360 MHz, CDCl3): δ 7.20 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.1 Hz, 2H), 2.89 (sept, J = 6.9 Hz, 1H), 2.52 (d, J = 7.0 Hz, 2H), 1.25 (d, J = 7.0 Hz, 6H), 1.03-0.94 (m, 1H), 0.54-0.49 (m, 2H), 0.22-0.18 (m, 2H); 13C NMR (90 MHz, CDCl3): δ 146.4, 139.6, 128.4 (2C), 126.4 (2C), 40.1, 33.9, 24.2 (2C), 12.0, 4.8 (2C); HRMS (EI) calcd for C13H18 [(M+.)] 174.1409; found: 174.1424; IR (neat) ν= 2959, 1731, 1514, 1460, 825 cm-1.
5-Cyclopropylmethyl-1,2,3-trimethoxybenzene (1f)
Following standard procedure A, the reaction was performed starting from methyl 3,4,5-trimethoxybenzyl chloride (433.3 mg, 2.0 mmol). After 4 h, the resulting crude was purified by preparative plate chromatography (heptanes : EtOAc 95 : 5), 1f was obtained as a yellow oil (248.4 mg, 56%). 1H NMR (360 MHz, CDCl3): δ 6.50 (s, 2H), 3.87 (s, 6H), 3.84 (s, 3H), 2.51 (d, J = 6.6 Hz, 2H), 1.05-0.94 (m, 1H), 0.58-0.53 (m, 2H), 0.20-0.24 (m, 2H); 13C NMR (90 MHz, CDCl3): δ 153.0 (2C), 137.9, 136.1, 105.2 (2C), 60.7, 56.0 (2C), 40.6, 11.7, 4.6 (2C); HRMS (EI) calcd for C13H18O3 [(M+.)] 222.1256; found: 222.1249; IR (neat) ν= 2997, 2936, 1588, 1237, 1127 cm-1.
1-Cyclopropylmethyl-4-nitrobenzene (1g)
Following standard procedure A, the reaction was performed starting from 4-nitrobenzyl chloride (343.2 mg, 2.0 mmol). After 3 h, the resulting crude was purified by preparative plate chromatography (heptane : EtOAc 85 : 15), 1g was obtained as a colorless oil (175.2 mg, 49%). 1H NMR (360 MHz, CDCl3): δ 8.16 (dt, J = 8.8, 2.2 Hz, 2H), 7.42 (dt, J = 8.1, 2.6 Hz, 2H), 2.65 (d, J = 7.0 Hz, 2H), 1.06-0.95 (m, 1H), 0.62-0.57 (m, 2H), 0.27-0.23 (m, 2H); 13C NMR (90 MHz, CDCl3): δ 150.2, 146.6, 129.2 (2C), 123.7 (2C), 40.3, 11.5, 5.0 (2C); HRMS (EI) calcd for C10H11NO2 [(M+.)] 177.0790; found: 177.0803; IR (neat) ν= 3079, 2933, 1599, 1516, 1344 cm-1.
1-Cyclopropylmethyl-3-nitrobenzene (1h)
Following standard procedure A, the reaction was performed starting from 3-nitrobenzyl chloride (353.8 mg, 2.0 mmol). After 7 h, the resulting crude was purified by preparative plate chromatography (heptane : EtOAc 85 : 15), 1h was obtained as a colorless oil (281.9 mg, 80%). 1H NMR (360 MHz, CDCl3): δ 8.14 (s, 1H), 8.07 (br d, J = 8.4 Hz, 1H), 7.60 (d, J = 7.3 Hz, 1H), 7.46 (t, J = 7.9 Hz, 1H), 2.65 (d, J = 7.0 Hz, 2H), 1.07-0.96 (m, 1H), 0.61-0.58 (m, 2H), 0.27-0.23 (m, 2H); 13C NMR (90 MHz, CDCl3): δ 148.4, 144.3, 134.7, 129.2, 123.2, 121.2, 34.0, 11.5, 4.9 (2C); HRMS (EI) calcd for C10H11NO2 [(M+.)] 177.0790; found: 177.0767; IR (neat) ν= 3078, 3002, 1524, 1349, 806 cm-1.
