

Abstract
The poly(ADP-ribose) (PAR) post-translational modification is essential for diverse cellular functions, including regulation of transcription, response to DNA damage, and mitosis. Cellular PAR is predominantly synthesized by the enzyme poly(ADP-ribose) polymerase-1 (PARP-1). PARP-1 is a critical node in the DNA damage response pathway, and multiple potent PARP-1 inhibitors have been described, some of which show considerable promise in the clinic for the treatment of certain cancers. Cellular PAR is efficiently degraded by poly(ADP-ribose) glycohydrolase (PARG), an enzyme for which no potent, readily accessible, and specific inhibitors exist. Herein we report the discovery of small molecules that effectively inhibit PARG in vitro and in cellular lysates. These potent PARG inhibitors can be produced in two chemical steps from commercial starting materials and have complete specificity for PARG over the other known PAR glycohydrolase (ADP-ribosylhydrolase 3, ARH3) and over PARP-1, and thus will be useful tools to study the biochemistry of PAR signaling.
Poly(ADP-ribosylation) is a post-translational modification critical to many cellular events, including DNA damage repair, transcription, RNA metabolism, and telomere function (1, 2). The poly(ADP-ribose) polymerase (PARP) family of enzymes, most notably PARP-1, use ß-NAD+ in the synthesis of poly(ADP-ribose) (PAR), a negatively-charged branched biopolymer of varying lengths, onto multiple acceptor proteins (e.g., histones, transcription factors, and PARP-1 itself) (1, 3). The presence of PAR is transient due to the high specific activity of poly(ADP-ribose) glycohydrolase (PARG), the main enzyme involved in the degradation of PAR. PARG catalyzes the hydrolysis of the ribosyl-ribose bond of PAR in both endo- and exo-glycosidic fashions, producing ADP-ribose monomers and shorter PAR chains (4, 5).
The PARP enzymes are emerging as targets for the treatments of various diseases; for example, PARP-1 inhibitors have shown promise in anticancer clinical trials (6, 7). PARG inhibitors also have potential as therapeutic agents, as PARG activity plays a key role in cellular response to insult and in the initiation of cell death (8, 9). PARG is an attractive pharmacological target due to its low cellular abundance (approximately 2,000 molecules per cell (10)), and conserved catalytic domain, as all four PARG isoforms are encoded by a single gene (11). A number of biochemical studies have investigated the consequences of loss of PARG function through knockdown and isoform-specific knockout. In general, cells with genetic depletion or RNAi silencing of PARG were protected from H2O2-induced cell death (12) and had increased susceptibility to radiation (13, 14), DNA-alkylating agents (15), and chemotherapeutics such as cisplatin and epirubicin (16). Additionally, apoptosis inducing factor (AIF) mediated cell death is specifically activated after ultraviolet treatment of PARG-null cells (9). Thus inhibition of PARG may be a viable strategy for cancer treatment and, given the embryonic lethality of PARG knockouts in mice (15), selective small molecule inhibitors of PARG would greatly aid in the interrogation of this interesting biological target.
Unfortunately, the lack of potent, specific, and easily synthesized small molecule inhibitors of PARG has limited the study of PARG’s function both in vitro and in vivo. PARG inhibitors can be grouped into three major categories: DNA intercalators, tannins, and ADP-ribose analogues (17, 18). However, the usefulness of the first two categories has been limited due to toxicity, lack of specificity, and high molecular weights (18, 19). Adenosine diphosphate (hydroxymethyl)pyrrolidinediol (ADP-HPD), an analog of the ADP-ribose product of the PARG-catalyzed reaction, is the most potent (IC50 = 120 nM) and widely-used PARG inhibitor (20). Despite its expense ($2600/mg), lengthy synthetic route (10 steps, multiple anion-exchange chromatography columns) (21, 22), lack of cell permeability (21, 23), and partial inhibition of PARG (20), ADP-HPD has found wide utility in the study of PAR: it has been used in vitro and in cell extracts to study PARG’s structure and function (24, 25), as an additive in Western blot analysis to evaluate PAR accumulation (26, 27), for the analysis of PAR levels in nuclear extracts (28), the measurement of PARP activity in permeabilized cells (29, 30), as well as the study of PAR’s role in spindle assembly (31), tankyrase-1 activity (32), and the Sir2 family of proteins (33). Described herein is the identification of a potent, specific, and easily synthesized class of small-molecule PARG inhibitors, compounds that will further facilitate the study of the biological properties of PARG and PAR.
Results and Discussion
The pyrophosphate moiety contained within PAR and ADP-HPD appears to play a key role in binding to PARG, as suggested by a recent X-ray structure (34). As this functional group can limit the stability of potential inhibitors and complicates synthetic routes, we sought to evaluate the PARG inhibition properties of compounds with functional groups known to inhibit enzymes with phosphate-containing substrates (35). The rhodanine scaffold, a 5-membered heterocycle, is present in potent inhibitors of phosphodiesterase type 4 enzymes (36), serotonin N-acetyltransferases (37), UDP-galactopyranose mutase (38), and the glycosyl transferase MurG (39), all of which bind to substrates containing phosphate groups. As rhodanine has been suggested as a phosphate surrogate, (35, 40) we chose to evaluate rhodanine-based small molecules as inhibitors of PARG.
