

Abstract
The ribonuclease H (RNase H) domain of retroviral reverse transcriptase (RT) plays a critical role in the life cycle by degrading the RNA strands of DNA/RNA hybrids. In addition, RNaseH activity is required to precisely remove the RNA primers from nascent (−) and (+) strand DNA. We report here three crystal structures of the RNase H domain of xenotropic murine leukemia virus-related virus (XMRV) RT, namely (i), the previously identified construct from which helix C was deleted, (ii), the intact domain, and (iii), the intact domain complexed with an active site α-hydroxytropolone inhibitor. Enzymatic assays showed that the intact RNase H domain retained catalytic activity, whereas the variant lacking helix C was only marginally active, corroborating the importance of this helix for enzymatic activity. Modeling of the enzyme-substrate complex elucidated the essential role of helix C in binding a DNA/RNA hybrid and its likely mode of recognition. The crystal structure of the RNase H domain complexed with β-thujaplicinol clearly showed that coordination by two divalent cations mediates recognition of the inhibitor.
Keywords: XMRV, retrovirus, RNase H, crystal structure, enzyme inhibition
1. Introduction
In contrast to a proposed role as an etiological agent in prostate cancer (Schlaberg et al., 2009) and chronic fatigue syndrome (Lombardi et al., 2009; Mikovits et al., 2010), a recent study has indicated that xenotropic murine leukemia virus-related virus (XMRV) arose through recombination following passaging human tumors in mice (Paprotka et al., 2011). Nevertheless, XMRV, which is very closely related to Moloney murine leukemia virus (Mo-MLV), provides a legitimate example of a replication-competent gammaretrovirus capable of infecting human cells. Despite a wealth of information derived from in vitro site-directed mutagenesis studies, the single-chain gammaretroviral reverse transcriptase (RT) has been less studied in structural terms than the counterpart p66/p51 heterodimeric enzyme from HIV-1 and -2, and thus is deserving of more attention.
Almost all XMRV-encoded proteins have been cloned and expressed (Gillette et al., 2010) and structures of its protease (Li et al., 2011) and RNase H polypeptide have been determined. The latter is located at the C-terminus of RT, and the equivalent domain of the Mo-MLV enzyme retains activity when expressed in isolation (Schultz and Champoux, 1996). The coordinates for a variant of XMRV RNase H have been deposited in the Protein Data Bank (PDB ID: 3P1G), but a corresponding publication became available on-line (Kirby et al., 2012) only after this manuscript was submitted. Previously, the structure of the very closely related Mo-MLV RNase H has been published (Lim et al., 2006). The constructs used in both investigations were modified by excision of helix C and the loop that follows, designated the “basic protrusion” (residues 595–605, numbered according to the sequence of XMRV RT). Although the structure of full-length Mo-MLV RT has been previously published, its RNase H domain was reported to be largely disordered and not contributing independent information about its conformation (Das and Georgiadis, 2004). In addition, amino acid differences between the Mo-MLV and XMRV RT accumulate at their C-terminal region, leading to possible structural differences in their RNase H domains. We have thus initiated structural studies of full-length XMRV RNase H and of its complex with an inhibitor, the results of which are described here.
2. Materials and methods
2.1. Cloning, expression, and purification
The clone of wild type XMRV RNase H domain was constructed by PCR using primers (P1 : 5'-GGA TCC GAG ATC TTG GCT GAG ACG CAC GGA ACC-3'; p2 : 5'-GAA TTC TCA GAG GAG TGT AGA GGT TTC TAG AAC TGC-3') containing BamH1 and EcoR1 restriction sites respectively. Amplified DNA was digested with BamH1 and EcoR1, gel purified, and ligated into BamH1/EcoRI digested pRSET-A expression vector. After transformation of E. coli strain DH5a, six colonies were selected for DNA analysis, and insert evaluation was by restriction analysis and DNA sequencing.
Gene constructs encoding two variants of XMRV RNase H were prepared starting from the clone encoding the intact enzyme. QuickChange Site-Directed Mutagenesis Kit (Agilent Technologies) was used to introduce in parallel variants in which helix C was deleted (RNase H ΔC1 with residues 593–603 deleted; and RNase H ΔC2, in which residues 595–605 were deleted). One of these variants was identical and the other similar to the construct used for crystallization of the variant already deposited in the PDB (ID: 3P1G). The starting constructs contained two different affinity tags: His-RNase H (pRSET A) and GST-RNase H (pGEX 3X). All mutations were confirmed by sequencing.