Methyl 4-Cyclopropylmethylbenzoate (1j)
Following standard procedure A, the reaction was performed starting from methyl 4-(chloromethyl)benzoate (380.7 mg, 2.0 mmol). After 4 h, the resulting crude was purified by preparative plate chromatography (heptane : EtOAc 70 : 30), 1j was obtained as a colorless oil (278.2 mg, 73%).1H NMR (360 MHz, CDCl3): δ 7.97 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H), 3.90 (s, 3H), 2.6 (d, J = 7.0 Hz, 2H), 1.05-0.94 (m, 1H), 0.57-0.52 (m, 2H), 0.24-0.20 (m, 2H); 13C NMR (90 MHz, CDCl3): δ 167.2, 147.7, 129.7 (2C), 128.4 (2C), 127.9, 52.0, 40.3, 11.6, 4.8 (2C); HRMS (EI) calcd for C12H14O2 [(M+.)] 190.0994; found: 190.0981; IR (neat) ν= 3001, 1719, 1434, 1277, 1108 cm-1.
4-Cyclopropylmethyl-benzonitrile (1k)
Following standard procedure A, the reaction was performed starting from 4-chloromethylbenzonitrile (309.4 mg, 2.0 mmol). After 3 h, the resulting crude was purified by preparative plate chromatography (heptane : EtOAc 80 : 20), 1k was obtained as a colorless oil (186.8 mg, 59%). 1H NMR (360 MHz, CDCl3): δ 7.59 (dt, J = 8.1, 1.8 Hz, 2H), 7.37 (d, J = 8.1 Hz, 2H), 2.61 (d, J = 7.0 Hz, 2H), 1.03-0.93 (m, 1H), 0.61-0.56 (m, 2H), 0.24-0.20 (m, 2H); 13C NMR (90 MHz, CDCl3): δ 148.0, 132.2 (2C), 129.2 (2C), 119.3, 109.9, 40.5, 11.4, 4.9 (2C); HRMS (EI) calcd for C11H11N [(M+.)] 157.0897; found: 157.0918; IR (neat) ν= 3079, 3003, 2227, 1608, 1020, 826 cm−1.
1-Chloro-4-cyclopropylmethylbenzene (1l)
Following standard procedure A, the reaction was performed starting from 4-chloro-benzyl chloride (332.1 mg, 2.0 mmol). After 10 h, the resulting crude was purified by preparative plate chromatography (heptanes : EtOAc 95 : 5), 1l was obtained as a colorless oil (112.9 mg, 34%). 1H NMR (360 MHz, CDCl3): δ 7.07 (dt, J = 8.4, 1.8 Hz, 2H), 7.00 (dt, J = 8.1, 1.8 Hz, 2H), 2.51 (d, J = 7.0 Hz, 2H), 1.00-0.89 (m, 1H), 0.55-0.50 (m, 2H), 0.21-0.17 (m, 2H); 13C NMR (90 MHz, CDCl3): δ 140.7, 131.6, 129.8 (2C), 128.4 (2C), 39.8, 11.9, 4.8 (2C); HRMS (EI) calcd for C10H11Cl [(M+.)] 166.0549; found: 166.0524; IR (neat) ν= 3002, 2918, 1491, 1088, 1016 cm−1.
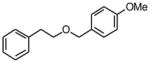
Procedure B: 1-Methoxy-4-phenethyloxymethylbenzene (2a)
A Biotage microwave vial was charged with benzyl chloride (255.7 mg, 2.0 mmol), potassium (4-methoxy)benzyltrifluoroborate (900.5 mg, 3.0 mmol), PEPPSI (69.5 mg, 0.1 mmol), and K2CO3 (552.8 mg, 4.0 mmol). The tube was sealed and purged with nitrogen. A degassed mixture of toluene : H2O 19 mL:1 mL was added under a nitrogen atmosphere. The reaction was stirred at 120 °C for 24 h. After cooling to room temperature, the reaction mixture was filtered through Celite and MgSO4. The solvent was removed in vacuo, and the residue was purified by preparative plate chromatography (silica gel, heptanes : EtOAc 70 : 30) to obtain 2a as a colorless oil (377.1 mg, 66%). The spectral data match those reported in the literature.33 1H NMR (360 MHz, CDCl3): δ 7.31-7.18 (m, 7H), 6.86 (dt, J = 8.4, 2.6 Hz, 2H), 4.46 (s, 2H), 3.80 (s, 3H), 3.66 (t, J = 7.3 Hz, 2H), 2.92 (t, J = 7.1 Hz, 2H); 13C NMR (90 MHz, CDCl3): δ 159.2, 139.1, 130.6, 129.2 (2C), 129.0 (2C), 128.4 (2C), 126.2, 113.8 (2C), 72.6, 71.0, 55.2, 36.4; HRMS (EI) calcd for C16H18O2 [(M+.)] 242.1307; found: 242.1288; IR (neat) ν= 2958, 2906, 1612, 1513, 1247, 1096 cm−1.