Screening and Lead Optimization
From an in-house collection of ~14,000 small molecules, 224 rhodanine-containing compounds were selected and screened for their ability to inhibit PARG in vitro at 200 μM. PARG enzymatic activity was evaluated by incubation of compounds with PARG for 10 min, followed by incubation with 32P-PAR for 2 h in order to identify compounds capable of prolonged PARG inhibition. Upon separation of intact 32P-PAR from 32P-ADP-ribose by thin-layer chromatography (TLC), the radiolabeled assay components were detected by phosphorimaging. Compounds that prevented PARG-mediated degradation of 32P-PAR were named rhodanine-based PARG inhibitors (RBPIs). We identified 16 primary hit compounds, and RBPI-1 (Figure 1), the most potent PARG inhibitor identified from this initial screen (IC50 = 46.5 ± 6.0 μM, Supplementary Figure S1), was chosen for further optimization.
Figure 1.

(A) Screening of 224 rhodanine-containing compounds reveals RBPI-1 as a PARG inhibitor, and a collection of >70 compounds was then synthesized based on this scaffold and evaluated for PARG inhibition. From this work RBPI-2, -3, -4, -5, and -6 were identified as potent PARG inhibitors. Inactive-1 and -2 are structurally-related compounds that do not inhibit PARG. (B) Representative dose-response PARG inhibition curves for each compound shown in A. For IC50 determination, compounds were incubated with PARG for 10 min, then 32P-PAR was added and incubated for 2 h at 37°C, at which point the extent of PAR degradation was assessed by separation on TLC plates and phosphorimaging. For TLC images and triplicate IC50 curves please see Supplementary Figure S7.
A library of RBPI-1 derivatives was readily produced using a two-step synthesis from commercial starting materials. Isatins (1, Supplementary Figure S2) were N-alkylated with benzyl halides to provide the desired intermediates (2, Supplementary Figure S2). Subsequent Knoevenagel condensation with N-substituted rhodanines with varying methylene chains produced 76 final compounds (Supplementary Figures S2 and S3), which were isolated by filtration in high purity (see characterization data in Supporting Information). All 76 compounds were then evaluated at 10 μM for their ability to inhibit PARG in vitro by the TLC assay described previously, and many compounds were also evaluated at 50 μM (Supplementary Figures S4, S5, and S6). Dose-response curves were generated for several compounds, including RBPI-2, -3, and -4 (Figure 1), all containing dihalogen substitution at the ortho positions on the benzyl group. These compounds were found to inhibit PARG with low micromolar IC50 values (2.9 ± 1.8, 5.8 ± 1.0, and 3.0 ± 1.6 μM, respectively, Figure 1 and Supplementary Figure S7).
In general, substitution at the 5-position of the isatin moiety with either bromine or chlorine and 2,6-dihalide substitution on the benzyl ring improves activity, while compounds lacking the benzyl ring are not active as PARG inhibitors (Supplementary Figure S6). Additionally, the length of the methylene linker from the rhodanine nitrogen to the carboxylic acid also affects activity, with two methylenes giving the best specificity and potency (Supplementary Figure S6). Since tetrazoles are a known isostere for carboxylic acids (41), the tetrazole-containing analogs of select compounds were prepared. Tetrazole-containing compounds RBPI-5 and -6 inhibit PARG at slightly reduced potencies (Figure 1 and Supplementary Figure S7) compared to their carboxylic acid counterparts, RBPI-2 and -3. Due to its reduced solubility relative to the other RBPIs, RBPI-2 was not used in further evaluations. Structurally related inactive derivatives (Inactive-1, and -2, Figure 1) were used as controls in subsequent experiments; these compounds have IC50 values greater than 100 μM for PARG inhibition (Figure 1 and Supplementary Figure S8).
RBPIs are specific, non-promiscuous PARG inhibitors
Many enzyme inhibitors that display excellent target specificity and activity in cell culture (42–46) and in vivo (47–49) have been developed with the rhodanine functional group. Additionally, the rhodanine-containing drug epalrestat, an aldose reductase inhibitor approved for use in Japan, is effective and safe even for long-term use (50, 51). Despite these successes, rhodanines are sometimes removed from screening libraries due to interference with colorimetric read-outs or potential reactivity and the inherent lipophilicity of rhodanines could make them susceptible to the formation of colloidal aggregates in buffered solution (52). Therefore, to rule out a promiscuous aggregation-based mode of enzyme inhibition (53–55), the RBPIs were analyzed in a series of enzymatic assays involving the addition of detergent, bovine serum albumin, and evaluation versus an unrelated enzyme.
Four of the potent PARG inhibitors (RBPI-3, -4, -5, -6) were tested for their ability to inhibit PARG in vitro in the presence or absence of detergent (Triton-X 100), which was used at a concentration (0.1%) high enough to disintegrate even “detergent hardy” aggregators (56). Inhibition of PARG by these four RBPIs at 50 μM was not altered by the addition of detergent (Figure 2). In contrast, inhibition of PARG by the known aggregating compound Congo Red was detergent-sensitive, with complete loss of inhibition observed in the presence of detergent (Figure 2).
Figure 2.

RBPI inhibition of PARG is insensitive to the presence of detergent. Compounds (50 μM) were incubated with PARG in the presence or absence of 0.1% Triton X-100 for 10 min, 32P-PAR was added and incubated for 2 h at 37°C, and the extent of PAR degradation was assessed by separation on TLC plates and phosphorimaging. (A) Representative TLC plate from this experiment; (B) Quantitation of the experiment described above by densitometry, n=3, error bars indicate standard error of the mean.