Recombinant plasmids were transformed into E. coli strain BL21 (DE3) codon plus ril for expression tests. Three constructs, His-RNase H, GST-RNase H ΔC1, and GST-RNase H ΔC2 expressed at high levels. Large-scale expression of these constructs followed a similar protocol, utilizing LB media incubated at 37 °C and shaken at 225 rev min−1. When the OD600 reached 0.4–0.6, the culture was induced with isopropyl-β-D-thiogalactopyranoside to a final concentration of 0.5 mM and incubated for another 12 h at 20 °C. Cells were harvested by centrifugation and resuspended in lysis buffer containing 50 mM Tris pH 8.0, 200 mM NaCl, 5% glycerol, and 6 mM β-mercaptoethanol, followed by lysis with a Microfluidizer. The supernatant was clarified by centrifugation, loaded onto a Histrap™ FF column (GE Healthcare) pre-equilibrated with binding buffer (identical to the lysis buffer), and eluted with a linear gradient of 0 – 0.75 M imidazole in binding buffer. Dilution buffer (50 mM Tris pH 8.0, 5% glycerol, and 6 mM β-mercaptoethanol) was added to fractions containing RNase H to bring the final salt concentration to 100 mM. Each sample was loaded onto a Hitrap™ Heparin HP column (GE Healthcare) and eluted with a buffer of 50 mM Tris pH 8.0, 1.2 M NaCl, 5% glycerol, and 6 mM β-mercaptoethanol. Fractions containing RNase H were collected and digested overnight at 20 °C with enterokinase (enteropeptidase) to remove the His-tag. Cleavage efficiency was assessed with SDS-PAGE and the His-tag and uncleaved protein were removed using the Histrap™ FF column. The flow-through was collected and dialyzed into storage buffer (20 mM Tris pH 8.0, 200 mM NaCl, 5% glycerol, and 1mM TCEP). Samples were concentrated to 15 mg/ml and stored for future use at −80 °C.
Purification of the RNase H variants ΔC1 and ΔC2 was performed using a slight variation of this protocol. Cells were harvested by centrifugation and resuspended in lysis buffer (20 mM Tris pH 8.0, 140 mM NaCl), followed by lysis with a Microfluidizer. The supernatant was clarified by centrifugation, loaded onto the GSTPrep™ FF 16/10 column (GE Healthcare) and eluted with a buffer of 50 mM Tris pH 8.0, 10 mM reduced glutathione. RNase H-containing fractions were combined and Factor Xa was added to cleave the GST tag. Proteolysis was performed at 20 °C for 24 h and cleavage efficiency assessed by SDS-PAGE. RNase H was further purified using Hitrap Q FF and Hiload 16/60 Superdex 75 (GE Healthcare) columns. Purified proteins were dialyzed into storage buffer and concentrated to 25 mg/ml and frozen at −80 °C for future use.
2.2. RNase H activity assay
RNase H activity was determined as previously reported (Budihas et al., 2005), using an 18-nt 3'-fluorescein-labeled RNA annealed to a complementary 18-nt 5'-dabsyl-labeled DNA. The reaction was initiated by adding 10 μL of RNA/DNA hybrid (2.5 μM) to a 96-well plate containing HIV-1 or XMRV RT variants in reaction buffer. Final assay conditions were 50 mM Tris·HCl, pH 8.0, 60 mM KCl, 1.0 mM dithiothreitol, 1.0 mM MnCl2, 250 nM substrate, 1% (v/v) DMSO, and increasing concentrations of inhibitor. Plates were incubated at 37 °C in a Spectramax Gemini EM fluorescence spectrometer for 10 minutes, and the fluorescence (λex = 475 nm; λem = 520 nm) was measured at 1-minute intervals such that linear initial rates could be measured.
2.3. Protein crystallization
Crystallization conditions reported for the previously published structure of Mo-MLV RNase H (Lim et al., 2006) and of a variant form of XMRV RNase H (PDB ID: 3P1G) were used in initial experiments involving the two deletion variants. However, despite extensive optimization efforts, crystals of RNase H ΔC1 were of uniformly poor quality and unsuitable for data collection. For the variant ΔC2 long, needle-shaped crystals were obtained from 40% PEG1500, 100 mM Na citrate pH 4.7, and 5 mM MgCl2. Crystallization was carried out by hanging-drop vapor-diffusion method at 20 °C by mixing 1 μl protein solution with 1 μl of mother liquor.
Unlike the variant forms of RNase H, full-length enzyme containing helix C has not been successfully crystallized in the past. An extensive search utilizing a variety of crystallization screens resulted in a hit from Precipitant Synergy 64 (Emerald BioSystems) condition 39 (40% PEG400, 10% PEG1000, and 0.15 M potassium phosphate dibasic/sodium phosphate monobasic pH 6.5). The initially obtained crystals were very small and irreproducible, but provided microseeds for further experiments. Diffraction quality crystals were obtained after seeding from 35% PEG3000, 15% MPD, 2% isopropanol, 0.2 M lithium sulfate, and 0.1 M imidazole pH 6.5. The microseeds, mother liquor, and protein solution were mixed at 1:2:3 volume ratio and incubated at 20 °C. Crystals grew to a full size in 2–3 weeks. To prepare an inhibitor complex, crystals were transferred into reservoir solution with added 20 mM MnCl2 and 2 mM β-thujaplicinol. An inhibitor soak was performed at room temperature for 2–4 hours.