{[4-(2-Phenylethoxy)butoxy]methyl}benzene (2b)
Following standard procedure B, the reaction was performed starting from potassium (4-methoxy)benzyloxymethyltrifluoroborate (900.5 mg, 3.0 mmol). After 24 h, the resulting crude was purified by preparative plate chromatography (heptanes : EtOAc 70: 30), 2b was obtained as a colorless oil (302.4 mg, 62%). 1H NMR (360 MHz, CDCl3): δ 7.37-7.18 (m, 10H), 4.49 (s, 2H), 3.62 (t, J = 7.1 Hz, 2H), 3.49-3.44 (m, 4H), 2.88 (t, J = 7.3 Hz, 2H), 1.68-1.65 (m, 4H); 13C NMR (90 MHz, CDCl3): δ 139.1, 138.7, 128.9 (2C), 128.3 (2C), 128.3 (2C), 127.6 (2C), 127.5, 126.1, 72.8, 71.8, 70.7, 70.1, 36.4, 28.5; HRMS (EI) calcd for C19H25O2 [(M+.)] 285.1855; found: 285.1839; IR (neat) ν= 2932, 2858, 1454, 1363, 1102 cm−1.
4-(2-Phenethyloxyethyl)-morpholine (2c)
Following standard procedure B, the reaction was performed starting from potassium [2-(morpholin-4-yl)ethoxy]methyltrifluoroborate (792.9 mg, 3.0 mmol). After 24 h, the resulting crude was purified by preparative plate chromatography [CH2Cl2 : ammonia (7 M in MeOH) : heptanes 60 : 2 : 38], 2c was obtained as a colorless oil (211.5 mg, 45%). 1H NMR (360 MHz, CDCl3): δ 7.28 (t, J = 7.3 Hz, 2H), 7.22-7.18 (m, 3H), 3.70-3.64 (m, 6H), 3.59 (t, J = 5.7 Hz, 2H), 2.88 (t, J = 7.1 Hz, 2H), 2.57 (t, J = 5.5 Hz, 2H), 2.46-2.45 (m, 4H); 13C NMR (90 MHz, CDCl3): δ 139.0, 129.0 (2C), 128.4 (2C), 126.3, 72.2, 68.5, 66.9 (2C), 58.3, 54.1 (2C), 36.3; HRMS (EI) calcd for C14H21NO2 [(M+.)] 235.1572; found: 235.1577; IR (neat) ν= 2854, 1453, 1115, 699 cm−1.
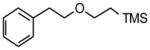
Trimethyl-(2-phenethyloxyethyl) silane (2d)
Following standard procedure B, the reaction was performed starting from (2-trimethylsilyl)-ethoxymethyl trifluoroborate (752.1 mg, 3.0 mmol). After 5 days, the resulting crude was purified by preparative plate chromatography (heptanes : EtOAc 70 : 30), 2d was obtained as a yellow oil (198.6 mg, 45%). 1H NMR (360 MHz, CDCl3): δ 7.32-7.19 (m, 5H), 3.64 (t, J = 7.3 Hz, 2H), 3.55-3.51 (m, 2H), 2.94-2.88 (m, 2H), 0.98-0.93 (m, 2H), 0.01 (s, 9H); 13C NMR (90 MHz, CDCl3): δ 139.3, 129.0 (2C), 128.5 (2C), 126.3, 71.4, 68.2, 36.6, 18.3, −1.2 (3C); HRMS (CI) calcd for C13H26NOSi [(MNH4+)] 240.1784; found: 240.1765; IR (neat) ν= 2951, 2855, 1248, 1102, 835 cm−1.
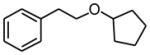
(2-Cyclopentyloxyethyl)benzene (2e)
Following standard procedure B, the reaction was performed starting from potassium cyclopentoxymethyltrifluoroborate (650.7 mg, 3.0 mmol). After 24 h, the resulting crude was purified by preparative plate chromatography (heptanes : EtOAc 70 : 30), 2e was obtained as a colorless oil (153.0 mg, 40%). 1H NMR (360 MHz, CDCl3): δ 7.31-7.18 (m, 5H), 3.93-3.88 (m, 1H), 3.58 (t, J = 7.3 Hz, 2H), 2.87 (t, J = 7.5 Hz, 2H), 1.73-1.48 (m, 8H); 13C NMR (90 MHz, CDCl3): δ 139.4, 129.1 (2C), 128.4 (2C), 126.2, 81.6, 70.0, 36.9, 32.4 (2C), 23.6 (2C); HRMS (EI) calcd for C13H16 [(M+.−H2O)] 172.1252; found:172.1274; IR (neat) ν= 2954, 2777, 1736, 1349, 1093 cm−1.