As another test for aggregation-based inhibition, RBPI-4 was tested for activity in the presence of a high concentration of bovine serum albumin (BSA, 0.1 mg mL−1), which binds non-specifically to small molecule aggregates; such BSA assays have previously been used to identify inhibitors acting through non-specific mechanisms (57). Consistent with the detergent results, RBPI-4 retained activity in the presence of BSA (Supplementary Figure S9).
The rhodanine-containing compounds were also tested against ß-lactamase to assess enzyme specificity; ß-lactamase inhibition is commonly used to evaluate compound promiscuity (58). Briefly, RBPI-3, -4, -5, -6 and Congo Red were incubated with ß-lactamase in a phosphate buffer for 0.5 h, followed by the addition of β-lactam substrate CENTA, and ß-lactamase activity was measured by monitoring the appearance of the cleavage product at 405 nm. None of the rhodanine-containing compounds showed appreciable inhibition of β-lactamase enzymatic activity at concentrations of 5 to 50 μM (all <10% inhibition, Figure 3A). In contrast, Congo Red inhibited ß-lactamase in a dose-dependent manner, with 100% inhibition at 50 μM (Figure 3A). Inhibition of ß-lactamase was also evaluated in the presence of detergent, as a further test for aggregation-based inhibition. In the presence of detergent, RBPIs remained inactive against ß-lactamase, whereas the addition of detergent eliminated the ability of Congo Red to inhibit the enzyme (<10% inhibition, Figure 3B). These collective results, which show that inhibition of PARG by RBPIs is not sensitive to addition of detergent or BSA and that the RBPIs do not inhibit β-lactamase, indicate that these compounds do not inhibit PARG via a promiscuous, aggregation-based mechanism.
Figure 3.

RBPIs do not inhibit β-lactamase. RBPIs and Congo Red (5-50 μM) were incubated with ß-lactamase (A) in the absence of detergent and (B) in the presence of 0.1% Triton X-100. After addition of the colorimetric substrate, enzyme activity was measured by recording absorbance at 405 nm, n=3, error bars indicate standard error of the mean.
RBPIs inhibit PARG with high efficiency
The time- and dose-dependence of PARG inhibition by the RBPIs was directly compared to ADP-HPD. PARG was treated with increasing concentrations of RBPI-3, RBPI-6, or ADP-HPD (0.5 – 25 μM), and the processing of 32P-labeled PAR was evaluated after 0.5 and 2 h. Although ADP-HPD concentrations as low as 0.5 μM partially inhibited PARG at 0.5 h, inhibition was incomplete even in the presence of 10 and 25 μM ADP-HPD (67% inhibition at 25 μM, Figure 4A and C). In contrast, 10 μM RBPI-3 and RBPI-6 showed complete inhibition of PARG after 0.5 h (>99% inhibition at 25 μM, Figure 4A and C). When PAR and PARG were incubated in the presence of compound for 2 h, ADP-HPD showed only minimal PARG inhibition; for example, PARG was able to almost completely process PAR into ADP-ribose in the presence of 25 μM ADP-HPD (Figure 4B and C).
Figure 4.

Inhibition of PARG by RBPIs persists even after 2 h incubation with substrate. RBPI-3, -6, and ADP-HPD were incubated with PARG for 10 min, then 32P-PAR was added and incubated at 37°C for (A) 0.5 h or (B) 2 h. The extent of PAR degradation was assessed by separation on TLC plates and phosphorimaging. (C) Quantitation of data shown in A and B, n=3, error bars indicate standard error of the mean.
The partial inhibition of PARG by ADP-HPD described in the literature (20) is consistent with our results in which ADP-HPD is able to retard, but not completely prevent, the degradation of PAR over 2 h (Figure 4). In contrast, under the same 2 h conditions, RBPI-3 and RBPI-6 prevented PAR degradation with only a slight shift in inhibition at 10 μM relative to the 0.5 h incubation (Figure 4A-C). Overall, these RBPIs were able to inhibit PARG more efficiently than ADP-HPD at prolonged incubation times. The long-lasting inhibition of PARG by RBPIs, coupled with their ease of synthesis suggests that the RBPIs are more tractable compounds for in vitro use.
RBPIs do not inhibit ARH3 or PARP-1
The specificity of the RBPIs and ADP-HPD for PARG was further evaluated by testing these compounds against the other known PAR glycohydrolase enzyme, ADP-ribosylhydrolase 3 (ARH3), and against PARP-1. ARH3 is a 39 kDa human protein known to have two enzymatic functions: deacetylation of O-acetyl ADP-ribose and PAR degradation, though deacetylation has been suggested as its main function (59-62). Although both enzymes can process PAR, ARH3 does not have a similar sequence or structure to PARG (34). To achieve a comparable rate of PAR degradation in these experiments, 1000-fold more ARH3 (256 nM) than PARG (0.24 nM) was required in the in vitro assay, along with the addition of 4 mM MgCl2. Upon incubating RBPI-3, -4, -5, -6 and Inactive-1 and -2 (25 μM) with ARH3 for 2 h, none of the rhodanine-containing compounds prevented ARH3-mediated degradation of PAR (<10% inhibition, Figure 5A), although the RBPIs inhibited PARG at the same concentration (>70% inhibition, Figure 5B). In contrast, ADP-HPD (25 μM) showed complete inhibition of ARH3 (98% inhibition, Figure 5A), but only partial inhibition of PARG at 2 h (39% inhibition, Figure 5B). Although ARH3 was thought to be unaffected by ADP-HPD (27), we determined the IC50 of ADP-HPD to be 14.3 ± 1.4 μM against ARH3 (Supplementary Figure S10). Thus ADP-HPD inhibits both PAR glycohydrolases, and is actually a more efficient inhibitor of ARH3, while the RBPIs potently and specifically inhibit PARG.