2.4. Data collection, processing, and structure refinement
Diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22-ID at the Advanced Photon Source, Argonne National Laboratory. Data obtained from a single crystal of RNase H ΔC2 were subsequently indexed, integrated, and scaled to 2.0 Å resolution with the program XDS (Kabsch, 1993). The crystal belongs to the space group P41 with one molecule in the asymmetric unit. The estimated Matthews coefficient (Matthews, 1968) is 1.87 Å3Da−1, corresponding to 34.5% solvent content. The structure was solved by molecular replacement with the program Phaser (Adams et al., 2002) using the XMRV RNase H variant structure (PDB ID: 3P1G) as a starting model. Further refinement was performed with REFMAC5 (Murshudov et al., 1997) and PHENIX (Adams et al., 2002), using all data between 20 and 2.0 Å, after setting aside 8% of randomly selected reflections (747 total) for calculation of Rfree (Brünger, 1992). Isotropic individual temperature factors were refined, with the TLS parameters added in the final stages of refinement. After several further rounds of automated refinement and manual correction using COOT (Emsley and Cowtan, 2004), the structural model was finally refined to an R-factor of 16.9% and Rfree of 19.8%.
Although the crystal of the wild-type RNase H used for structure determination was not single but consisted of several crystallites, it was possible to index the predominant lattice with HKL2000 (Otwinowski and Minor, 1997). Diffraction data were indexed, integrated, and scaled to 1.9 Å resolution. The space group was C2221 with one molecule in the asymmetric unit. The estimated Matthews coefficient was 1.95 Å3Da−1, corresponding to 37% solvent content. The structure was solved by molecular replacement with the program Phaser using our coordinates of RNase H ΔC2 as a search model. Further refinement was performed with REFMAC5 and PHENIX, using all data between 30 and 1.9 Å, after setting aside 7% of randomly selected reflections (815 total) for calculation of Rfree. Isotropic individual temperature factors were refined, with the TLS parameters added in the final stages of refinement. After several further rounds of automated refinement and manual correction using COOT, the structural model was finally refined to an R-factor of 17.3% and Rfree of 21.7%.
Diffraction data for the enzyme-inhibitor complex were collected and processed using a protocol similar to the one reported for the uninhibited enzyme. The final model was refined to 2.8 Å resolution, resulting in an R-factor of 19.5% and Rfree of 27.2%. Data processing and refinement statistics for all three structures of RNase H are shown in Table 1.
Table 1.
Data collection and structure refinement
| Data collection | |||
| wt RNase H | RNase H ΔC2 | wt RNase H/inhibitor | |
| Space group | C2221 | P41 | C2221 |
| Molecules/a.u. | 1 | 1 | 1 |
| Unit cell a, b, c (Å); | 53.3, 84.2, 70.5 | 37.5, 37.5, 99.9 | 53.7, 84.75, 70.8 |
| α=β=γ (°) | 90 | 90 | 90 |
| Resolution (Å)* | 50.0–1.90 (1.97–1.90) | 20.0–2.0 (2.3–2.0) | 30.0–2.80 (2.90–2.80) |
| Rmerge† (%) | 7.9 (34.9) | 14.6 (124.0) | 13.4 (35.4) |
| No. of reflections (measured/unique) | 49,240/11,792 | 70,320/9,361 | 15,027/4,042 |
| <I / σI> | 27.0 (3.5) | 9.73 (1.7) | 12.9 (2.7) |
| Completeness (%) | 89.7 (57.7) | 99.7 (99.7) | 96.2 (79.4) |
| Redundancy | 4.2 (2.1) | 7.5 (7.5) | 3.7 (3.1) |
| Refinement | |||
| Resolution (Å) | 27.7–1.90 | 18.4–2.0 | 27.2–2.80 |
| No. of reflections (refinement/ Rfree) | 11,760/815 | 9,334/747 | 4,020/496 |
| R / Rfree‡ | 0.173/0.217 | 0.169/0.198 | 0.195/0.277 |
| No. atoms | |||
| Protein | 1278 | 1207 | 1278 |
| Ligands | 34 | 0 | 43 |
| Water | 71 | 80 | 12 |
| R.m.s. deviations from ideal | |||
| Bond lengths (Å) | 0.010 | 0.015 | 0.013 |
| Bond angles (°) | 1.265 | 1.561 | 0.748 |
| PDB code | 3V1O | 3V1Q | 3V1R |
The highest resolution shell is shown in parentheses.
Rmerge = ΣhΣi|Ii−〈I〉|/ΣhΣiIi, where Ii is the observed intensity of the i-th measurement of reflection h, and 〈I〉 is the average intensity of that reflection obtained from multiple observations.
R = Σ||Fo|−|Fc||/Σ|Fo, where Fo and Fc are the observed and calculated structure factors, respectively, calculated for all data. Rfree was defined in (Brünger, 1992).