tert-Butyl 4-(2-Phenylethoxy)piperidine-1-carboxylate (2f)
Following standard procedure B, the reaction was performed starting from potassium (1-Boc-4-piperidinylmethoxyoxy)methyltrifluoroborate (1.01 g, 3.0 mmol). After 24 h, the resulting crude was purified by preparative plate chromatography (CH2Cl2 : heptane : ammonia 60 : 37 : 3), 2f was obtained as a yellow oil (253.9 mg, 42%). 1H NMR (360 MHz, CDCl3): δ 7.31-7.19 (m, 5H), 3.71-3.64 (m, 4H), 3.10 (sept, J = 3.7 Hz, 1H), 3.07 (ddd, J = 13.1, 9.2, 3.3 Hz, 2H), 2.88 (t, J = 7.3 Hz, 2H), 1.81-1.53 (br m, 2H), 1.54-1.46 (m, 2H), 1.45 (s, 9H); 13C NMR (90 MHz, CDCl3): δ 155.0, 139.2, 129.1 (2C), 128.4 (2C), 126.3, 79.5, 77.4, 74.7, 69.2, 41.2, 36.9, 31.0, 28.6 (3C); HRMS (EI) calcd for C14H18NO3 [(M+.–C4H9)] 248.1287; found: 248.1290; IR (neat) ν= 2929, 2861, 1690, 1420, 1169, 1028 cm−1.
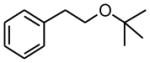
(2-tert-Butoxy-ethyl)-benzene (2g)
Following standard procedure B, the reaction was performed starting from potassium tert-butoxymethyltrifluoroborate (612.8 mg, 3.0 mmol). After 24 h, the resulting crude was purified by preparative plate chromatography (heptanes : EtOAc 95 : 5). 2g was obtained as a colorless oil (104.0 mg, 29%). 1H NMR (360 MHz, CDCl3): δ 7.31-7.19 (m, 5H), 3.54 (t, J = 7.7 Hz, 2H), 2.83 (t, J = 7.7 Hz, 2H), 1.18 (s, 9H); 13C NMR (90 MHz, CDCl3): δ 139.5, 129.1 (2C), 128.4 (2C), 126.2, 73.0, 63.2, 37.6, 27.7 (3C); HRMS (EI) calcd for C12H18O [(M+.)] 178.1358; found: 178.1361; IR (neat) ν= 2973, 1735, 1362, 1197, 1080 cm−1.
Supplementary Material
Acknowledgments
We acknowledge the National Institutes of Health (R01 GM035249) and the Neuroscience Medicinal Chemistry Department of Janssen Pharmaceutica for their generous support of this work. Additionally, we thank Dr. Andrés Trabanco (Janssen Pharmaceutica) and Dr. Marc Presset (University of Pennsylvania) for their helpful advice and suggestions.
Footnotes
Supporting Information. 1H and 13C NMR spectra of compounds 1b-1l and 2a–2g are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wessjohann LA, Brandt W. Chem Rev. 2003;103:1625–1647. doi: 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]
- 2.de Meijere A, Kozhushkov SI. Mendeleev Commun. 2010;20:301–311. [Google Scholar]
- 3.Nudelman A, Elisheva G, Katz Y, Azulai R, Cohen-Ohana M, Zhuk R, Sampson SR, Langzam L, Fibach E, Prus E, Pugach V, Raphaeli A. Eur J Med Chem. 2001;36:63–74. doi: 10.1016/s0223-5234(00)01199-5. [DOI] [PubMed] [Google Scholar]
- 4.Shirasaki Y, Miyashita H, Yamagushi M, Inoue J, Nakamura M. Bioorg Med Chem. 2005;13:4473–4484. doi: 10.1016/j.bmc.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 5.Hartz RA, Ahuja VT, Mattson RJ, Denhart DJ, Deskus JA, Vrudhula VM, Pan S, Ditta JL, Shu Y-Z, Grace JE, Lentz KA, Lelas S, Li Y-W, Molski TF, Krishnananthan S, Wong H, Qian-Cutrone J, Schartman R, Denton R, Lodge NJ, Zaczek R, Macor JE, Bronson JJ. J Med Chem. 2009;52:7653–7668. doi: 10.1021/jm900716v. [DOI] [PubMed] [Google Scholar]
- 6.Hills ID, Netherton MR, Fu GC. Angew Chem Int Ed. 2003;42:5749–5752. doi: 10.1002/anie.200352858. [DOI] [PubMed] [Google Scholar]