Figure 5.
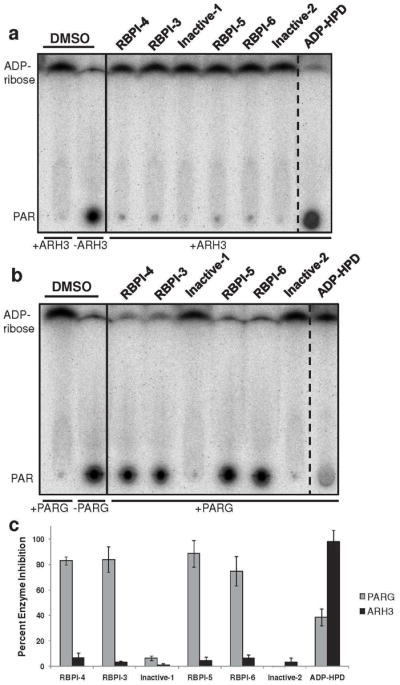
ADP-HPD, but not RBPIs, inhibits ARH3 PAR glycohydrolase activity. Compounds were incubated with (A) ARH3 (256 nM) or (B) PARG (0.24 nM) for 10 min, and then 32P-PAR was added and incubated at 37°C for 2 h. The extent of PAR degradation was visualized by separation on a TLC plate and phosphorimaging. (C) Quantitation of data from part A and B, n=3, error bars indicate standard error of the mean. See Supplementary Figure S10 for IC50 data of ADP-HPD inhibition of ARH3.
RBPI-6 was further evaluated for its ability to inhibit the synthesis of 32P-PAR from 32P-NAD+ by PARP-1. The production of 32P-PAR in the presence of 25 μM RBPI-6 or PJ34, a potent PARP inhibitor (63), was evaluated after 0.5 h. As shown in Supplementary Figure S11, no significant inhibition of PARP-1 was observed with RBPI-6, while the same concentration of PJ34 showed complete PARP-1 inhibition. PARP-1 enzymatic activity was also not affected by RPBI-6 at 100 μM, the highest concentration evaluated. Thus the combined data shows that the RBPIs potently inhibit PARG, but do not inhibit other enzymes such as β-lactamase, ARH3, and PARP-1.
Comparison of RBPIs, ADP-HPD and salicylanilides as PARG inhibitors
A class of modified salicylanilide PARG inhibitors was recently reported (64), and the ability of these compounds to inhibit PARG in vitro was directly compared to that of the RBPIs and ADP-HPD. Salicylanilide 6a, with a reported IC50 for PARG inhibition of 12 ± 2 μM, was synthesized according to the published route (See Supporting Information for characterization). Using the assay conditions in which compound, PARG, and PAR are incubated for 2 h, salicylanilide 6a did not inhibit PARG at concentrations up to 625 μM (<10% inhibition), while complete inhibition by RBPI-4 (95% inhibition at 12.5 μM), and partial inhibition by ADP-HPD (33% inhibition at 25 μM) was observed (Supplementary Figure S12). Using the PARG assay conditions in which the salicylanilides were originally assessed (64), which employ a phosphate buffer and incubation period of 5 min, inhibition of PARG by 6a was observed as low as 50 μM (54% inhibition), which is similar to the reported efficacy (Supplementary Figure S13) (64). When 0.1% Triton X-100 was added to this assay, no inhibition of PARG by 6a was observed up to 500 μM (<5% inhibition, Supplementary Figure S13). In contrast, both ADP-HPD (10 μM) and RBPI-4 (50 μM) displayed complete inhibition of PARG both in the absence and presence of 0.1% Triton X-100 (Supplementary Figure S13). Therefore, the RBPIs are more efficient PARG inhibitors than the recently reported salicylanilides.
RBPIs effectively inhibit PARG in cellular lysate
To determine the efficacy of RBPIs in complex mixtures, we evaluated their ability to inhibit endogenous PARG in whole cell lysate generated from mouse embryonic fibroblasts (MEFs). Lysate (1.5 μg) was treated with vehicle (DMSO), RBPI-4, or ADP-HPD (each at 25 μM), then 32P-PAR was added and its degradation was evaluated over time. As shown in Figure 6, the vehicle-treated lysate rapidly degraded 32P-PAR with a loss in signal as early as 5 min and the majority of PAR cleaved within 30 min. In contrast, 32P-PAR remained mostly intact in the RBPI-4-treated lysate for the duration of the 60 min time course (Figure 6 and Supplementary Figure S14). When compared directly to ADP-HPD in this cell lysate experiment, RBPI-4 displays similar potency (approx. 80% intact PAR at 60 min, Figure 6 and Supplementary Figure S14).
Figure 6.