3. Results and discussion
Several constructs of the XMRV RNase H domain were prepared for this study. These included either a cleavable (His)6 or GST tag fused to full-length protein and two of its variants. All three forms of the protein were stably overexpressed in E. coli and highly soluble. The full-length RNase H exhibited full RNase H activity, whereas the ΔC1 and ΔC2 variants were virtually inactive. Earlier reports indicated that it would be difficult to crystallize the intact RNase H domain including helix C, which plays an essential role in DNA/RNA substrate binding (Lim et al., 2006). The earlier studies also suggested that helix C may play a specific role in vivo other than simple nonspecific substrate binding (Boyer et al., 2001; Telesnitsky et al., 1992). Thus, although we obtained crystals of the two deletion variants identical or similar to those described previously, we continued our efforts to crystallize an intact RNase H domain. Such efforts were ultimately successful, resulting in diffraction-quality crystals that allowed data collection and processing to 1.9 Å resolution, although the crystal used to obtain such data was not single. The refined model consists of residues 503–667 (XMRV RT numbering). The structure of the variant ΔC2, comprising residues 500–594 and 606–667, was refined to 2.0 Å. Both models were validated with the program PROCHECK (Laskowski et al., 1993) which showed more than 95% residues falling within the most favored regions and no residues in the disallowed regions, indicating acceptable quality. In addition, the structure of a complex of the full-length RNase H domain with β-thujaplicinol was refined at a lower resolution of 2.8 Å.
3.1. The structures of XMRV RNase H
The structures of XMRV RNase H with (Fig. 1a) or without (Fig. 1b) helix C are very similar, with the RMSD of 0.292 Å between the coordinates of the two proteins, calculated for 114 pairs of superimposed Cα atoms. Significant differences are limited to the region at the N terminus and, not surprisingly, the area adjacent to the deleted α-helix. Unless otherwise specified, the structure of the full-length enzyme will be discussed in more detail below.
Figure 1.
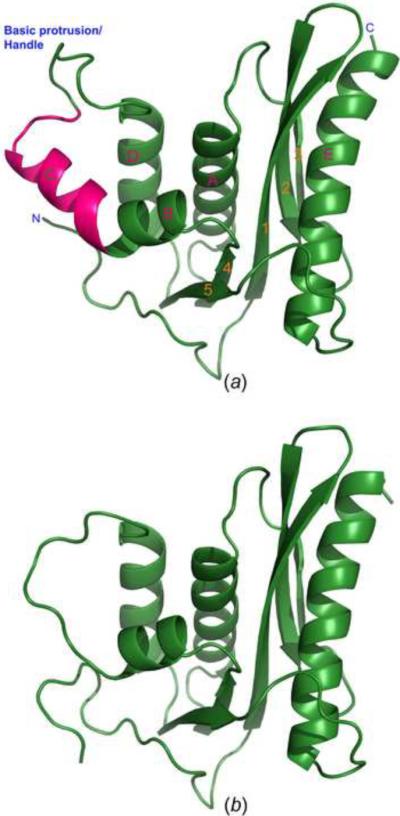
Crystal structures of the full-length XMRV RNase H and its deletion variant ΔC2. (A) Ribbon representation of the overall three-dimensional structure of full-length RNase H. Helix C is highlighted in hot pink, the other parts of the structure in green. Identification of the secondary structure elements (used throughout the manuscript) follows the precedent from related structures. (B) Overall structure of the deletion variant ΔC2 of XMRV RNase H in which helix C was removed. The model is oriented in the same way as in panel A.
A molecule of XMRV RNase H contains a central β-sheet of four parallel strands (1, 3, 4, and 5) and one antiparallel strand (2), with five α-helices arranged on both sides (Fig. 1a). Helices A, B, C, and D are located on one side of the sheet, whereas the long helix E is found on the other side. The overall structure of XMRV RNase H closely resembles its Mo-MLV (Lim et al., 2006), E. coli (Katayanagi et al., 1990), human (Nowotny et al., 2007), B. halodurans (Nowotny et al., 2005), and HIV-1 (Yang et al., 1990) orthologs (Fig. 2), the main difference being the absence of helix C in HIV-1 and B. halodurans RNases H. The corresponding residues were deleted in variants of Mo-MLV and XMRV RNase H for which structures were previously determined (Lim et al., 2006). Superposition of XMRV and E. coli RNases H (Fig. 2a) results in RMSD of 2.73 Å for 94 pairs of Cα atoms, whereas a corresponding superposition with the human RNase H results in RMSD of 3.03 Å for 103 Cα pairs. Helix E is longer in XMRV RNase H than its counterpart in the E. coli enzyme, whereas β-strands 3 and 2 are shorter. Differences also are seen in several loop regions, particularly those between β-strands 1 and 2, as well as 2 and 3. Whereas helix C in both enzymes superimposes well, the basic protrusion that follows it is significantly different, even though of the same length (Fig. 2c). The direction of the chains diverges, with the distance of 13.4 Å between the equivalent residues in XMRV and E. coli RNase H (Thr605 in the former and Ala93 in the latter). Minor differences are also evident in the loops between β-strand 3 and helix A, as well as β-strand 5 and helix E. The loop between β-strand 5 and helix E, designated the “His-loop”, is close to the active site, thus the significant positional deviation between XMRV and E. coli enzyme leads to slightly different configuration of the catalytic core.