- 7.Cardenas DJ. Angew Chem Int Ed. 1999;38:3018–3020. doi: 10.1002/(sici)1521-3773(19991018)38:20<3018::aid-anie3018>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 8.Cardenas DJ. Angew Chem Int Ed. 2003;42:384–387. doi: 10.1002/anie.200390123. [DOI] [PubMed] [Google Scholar]
- 9.Kirchhoff JH, Netherton MR, Hills ID, Fu GC. J Am Chem Soc. 2002;124:13662–13663. doi: 10.1021/ja0283899. [DOI] [PubMed] [Google Scholar]
- 10.Brenstrum T, Gerristma DA, Adjabeng GM, Frampton CS, Britten J, Robertson AJ, McNulty J, Capretta A. J Org Chem. 2004;69:7635–7639. doi: 10.1021/jo048875+. [DOI] [PubMed] [Google Scholar]
- 11.Lou S, Fu GC. Org Syn. 2010;87:299–309. doi: 10.15227/orgsyn.087.0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Netherton MR, Dai C, Neuschütz K, Fu GC. J Am Chem Soc. 2001;123:10099–10100. doi: 10.1021/ja011306o. [DOI] [PubMed] [Google Scholar]
- 13.Peh G-R, Kantchev EAB, Er J-C, Ying JY. Chem Eur J. 2010;16:4010–4017. doi: 10.1002/chem.200902842. [DOI] [PubMed] [Google Scholar]
- 14.Ishiyama T, Abe S, Miyaura N, Suzuki A. Chem Lett. 1992:691–694. [Google Scholar]
- 15.Netherton MR, Fu GC. Angew Chem Int Ed. 2002;41:3910–3912. doi: 10.1002/1521-3773(20021018)41:20<3910::AID-ANIE3910>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 16.Kirchhoff JH, Dai C, Fu GC. Angew Chem Int Ed. 2002;41:1945–1947. [PubMed] [Google Scholar]
- 17.Kambe N, Iwasaki T, Terao J. Chem Soc Rev. 2011;40:4937–4947. doi: 10.1039/c1cs15129k. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Deng M-Z. J Chem Soc, Perkin Trans. 2000;1:1609–1613. [Google Scholar]
- 19.Charette AB, De Freitas-Gill RP. Tetrahedron Lett. 1997;38:2809–2812. [Google Scholar]
- 20.Chen H, Deng M-Z. J Org Chem. 2000;65:4444–4446. doi: 10.1021/jo0000933. [DOI] [PubMed] [Google Scholar]
- 21.Moriconi A, Cesta MC, Cervellera MN, Aramini A, Coniglio S, Colagioia S, Beccari AR, Bizzarri C, Cavicchia MR, Locati M, Galliera E, Benedetto PD, Vigilante P, Bertini R, Allegretti M. J Med Chem. 2007;50:3984–4002. doi: 10.1021/jm061469t. [DOI] [PubMed] [Google Scholar]
- 22.De Lang R–J, Brandsma L. Synth Commun. 1998;28:225–232. [Google Scholar]
- 23.Molander GA, Canturk B. Org Lett. 2008;7:2135–3138. doi: 10.1021/ol800532p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molander GA, Elia MD. J Org Chem. 2006;71:9198–9202. doi: 10.1021/jo061699f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miura M. Angew Chem Int Ed. 2004;43:2201–2203. doi: 10.1002/anie.200301753. [DOI] [PubMed] [Google Scholar]
- 26.Martin R, Buchwald S. Acc Chem Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amatore C, Jutand A. Coord Chem Rev. 1998;178–180:511–528. [Google Scholar]
- 28.Macé Y, Kapdi AR, Fairlamb IJS, Jutand A. Organometallics. 2006;25:1795–1800. [Google Scholar]
- 29.Öfele K. J Organomet Chem. 1968;12:42–43. [Google Scholar]
- 30.Herrmann W. Angew Chem Int Ed. 2002;41:1290–1309. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 31.Valente C, Baglione S, Candito D, O’Brien CJ, Organ MG. Chem Commun. 2008:735–737. doi: 10.1039/b715081d. [DOI] [PubMed] [Google Scholar]
- 32.O’Brien CJ, Kantchev EAB, Valente C, Hadei N, Chass GA, Lough A, Hopkinson AC, Organ MG. Chem Eur J. 2006;12:4743–4748. doi: 10.1002/chem.200600251. [DOI] [PubMed] [Google Scholar]
- 33.Shintou T, Mukaiyama T. J Am Chem Soc. 2004;126:7359–7367. doi: 10.1021/ja0487877. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.