Processing of 32P-PAR by cellular lysate (MEF, 1.5 μg) over 60 min in the presence of DMSO, 25 μM RBPI-4, or 25 μM ADP-HPD. Compounds were incubated with lysate for 10 min, then 32P-PAR was added and incubated at 37°C for 0-60 min. The extent of PAR degradation was visualized by separation on a TLC plate and phosphorimaging. Graph is representative of three independent experiments, see Supplementary Figure S13 for triplicate data.
Evaluation of RBPI-3, -5, and -6 in MEF cellular lysate gave similar results (Supplementary Figure S15) while Inactive-1 had no inhibitory effect (Supplementary Figure S16). This persistence of PAR in RBPI-treated cell lysate indicates effective inhibition of endogenous PARG by RBPIs. Inhibition of PARG by RBPI-4 was also evaluated in lysates from five additional cell lines, representing a diverse sampling of cell types. RBPI-4 was able to delay 32P-PAR degradation in all cases, although the extent of inhibition varied with the different cell lines (Supplementary Figures S17-21). While some rhodanine rings with exocyclic double bonds are susceptible to conjugate addition by cellular thiols (65), the activity of RBPIs in cellular lysate indicates that cellular nucleophiles do not affect the RBPI-mediated inhibition of PARG. These results also show that RBPIs are specific for PARG and are able to inhibit endogenous PARG in the presence of an abundance of cellular proteins. As these RBPIs do not significantly inhibit PAR degradation in whole cell experiments, at the present time their utility is restricted to in vitro and cell lysate experiments.
Conclusion
Small molecule PARG inhibitors are needed not only to study the function of PARG and to investigate the therapeutic possibilities of PARG inhibition. While ADP-HPD has facilitated many studies on the in vitro inhibition of PARG, readily accessible, specific, drug-like inhibitors of PARG are lacking. Through targeted screening and subsequent chemical optimization, we identified the RBPIs as potent, easy-to-synthesize, selective PARG inhibitors with persistent inhibition in vitro, strong activity in cellular lysate, and high specificity for PARG over ARH3 and PARP-1. The head-to-head evaluations of PARG inhibitors of various classes highlights the attractive properties of the RBPIs, and these compounds should facilitate further studies on PAR and PARG.
Materials and Methods
ß-Lactamase assay
ß-lactamase activity was assessed as previously reported (53).
PAR Synthesis
32P-PAR was synthesized in a similar manner to previous reports (66) with slight modifications. After the PARP reaction was finished, the polymer was precipitated with sodium acetate and isopropanol, washed twice with 80% ethanol, and used as a solution in water without further purification.
PARG IN VITRO assay
PARG activity was assessed by pre-incubation of PARG with compound in Trevigen PARG buffer, then 32P-PAR was added and incubated at 37 °C for 2 h. The assay was quenched either by heating to 90 °C for 2 min or addition of 1% (v/v) SDS. Two aliquots were spotted on a silica gel TLC plate which was developed in 70:30 iPrOH:0.2% (v/v) NH4OH(aq) eluent. The results were imaged by phosphorimaging and densitometry was used to quantify the amount of either PAR or ADP-ribose present. Experiments involving the addition of detergent or BSA were carried out in a phosphate buffer containing 2-mercaptoethanol.
Cell Lysate Activity
Cellular lysate was diluted to the appropriate concentrations in 1X Trevigen PARG buffer before use in the radiometric PARG assay.
ARH3 IN VITRO Activity
Recombinant ARH3 was diluted in 2X Trevigen PARG buffer supplemented with 4 mM MgCl2 before use in the radiometric PARG assay.
PARP-1 IN VITRO Activity
PARP-1 activity was assessed by addition of PARP-1 to a solution of compound, NAD+, 32P-NAD+, and calf thymus DNA in Trevigen PARG buffer. After incubation for 30 min at rt, PAR was precipitated with iPrOH and NaOAc, washed once with 80% EtOH and applied to a cellulose PEI TLC plate. After separation of PAR from NAD+, the PAR-containing spot was excised and quantified by scintillation counting.
Supplementary Material
Acknowledgments
This work was supported by the Michael J. Fox Foundation and the University of Illinois. K.E.F. was partially supported by the NIH Chemistry-Biology Interface Training Grant (NRSA 1-T32-GM070421). C.E.K is a National Science Foundation predoctoral fellow.