Figure 2.

Comparisons of the structures of RNase H from different species. (a) Superposition of RNase H from XMRV (green) and E. coli (light pink). (b) Superposition of RNase H from XMRV (green) and HIV-1 (orange). (c) Structure-based sequence alignment of XMRV, human, E. coli, and HIV-1 RNase H enzymes. Catalytic amino acids are highlighted in red, and conserved residues located near the active site and might contribute to catalysis in cyan, whereas other identical residues are highlighted in green. Yellow highlight indicates the sequence corresponding to the helix C and the basic protrusion.
A comparison of the XMRV and HIV-1 RNase H (Fig. 2b) reveals that the largest difference occurs in the vicinity of helix C. The HIV-1 RNase H domain does not contain such a helix and the following basic protrusion, whose functional role was thought to be provided by the connection subdomain of the p66 RT subunit (Lim et al., 2006). Helix E is longer in the XMRV RNase H than its counterpart in the HIV-1 enzyme. However, these two enzymes are quite similar in loop regions such as the His-loop, as well as in their N termini. The RMSD between these two enzymes is 2.3 Å for 95 pairs of superimposed Cα atoms.
Due to the deletion, the ΔC2 variant structure does not contain helix C, and the adjacent basic protrusion loop (Cerritelli and Crouch, 1998) is smoother. This difference affects interactions between the enzyme and substrates (see below). A minor difference occurs at the N-terminal loop whose orientation differs compared to the full-length type enzyme. Since the N-terminal loop is close to helix C, and involved in interactions with this helix, its deletion obviously changed contacts among these residues and affected the structure at the N terminus. The interactions between N-terminal residues and helices C, D, β-strand 4, 5, and the loop between helix D and β-strand 5 stabilize the conformation of N-terminal loop. In particular, the side chain of Arg506 can form three hydrogen bonds with the backbone atoms of Leu624, Pro627 and Lys628 (Fig. 3), decreasing the flexibility of the linker between the DNA polymerase and RNase H domains.
Figure 3.
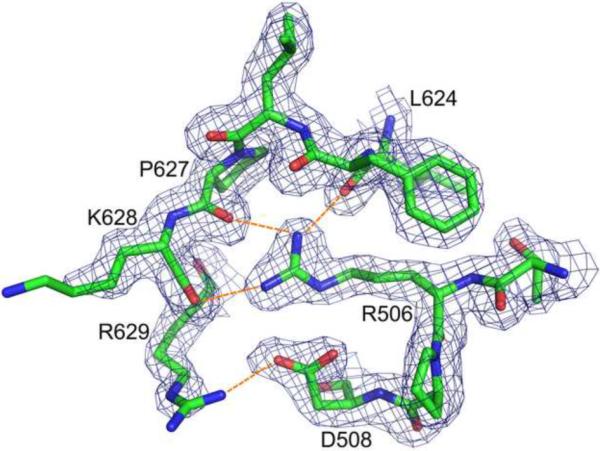
Interactions between the N-terminal loop and the catalytic domain. Arg506 and Asp508 form multiple hydrogen bonds (orange dashed lines) with the residues located in the loop located between helix D and β-strand 5. The 2Fo-Fc electron density map was contoured at 1.0 σ.
3.2. Enzymatic activity and the structure of the active site
Fig. 4a compares Mn2+-dependent RNase H activities of the p66/p51 HIV-1 RT heterodimer, the full-length XMRV enzyme and three variants of the XMRV RNase H domain. Under conditions used here, full-length XMRV RT was ~5-fold more active than its HIV-1 counterpart. Activity of the intact RNase H domain was ~70% of that of the parental XMRV enzyme and 3-fold greater than HIV-1 RT. In keeping with the proposed role for helix C in substrate binding, RNase H variants ΔC1 and RH ΔC2 were 10- to 25-fold less active than the intact RNase H domain. As an independent assessment of RNase H active site architecture, sensitivity of all enzymes to the natural product RNase H inhibitor β-thujaplicinol (Himmel et al., 2009) was determined in the presence of Mn2+ (Fig. 4b). For the full length enzymes and the intact XMRV RNase H domain IC50 values ranged from 21 to 67 nM. In contrast, XMRV variants lacking helix C were considerably less sensitive to β-thujaplicinol, with IC50 values of 0.68 μM (ΔC2) and 1.9 μM (ΔC1). Thus, data presented in Fig. 4 collectively illustrate that the integrity of the XMRV RNase H domain can be maintained in the absence of the DNA polymerase domain.
Figure 4.

(a) Mn2+-dependent RNase H activities of intact HIV-1 and XMRV RT, and a comparison with XMRV variants of the isolated RNase H domain. (b) Inhibition of HIV-1 and XMRV RNase H variants by the active site inhibitor β-thujaplicinol. All enzymes were evaluated in the presence of Mn2+, and IC50 values are the average of triplicate analysis.
The active site, comprising several acidic residues, is highly conserved in the RNase H superfamily. The catalytic core of XMRV RNase H comprises four carboxylate-bearing residues (Asp524, Glu562, Asp583, and Asp653), capable of coordinating two divalent cations. However, despite inclusion of 5 mM MgCl2 in the crystallization mother liquor, no enzyme-bound ions could be detected in either the full-length RNase H or its ΔC2 variant. However, two Mn2+ ions were clearly seen in the complex with the natural product RNase H inhibitor β-thujaplicinol, although the inhibitor itself was not well ordered. Previous investigations have shown that all four acidic residues are involved in coordinating the two metal ions, although only Asp524, Glu562, and Asp653 are essential for the catalytic activity (Kanaya et al., 1990). The adjacent residues include His638, Asn649, and Arg657. The distance between the side chains of His638 and Asp653 (CE1 to OD1) is 3.79Å, 2.6 Å between Asn649 and Asp583 side chains (NH1 to OD1), and 2.26 Å between Asp653 and Arg657 side chains (ND2 to OD2). Previous studies revealed that mutating the equivalent residues of E. coli RNase H (His124 and Asn130) did not completely inactivate the enzyme, but rather resulted in a decrease of substrate binding affinity and catalytic activity (Kanaya et al., 1990; Yang et al., 1990), suggesting these two residues contribute to catalysis. Arg657 is not conserved among all RNases H (Fig. 2c) and the contribution of this residue to catalysis remains unclear. According to the two-metal-ion catalysis mechanism (Nowotny et al., 2005), two divalent cations (Mg2+ or Mn2+) must be present in the catalytic site for activity. In this study, the ΔC2 variant was crystallized from a solution that contained 5 mM MgCl2 and the wild type RNase H was crystallized in the absence of divalent cations. Not surprisingly, we failed to observe divalent cations in the active site of the full-length enzyme, but no metal ions were seen also in the ΔC2 variant. The latter result agrees with previous studies indicating that, in the absence of a substrate of inhibitor, divalent cations were observed only when their concentration exceeded 10 mM (Nowotny et al., 2005).
3.3. β-Thujaplicinol binds at the active site
Previous studies revealed that β-thujaplicinol is a noncompetitive inhibitor of RNase H (Budihas et al., 2005; Himmel et al., 2009), whereas data of Fig. 4 demonstrate that the enzymatically-active XMRV RNase H domain is also highly sensitive to inhibition. We therefore attempted to obtain a complex of the intact XMRV RNase H with β-thujaplicinol by soaking the crystals of the enzyme in solution containing 2 mM inhibitor and 20 mM MnCl2. Two manganese ions were clearly seen in the inhibitor complex, although the inhibitor itself was not well ordered. The distance between the two Mn2+ ions is 3.94 Å and their positions correspond to the canonical A and B sites described for the related enzymes (Fig. 5). The two metal ions were coordinated by the four carboxylate-bearing residues in the active site. Asp524 and Asp653 coordinated the metal ion present in site A, whereas Asp524, Asp583, and Glu562 coordinated the ion in site B. β-Thujaplicinol contains a tropolone ring and an acidic hydroxyl, carries a partial positive charge on the tropolone ring, and has a corresponding partial negative charge shared by the resonance involving its carbonyl and hydroxyls (Himmel et al., 2009). The partially negatively charged carbonyl and hydroxyls of the tropolone ring chelated the two divalent metal ions, and also formed ionic interactions or hydrogen bonds with the side chains of Asp524, Glu562, Asp583, His638, Asp653, and Arg657.
Figure 5.

Binding of the α-hydroxytropolone inhibitor β-thujaplicinol at the XMRV RNase H active site. Direct contacts between the protein (green), inhibitor (gray), and manganese cations (purple) are shown in blue dashed lines.