Footnotes
Full details for all assays as well as preparation and characterization of all compounds are provided in the Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 2.d'Adda di Fagagna F, Hande MP, Tong WM, Lansdorp PM, Wang ZQ, Jackson SP. Functions of poly(ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nat Genet. 1999;23:76–80. doi: 10.1038/12680. [DOI] [PubMed] [Google Scholar]
- 3.Aguilar-Quesada R, Munoz-Gamez JA, Martin-Oliva D, Peralta-Leal A, Quiles-Perez R, Rodriguez-Vargas JM, Ruiz de Almodovar M, Conde C, Ruiz-Extremera A, Oliver FJ. Modulation of transcription by PARP-1: consequences in carcinogenesis and inflammation. Curr Med Chem. 2007;14:1179–1187. doi: 10.2174/092986707780597998. [DOI] [PubMed] [Google Scholar]
- 4.Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- 5.Malanga M, Althaus FR. Poly(ADP-ribose) molecules formed during DNA repair in vivo. J Biol Chem. 1994;269:17691–17696. [PubMed] [Google Scholar]
- 6.Graziani G, Szabo C. Clinical perspectives of PARP inhibitors. Pharmacol Res. 2005;52:109–118. doi: 10.1016/j.phrs.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Kling J. PARP inhibitors blaze a trail in difficult-to-treat cancers. Nat Biotechnol. 2009;27:784–786. doi: 10.1038/nbt0909-784. [DOI] [PubMed] [Google Scholar]
- 8.Koh DW, Dawson VL, Dawson TM. The road to survival goes through PARG. Cell Cycle. 2005;4:397–399. doi: 10.4161/cc.4.3.1559. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Y, Feng X, Koh DW. Activation of cell death mediated by apoptosis-inducing factor due to the absence of poly(ADP-ribose) glycohydrolase. Biochemistry. 2011;50:2850–2859. doi: 10.1021/bi101829r. [DOI] [PubMed] [Google Scholar]
- 10.Hatakeyama K, Nemoto Y, Ueda K, Hayaishi O. Purification and characterization of poly(ADP-ribose) glycohydrolase. Different modes of action on large and small poly(ADP-ribose) J Biol Chem. 1986;261:14902–14911. [PubMed] [Google Scholar]
- 11.Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp Cell Res. 2004;297:521–532. doi: 10.1016/j.yexcr.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 12.Blenn C, Althaus FR, Malanga M. Poly(ADP-ribose) glycohydrolase silencing protects against H2O2-induced cell death. Biochemical J. 2006;396:419–429. doi: 10.1042/BJ20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.St-Laurent JF, Gagnon SN, Dequen F, Hardy I, Desnoyers S. Altered DNA damage response in Caenorhabditis elegans with impaired poly(ADP-ribose) glycohydrolases genes expression. DNA Repair. 2007;6:329–343. doi: 10.1016/j.dnarep.2006.10.027. [DOI] [PubMed] [Google Scholar]
- 14.Ame JC, Fouquerel E, Gauthier LR, Biard D, Boussin FD, Dantzer F, de Murcia G, Schreiber V. Radiation-induced mitotic catastrophe in PARG-deficient cells. J Cell Sci. 2009;122:1990–2002. doi: 10.1242/jcs.039115. [DOI] [PubMed] [Google Scholar]
- 15.Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, Stoger T, Poirier GG, Dawson VL, Dawson TM. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc Natl Acad Sci USA. 2004;101:17699–17704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y, Feng X, Koh DW. Enhanced DNA accessibility and increased DNA damage induced by the absence of poly(ADP-ribose) hydrolysis. Biochemistry. 2010;49:7360–7366. doi: 10.1021/bi100979j. [DOI] [PubMed] [Google Scholar]
- 17.Nottbohm AC, Hergenrother PJ. Drug Discovery Research: New Frontiers in the Post-Genomic Era. Wiley & Sons, Ltd; 2007. The Promises and Pitfalls of Small-Molecule Inhibition of Poly(ADP-Ribose) Glycohydrolase (PARG) pp. 163–185. [Google Scholar]
- 18.Blenn C, Wyrsch P, Althaus FR. The ups and downs of tannins as inhibitors of poly(ADP-ribose)glycohydrolase. Molecules. 2011;16:1854–1877. doi: 10.3390/molecules16021854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Falsig J, Christiansen SH, Feuerhahn S, Burkle A, Oei SL, Keil C, Leist M. Poly(ADP-ribose) glycohydrolase as a target for neuroprotective intervention: assessment of currently available pharmacological tools. Eur J Pharmacol. 2004;497:7–16. doi: 10.1016/j.ejphar.2004.06.042. [DOI] [PubMed] [Google Scholar]
- 20.Slama JT, Aboul-Ela N, Jacobson MK. Mechanism of Inhibition of Poly(ADP-ribose) Glycohydrolase by Adenosine Diphosphate (Hydroxymethyl)pyrrolidinediol. J Med Chem. 1995;38:4332–4336. doi: 10.1021/jm00021a023. [DOI] [PubMed] [Google Scholar]
- 21.Slama JT, Aboul-Ela N, Goli DM, Cheesman BV, Simmons AM, Jacobson MK. Specific inhibition of poly(ADP-ribose) glycohydrolase by adenosine diphosphate (hydroxymethyl)pyrrolidinediol. J Med Chem. 1995;38:389–393. doi: 10.1021/jm00002a021. [DOI] [PubMed] [Google Scholar]
- 22.Goli DM, Cheesman BV, Hassan ME, Lodaya R, Slama JT. Synthesis of (2R,3R,4S)-2-hydroxymethylpyrrolidine-3,4-diol from (2S)-3,4-dehydroproline derivatives. Carbohyd Res. 1994;259:219–241. doi: 10.1016/0008-6215(94)84059-8. [DOI] [PubMed] [Google Scholar]