3.4. Modeling the XMRV RNase H domain complexed with a DNA/RNA hybrid
In the absence of an experimental structure of such a complex we created a model based on data obtained in this and previous studies. Although the crystal structures of human and Bacillus halodurans RNase H complexed with DNA/RNA hybrids are available (Nowotny et al., 2005; Nowotny et al., 2007), no equivalent structure is available for the isolated RNase H domain of retroviral RT. Since the structure reported here was obtained without a DNA/RNA hybrid, no direct identification of the contacts between enzyme and substrate could be made. However, due to the high overall folding similarity, a superposition of XMRV RNase H and human RNase H/hybrid complex provides clues on how retroviral RNase H might bind its substrate. The superposition of the XMRV and human RNases H indicated that helices A, B, C, and D are well aligned with the exception of the basic protrusion and the loop between β-strands 1 and 2. However, the basic protrusion of XMRV RNase H is involved in crystal contacts which may affect its conformation. In human RNase H the majority of enzyme-substrate contacts occur along the minor groove of the DNA/RNA hybrid, whereas the tip of the basic protrusion binds the major groove. The DNA and RNA backbones fit in two grooves of the protein surface (Nowotny et al., 2007). According to the results of our superposition, the general enzyme-substrate binding mode also applies to XMRV RNase H, although we observed some clashes between the substrate and the side chains of the protein. Despite small differences in the binding modes for XMRV and human RNases H, the two structures are close enough that a convincing model can be built. Fig. 6 illustrates the substrate binding motif in the model of the complex. Similar to human RNase H, the surface of XMRV RNase H also contains two obvious grooves that should accommodate the backbones of RNA and DNA. The active site is located in the RNA binding groove (Fig. 6a), and the distances between the four catalytic residues and the RNA backbone are in the range of 4–6.5 Å, which is reasonable for divalent cation-mediated catalysis. The binding regions interacting with the DNA/RNA hybrid minor groove include helices A, B, C, E, and β-strand 1. The His-loop and the loop between β-strand 4 and helix B make contacts with the RNA backbone, and the tip of the basic protrusion binds its major groove. Five consecutive ribonucleotides are potentially able to make contact with the protein. Critical interactions occur around the catalytic core, mediated by the side chain or main chain atoms of Ser526, Phe528, Leu529, Glu562, Arg585, Gly637, His638, and Arg657. DNA-protein interactions mainly occur at two regions. The first region is the so-called “phosphate-binding pocket” (Nowotny et al., 2005), which is formed by Ser557, Gln559, and Asn613. Ser557 and Asn613 are equivalent, respectively, to Thr181 and Asn240 in human RNase H, which were proposed to form the phosphate-binding pocket. The second prominent binding site is located in the vicinity of helices B, C and the basic protrusion loop. The key residues Tyr586, His594, and Tyr598 provide stacking and hydrogen-bonded interactions. This indicates the key role of helix C in substrate binding and explains why the two variants of XMRV RNase H with that helix deleted only exhibit low or no enzymatic activity. Besides these six residues, other residues contributing to DNA-protein interactions include Arg534, Glu610, Lys612, and Lys640. Arg534 is located in β-strand 2, Glu610 and Lys612 are located in the basic protrusion loop, whereas Lys640 is located in the His-loop. These residues contribute their charge to the protein-nucleic acid interaction region (Fig. 6b). Our proposed model of a complex between XMRV RNase H and a DNA/RNA hybrid thus provides a reasonable explanation of substrate recognition.
Figure 6.
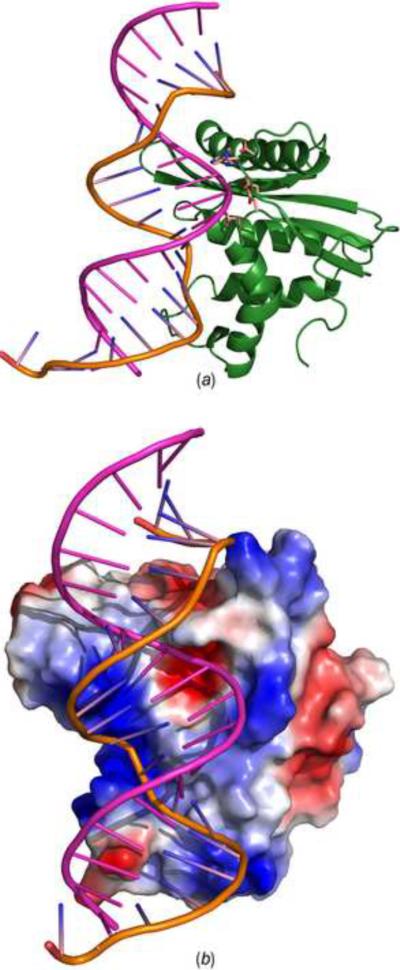
A model of the XMRV RNase H-substrate complex. (a) Docking of a 20-mer DNA/RNA hybrid from the experimental structure of its complex with human RNase H (Nowotny et al., 2007) onto XMRV RNase H. The active site residues are shown in sticks. The DNA strand is in orange and the RNA strand is in pink. (b) Surface potential representation of XMRV RNase H complexed to a 20-mer DNA/RNA hybrid (lines). The DNA and RNA strands are colored as in panel A.
In conclusion, we presented here the first structure of an isolated full-length RNase H domain of the XMRV RT, providing experimental evidence for the conformation of helix C and the basic protrusion loop which was not available in the previous studies. The enzyme-inhibitor complex structure clearly elucidates the coordination of two metal ions and the recognition between the inhibitor and protein mediated by the two divalent cations. A model of the enzyme-substrate complex postulated a possible recognition mechanism between DNA/RNA hybrid and XMRV RNase H.
4. PDB accession codes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 3V1O, 3V1Q and 3V1R for the full-length RNase H, variant ΔC2, and the enzyme-inhibitor complex, respectively.
Acknowledgments
We thank Dr. Zbyszek Dauter and Mi Li for help with data processing, and Dr. Genbin Shi for help with structure modeling. This project was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Boyer PL, Gao HQ, Frank P, Clark PK, Hughes SH. The basic loop of the RNase H domain of MLV RT is important both for RNase H and for polymerase activity. Virol. 2001;282:206–213. doi: 10.1006/viro.2000.0827. [DOI] [PubMed] [Google Scholar]
- Brünger AT. The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Budihas SR, Gorshkova I, Gaidamakov S, Wamiru A, Bona MK, et al. Selective inhibition of HIV-1 reverse transcriptase-associated ribonuclease H activity by hydroxylated tropolones. Nucleic Acids Res. 2005;33:1249–1256. doi: 10.1093/nar/gki268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerritelli SM, Crouch RJ. Cloning, expression, and mapping of ribonucleases H of human and mouse related to bacterial RNase HI. Genomics. 1998;53:300–307. doi: 10.1006/geno.1998.5497. [DOI] [PubMed] [Google Scholar]
- Das D, Georgiadis MM. The crystal structure of the monomeric reverse transcriptase from Moloney murine leukemia virus. Structure. 2004;12:819–829. doi: 10.1016/j.str.2004.02.032. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Gillette WK, Esposito D, Taylor TE, Hopkins RF, Bagni RK, Hartley JL. Purify First: Rapid expression and purification of proteins from XMRV. Protein Expr. Purif. 2010;76:238–247. doi: 10.1016/j.pep.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Himmel DM, Maegley KA, Pauly TA, Bauman JD, Das K, et al. Structure of HIV-1 reverse transcriptase with the inhibitor beta-Thujaplicinol bound at the RNase H active site. Structure. 2009;17:1625–1635. doi: 10.1016/j.str.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- Kanaya S, Kohara A, Miura Y, Sekiguchi A, Iwai S, et al. Identification of the amino acid residues involved in an active site of Escherichia coli ribonuclease H by site-directed mutagenesis. J Biol. Chem. 1990;265:4615–4621. [PubMed] [Google Scholar]
- Katayanagi K, Miyagawa M, Matsushima M, Ishikawa M, Kanaya S, et al. Three-dimensional structure of ribonuclease H from E. coli. Nature(London) 1990;347:306–309. doi: 10.1038/347306a0. [DOI] [PubMed] [Google Scholar]
- Kirby KA, Marchand B, Ong YT, Ndongwe TP, Hachiya A, et al. Structural and Inhibition Studies of the RNase H Function of Xenotropic Murine Leukemia Virus-Related Virus Reverse Transcriptase. Antimicrob. Agents Chemother. 2012 doi: 10.1128/AAC.06000-11. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- Li M, DiMaio F, Zhou D, Gustchina A, Lubkowski J, et al. Crystal structure of XMRV protease differs from the structures of other retropepsins. Nat. Struct. Mol. Biol. 2011;18:227–229. doi: 10.1038/nsmb.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim D, Gregorio GG, Bingman C, Martinez-Hackert E, Hendrickson WA, Goff SP. Crystal structure of the moloney murine leukemia virus RNase H domain. J Virol. 2006;80:8379–8389. doi: 10.1128/JVI.00750-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi VC, Ruscetti FW, Das GJ, Pfost MA, Hagen KS, et al. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science. 2009;326:585–589. doi: 10.1126/science.1179052. [DOI] [PubMed] [Google Scholar]
- Matthews BW. Solvent content of protein crystals. J. Mol. Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- Mikovits JA, Lombardi VC, Pfost MA, Hagen KS, Ruscetti FW. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Virulence. 2010;1:386–390. doi: 10.4161/viru.1.5.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016. doi: 10.1016/j.cell.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Nowotny M, Gaidamakov SA, Ghirlando R, Cerritelli SM, Crouch RJ, Yang W. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell. 2007;28:264–276. doi: 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Paprotka T, Delviks-Frankenberry KA, Cingoz O, Martinez A, Kung HJ, et al. Recombinant Origin of the Retrovirus XMRV. Science. 2011;333:97–101. doi: 10.1126/science.1205292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaberg R, Choe DJ, Brown KR, Thaker HM, Singh IR. XMRV is present in malignant prostatic epithelium and is associated with prostate cancer, especially high-grade tumors. Proc. Natl. Acad. Sci. U. S. A. 2009;106:16351–16356. doi: 10.1073/pnas.0906922106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Schultz SJ, Champoux JJ. RNase H domain of Moloney murine leukemia virus reverse transcriptase retains activity but requires the polymerase domain for specificity. J Virol. 1996;70:8630–8638. doi: 10.1128/jvi.70.12.8630-8638.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telesnitsky A, Blain SW, Goff SP. Defects in Moloney murine leukemia virus replication caused by a reverse transcriptase mutation modeled on the structure of Escherichia coli RNase H. J Virol. 1992;66:615–622. doi: 10.1128/jvi.66.2.615-622.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Hendrickson WA, Crouch RJ, Satow Y. Structure of ribonuclease H phased at 2 Å resolution by MAD analysis of the selenomethionyl protein. Science. 1990;249:1398–1405. doi: 10.1126/science.2169648. [DOI] [PubMed] [Google Scholar]