- 23.Cuzzocrea S, Wang ZQ. Role of poly(ADP-ribose) glycohydrolase (PARG) in shock, ischemia and reperfusion. Pharmacol Res. 2005;52:100–108. doi: 10.1016/j.phrs.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Cortes U, Tong WM, Coyle DL, Meyer-Ficca ML, Meyer RG, Petrilli V, Herceg Z, Jacobson EL, Jacobson MK, Wang ZQ. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol Cell Biol. 2004;24:7163–7178. doi: 10.1128/MCB.24.16.7163-7178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel CN, Koh DW, Jacobson MK, Oliveira MA. Identification of three critical acidic residues of poly(ADP-ribose) glycohydrolase involved in catalysis: determining the PARG catalytic domain. Biochem J. 2005;388:493–500. doi: 10.1042/BJ20040942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang P, Coughlin M, Mitchison TJ. Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat Cell Biol. 2005;7:1133–1139. doi: 10.1038/ncb1322. [DOI] [PubMed] [Google Scholar]
- 27.Zhu G, Chang P, Lippard SJ. Recognition of Platinum-DNA Damage by Poly(ADP-ribose) Polymerase-1. Biochemistry. 2010;49:6177–6183. doi: 10.1021/bi100775t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asher G, Reinke H, Altmeyer M, Gutierrez-Arcelus M, Hottiger MO, Schibler U. Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell. 2010;142:943–953. doi: 10.1016/j.cell.2010.08.016. [DOI] [PubMed] [Google Scholar]
- 29.Kanai M, Tong WM, Wang ZQ, Miwa M. Haploinsufficiency of poly(ADP-ribose) polymerase-1-mediated poly(ADP-ribosyl)ation for centrosome duplication. Biochem Bioph Res Co. 2007;359:426–430. doi: 10.1016/j.bbrc.2007.05.108. [DOI] [PubMed] [Google Scholar]
- 30.Leung AK, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol Cell. 2011;42:489–499. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang P, Jacobson MK, Mitchison TJ. Poly(ADP-ribose) is required for spindle assembly and structure. Nature. 2004;432:645–649. doi: 10.1038/nature03061. [DOI] [PubMed] [Google Scholar]
- 32.Dregalla RC, Zhou J, Idate RR, Battaglia CL, Liber HL, Bailey SM. Regulatory roles of tankyrase 1 at telomeres and in DNA repair: suppression of T-SCE and stabilization of DNA-PKcs. Aging. 2010;2:691–708. doi: 10.18632/aging.100210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanders BD, Zhao K, Slama JT, Marmorstein R. Structural basis for nicotinamide inhibition and base exchange in Sir2 enzymes. Mol Cell. 2007;25:463–472. doi: 10.1016/j.molcel.2006.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, Ahel I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature. 2011;477:616–620. doi: 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nottbohm AC, Hergenrother PJ. Wiley. Wiley Encyclopedia of Chemical Biology. 2008. Replacing the Irreplaceable: Cyclic compounds as Novel Phosphate Mimics; pp. 1–16. [Google Scholar]
- 36.Irvine MW, Patrick GL, Kewney J, Hastings SF, MacKenzie SJ. Rhodanine derivatives as novel inhibitors of PDE4. Bioorg Med Chem Lett. 2008;18:2032–2037. doi: 10.1016/j.bmcl.2008.01.117. [DOI] [PubMed] [Google Scholar]
- 37.Szewczuk LM, Saldanha SA, Ganguly S, Bowers EM, Javoroncov M, Karanam B, Culhane JC, Holbert MA, Klein DC, Abagyan R, Cole PA. De novo discovery of serotonin N-acetyltransferase inhibitors. J Med Chem. 2007;50:5330–5338. doi: 10.1021/jm0706463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dykhuizen EC, May JF, Tongpenyai A, Kiessling LL. Inhibitors of UDP-galactopyranose mutase thwart mycobacterial growth. J Am Chem Soc. 2008;130:6706–6707. doi: 10.1021/ja8018687. [DOI] [PubMed] [Google Scholar]
- 39.Hu Y, Helm JS, Chen L, Ginsberg C, Gross B, Kraybill B, Tiyanont K, Fang X, Wu T, Walker S. Identification of selective inhibitors for the glycosyltransferase MurG via high-throughput screening. Chem Biol. 2004;11:703–711. doi: 10.1016/j.chembiol.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 40.Andres CJ, Bronson JJ, D'Andrea SV, Deshpande MS, Falk PJ, Grant-Young KA, Harte WE, Ho HT, Misco PF, Robertson JG, Stock D, Sun Y, Walsh AW. 4-Thiazolidinones: novel inhibitors of the bacterial enzyme murB. Bioorg Med Chem Lett. 2000;10:715–717. doi: 10.1016/s0960-894x(00)00073-1. [DOI] [PubMed] [Google Scholar]
- 41.Matta CF, Arabi AA, Weaver DF. The bioisosteric similarity of the tetrazole and carboxylate anions: clues from the topologies of the electrostatic potential and of the electron density. Eur J Med Chem. 2010;45:1868–1872. doi: 10.1016/j.ejmech.2010.01.025. [DOI] [PubMed] [Google Scholar]
- 42.Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, Gautier J. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol. 2008;4:119–125. doi: 10.1038/nchembio.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strittmatter T, Bareth B, Immel TA, Huhn T, Mayer TU, Marx A. Small Molecule Inhibitors of Human DNA Polymerase lambda. ACS Chem Biol. 2011;6:314–319. doi: 10.1021/cb100382m. [DOI] [PubMed] [Google Scholar]
- 44.Katritzky AR, Tala SR, Lu H, Vakulenko AV, Chen QY, Sivapackiam J, Pandya K, Jiang S, Debnath AK. Design, synthesis, and structure-activity relationship of a novel series of 2-aryl 5-(4-oxo-3-phenethyl-2-thioxothiazolidinylidenemethyl)furans as HIV-1 entry inhibitors. J Med Chem. 2009;52:7631–7639. doi: 10.1021/jm900450n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 46.Gonsalves FC, Klein K, Carson BB, Katz S, Ekas LA, Evans S, Nagourney R, Cardozo T, Brown AMC, DasGupta R. An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1017496108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forino M, Johnson S, Wong TY, Rozanov DV, Savinov AY, Li W, Fattorusso R, Becattini B, Orry AJ, Jung D, Abagyan RA, Smith JW, Alibek K, Liddington RC, Strongin AY, Pellecchia M. Efficient synthetic inhibitors of anthrax lethal factor. Proc Natl Acad Sci USA. 2005;102:9499–9504. doi: 10.1073/pnas.0502733102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma L, Xie C, Ma Y, Liu J, Xiang M, Ye X, Zheng H, Chen Z, Xu Q, Chen T, Chen J, Yang J, Qiu N, Wang G, Liang X, Peng A, Yang S, Wei Y, Chen L. Synthesis and biological evaluation of novel 5-benzylidenethiazolidine-2,4-dione derivatives for the treatment of inflammatory diseases. J Med Chem. 2011;54:2060–2068. doi: 10.1021/jm1011534. [DOI] [PubMed] [Google Scholar]
- 49.Ramirez MA, Borja NL. Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy. 2008;28:646–655. doi: 10.1592/phco.28.5.646. [DOI] [PubMed] [Google Scholar]
- 50.Hotta N, Akanuma Y, Kawamori R, Matsuoka K, Oka Y, Shichiri M, Toyota T, Nakashima M, Yoshimura I, Sakamoto N, Shigeta Y tAS Group. Long-Term Clinical Effects of Epalrestat, an Aldose Reductase Inhibitor, on Diabetic Peripheral Neuropathy. Diabetes Care. 2006;29:1538–1544. doi: 10.2337/dc05-2370. [DOI] [PubMed] [Google Scholar]
- 51.Hotta N, Sakamoto N, Shigeta Y, Kikkawa R, Goto Y. Clinical investigation of epalrestat, an aldose reductase inhibitor, on diabetic neuropathy in Japan: Multicenter study. J Diabetes Complicat. 2006;10:168–172. doi: 10.1016/1056-8727(96)00113-4. [DOI] [PubMed] [Google Scholar]
- 52.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 53.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat Protocols. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shoichet BK. Interpreting steep dose-response curves in early inhibitor discovery. J Med Chem. 2006;49:7274–7277. doi: 10.1021/jm061103g. [DOI] [PubMed] [Google Scholar]
- 55.Shoichet BK. Screening in a spirit haunted world. Drug Discov Today. 2006;11:607–615. doi: 10.1016/j.drudis.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jadhav A, Ferreira RS, Klumpp C, Mott BT, Austin CP, Inglese J, Thomas CJ, Maloney DJ, Shoichet BK, Simeonov A. Quantitative analyses of aggregation, autofluorescence, and reactivity artifacts in a screen for inhibitors of a thiol protease. J Med Chem. 2010;53:37–51. doi: 10.1021/jm901070c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coan KE, Shoichet BK. Stability and equilibria of promiscuous aggregates in high protein milieus. Mol Biosyst. 2007;3:208–213. doi: 10.1039/b616314a. [DOI] [PubMed] [Google Scholar]
- 58.Seidler J, McGovern SL, Doman TN, Shoichet BK. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J Med Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 59.Oka S, Kato J, Moss J. Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J Biol Chem. 2006;281:705–713. doi: 10.1074/jbc.M510290200. [DOI] [PubMed] [Google Scholar]
- 60.Ono T, Kasamatsu A, Oka S, Moss J. The 39-kDa poly(ADP-ribose) glycohydrolase ARH3 hydrolyzes O-acetyl-ADP-ribose, a product of the Sir2 family of acetyl-histone deacetylases. Proc Natl Acad Sci U S A. 2006;103:16687–16691. doi: 10.1073/pnas.0607911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niere M, Kernstock S, Koch-Nolte F, Ziegler M. Functional Localization of Two Poly(ADP-Ribose)-Degrading Enzymes to the Mitochondrial Matrix. Mol Cell Biol. 2007;28:814–824. doi: 10.1128/MCB.01766-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mueller-Dieckmann C, Kernstock S, Lisurek M, von Kries JP, Haag F, Weiss MS, Koch-Nolte F. The structure of human ADP-ribosylhydrolase 3 (ARH3) provides insights into the reversibility of protein ADP-ribosylation. Proc Natl Acad Sci USA. 2006;103:15026–15031. doi: 10.1073/pnas.0606762103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abdelkarim GE, Gertz K, Harms C, Katchanov J, Dirnagl U, Szabo C, Endres M. Protective effects of PJ34, a novel, potent inhibitor of poly(ADP-ribose) polymerase (PARP) in in vitro and in vivo models of stroke. Int J Mol Med. 2001;7:255–260. [PubMed] [Google Scholar]
- 64.Steffen JD, Coyle DL, Damodaran K, Beroza P, Jacobson MK. Discovery and Structure-Activity Relationships of Modified Salicylanilides as Cell Permeable Inhibitors of Poly(ADP-ribose) Glycohydrolase (PARG) J Med Chem. 2011 doi: 10.1021/jm200325s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carlson EE, May JF, Kiessling LL. Chemical probes of UDP-galactopyranose mutase. Chem Biol. 2006;13:825–837. doi: 10.1016/j.chembiol.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 66.Menard L, Poirier GG. Rapid assay of poly(ADP-ribose) glycohydrolase. Biochem Cell Biol. 1987;65:668–673. doi: 10.1139/o87-088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.